Abstract
Studies using cultured melanoma cells and patient tumor biopsies have demonstrated deregulated PI3 kinase-Akt3 pathway activity in ~70% of melanomas. Furthermore, targeting Akt3 and downstream PRAS40 has been shown to inhibit melanoma tumor development in mice. Although these preclinical studies and several other reports using small interfering RNAs and pharmacological agents targeting key members of this pathway have been shown to retard melanoma development, analysis of early Phase I and Phase II clinical trials using pharmacological agents to target this pathway demonstrate the need for (1) selection of patients whose tumors have PI3 kinase-Akt pathway deregulation, (2) further optimization of therapeutic agents for increased potency and reduced toxicity, (3) the identification of additional targets in the same pathway or in other signaling cascades that synergistically inhibit the growth and progression of melanoma, and (4) better methods for targeted delivery of pharmaceutical agents inhibiting this pathway. In this review we discuss key potential targets in PI3K-Akt3 signaling, the status of pharmacological agents targeting these proteins, drugs under clinical development, and strategies to improve the efficacy of therapeutic agents targeting this pathway.
Introduction
Aberrant expression and activity of the PI3K-Akt pathway proteins has been shown to promote melanomagenesis.Citation1–Citation7 The PI3K-Akt3 signaling cascade regulates initiation, progression and invasion by inhibiting cell senescence and apoptosis pathways, and by inducing cell survival cascades in melanoma cells ().Citation1,Citation8–Citation10 Therefore, members of this signaling cascade are attractive targets for inhibiting melanoma.Citation11–Citation13 Prior studies using siRNA and pharmacological inhibitors have demonstrated that targeting Akt3 and downstream PRAS40 inhibits melanoma development in mice by triggering apoptosis.Citation6,Citation14 Furthermore, immunohistochemical staining methods comparing pAkt levels in advanced stage patient tumors with matched normal controls demonstrated elevated pAkt expression in ∼77% of melanomas.Citation15 Similarly, enhanced expression of p85 and p110a subunits of PI3K and pPRAS40 have also been reported in melanomas compared with melanocytes.Citation14,Citation16 Therefore strategies targeting members of PI3K-Akt pathways represent a promising approach to develop clinically effective therapies for the treatment of melanoma.
Overview of the PI3K-Akt Signaling in Melanomas
The PI3K-Akt pathway is activated by (1) nutrients, (2) hormones, or (3) growth factors ().Citation2,Citation3,Citation17 Binding of mitogens to G-protein-coupled receptors activate PI3K, which in turn triggers the generation of PtdIns(3,4,5)P3 molecules by phosphorylating phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] on the 3-OH group.Citation17,Citation18 PtdIns(3,4,5)P3 binds to the PH domain of Akt, thereby facilitating its translocation to the plasma membraneCitation2,Citation3,Citation17 (). Akt activation is initiated when the translocated Akt is phosphorylated on T308 residue by membrane localized phosphoinositide-dependent kinase-1 (PDK-1).Citation2,Citation3,Citation17 However, complete activation is achieved only when PDK2 the second phosphorylation site S473 is phosphorylated by rapamycin-insensitive mTORC2.Citation19,Citation20 Additional studies have shown that prior phosphorylation of tyrosine 315 (Y315) and 326 (Y326) residues is a pre-requisite for the activation of Akt kinases.Citation21 Although it is known that activated Akt is present in both the cytosol and nucleus, further studies are necessary to delineate the differential contributions of Akt in the cytosol vs. nucleus and how they affect melanoma cell growth. Activated Akt phosphorylates GSK3α/β, PRAS40, NFκB and many other proteins harboring the Arg-X-Arg-X-X-[Ser/Thr]-Hyd (where X is any amino acid and Hyd is a bulky hydrophobic amino acid) consensus sequence ()Citation2,Citation3,Citation7,Citation17. Even though several Akt substrate proteins have been identified, few have been implicated in the genesis or progression of melanoma.Citation2,Citation3,Citation17 Furthermore, while phosphorylation of some substrates induces catalytic activity, phosphorylation of other substrates may inhibit it. Therefore, care must be taken in relating the phosphorylation status of Akt substrates to the molecular mechanisms underlying the development of melanoma.
PI3K-Akt pathway activity is negatively regulated by phosphatase and tensin homolog deleted on chromosome 10, referred as PTEN. PTEN is a unique dual specificity phosphatase that dephosphorylates proteinsCitation22–Citation24 and hydrolyzes the secondary messenger inositol phosphates [PtdIns(3,4,5)P3].Citation23,Citation25 Preclinical and clinical studies have demonstrated inactivation, loss or decreased protein expression of PTEN, in 29–43% of melanoma cell lines and in biopsy specimens from melanoma patients.Citation26,Citation27 Loss of PTEN promotes Akt activity, increases resistance to chemotherapeutic agents and inhibits cellular apoptosis.Citation5,Citation14,Citation28 Therefore, restoring functionally active PTEN expression in melanomas has the potential to improve the therapeutic efficacy of agents triggering apoptosis.Citation5,Citation14,Citation28 For example, transfer of a portion of chromosome-10 containing PTEN gene has been shown to decrease pAkt3 and downstream pPRAS40 and to sensitize melanoma cells to the apoptosis inducing agent staurosporine.Citation5,Citation14 Another study showed elevated phosphorylation of Akt3, induction of BCl2 and enhanced chemoresistance in melanocytes and early melanoma WM35 cells only when PTEN was inhibited.Citation5,Citation6,Citation14,Citation29,Citation30 Furthermore, mice lacking PTEN but expressing conditionally induced V600EB-Raf form spontaneous melanomas, whereas induction of PTEN expression reduced tumor-forming ability.Citation31 In addition, loss of PTEN increased melanoma cell invasion and migration by shifting the phosphorylation status from Akt3 to Akt2 and by downregulating E-cadherin.Citation32 Therefore, an unanswered question remains as to why inhibiting PTEN exerts differential effects on Akt3 and Akt2 activities in melanomas.
PTEN also regulates the synthesis of proteins involved in melanoma proliferation and survival.Citation33 For example, PTEN mediates regulation of phosphorylation of eukaryotic initiation factor-2α (eIF2α) and induces anti-proliferation and apoptotic signals in isogenic melanoma cells.Citation33 Melanoma cells lacking PTEN have low levels of phosphorylated eIF2 compared with cells expressing wild-type PTEN. Reconstitution of wild-type or phosphatase-defective PTEN in PTEN-null human glioblastoma cells enhances phosphorylation of eIF2α by binding to the PDZ domain of PTEN.Citation33 Phosphorylated eIF2α inhibits eIF2B and blocks the initiation of translation and overall protein synthesis.Citation33 PTEN can also inhibit cell proliferation by altering cell cycle progression through G1 to S phase.Citation30,Citation34–Citation36 Cells containing functionally active PTEN express high levels of p27 and low levels of cyclin-D1 and cyclin-D2 proteins.Citation36–Citation38
Therapeutic Targets in the Akt Pathway
There has been significant interest in identifying key members of the PI3K-Akt signaling cascade promoting melanoma development, since downregulation of this pathway appears to be a promising therapeutic strategy. In this section of review an overview of several attractive targets in this signaling cascade is provided ().
Phosphotidyl inositol 3 kinases (PI3K).
PI3K is one of the key targets in the PI3K-Akt3 signaling cascades.Citation39 PI3Ks are a family of intracellular lipid kinases that phosphorylate the 3′ hydroxyl group of phosphatidylinositols (Pis) and phosphoinositides.Citation16,Citation40 Based on substrate specificity and structure, PI3K proteins have been categorized into class-I, class-II and class-III kinases.Citation40,Citation41 Whereas activity of class-Ia PI3K is triggered by growth factor receptor tyrosine kinases, class-Ib proteins are activated by G protein-coupled receptors. Class-Ia PI3K is a heterodimer comprising a p85 regulatory and p110 catalytic subunits.Citation40,Citation41 Upon growth factor stimulation class-Ia PI3Ks phosphorylates PtdIns (4,5)P2 at the 3′ position converting it into PtdIns(3,4,5)P3, which binds to the PH domain containing PDK1 and Akt proteins, thereby facilitating translocation of these proteins to the cell membrane. Class-I PI3Ks also exhibit protein kinase activity, but activation of each of the two class-I isoforms has differing consequences.Citation42,Citation43 For example, class-Ia PI3K phosphorylates insulin receptor substrate-1 (IRS-1), whereas class-Ib PI3K activates the MAPK signaling cascade.Citation40,Citation41
Activation of PI3K is a key event for the initiation of Akt signaling in melanomas.Citation16,Citation44 PI3K is activated by ligand-dependent activation of tyrosine kinase receptors, G-protein-coupled receptors, or integrins (). Several studies have shown that these receptors are overexpressed in many cancers including melanomas.Citation45,Citation46 However, in the 10–20% of melanomas that express constitutively active Ras proteins, PI3K-Akt signaling cascade is also activated through a receptor-independent mechanism.Citation47–Citation49 Therefore, targeting activated PI3K inhibits melanoma development.Citation44,Citation50 For example, overexpression of a deleted subunit of PI3K (Deltap85) reduced PI3K signaling in the melanoma cell line G361.Citation51 Similarly, targeting PI3 kinase using siRNAs or pharmacological agent ZSTK474 inhibited melanoma tumor development.Citation40,Citation41 Likewise, targeting PI3K using nanoparticle encapsulated LY-294002 inhibited xenografted mouse melanoma cell growth.Citation52 Although these and other studies demonstrate the critical role of PI3K in the regulation of melanoma development, studies have failed to demonstrate consistently elevated expression of this protein in tumor specimens from melanoma patients. Furthermore, several contradictory results have been reported regarding PI3K activity in melanomas with some studies reporting little or no expression of PI3K in patient samples, while others have demonstrated high expression levels.Citation40,Citation41 In a recent study, investigators quantitatively assessed expression of PI3K in 523 melanoma and 540 nevi specimens and found that p85 and p110 subunit expression was higher in melanomas than in nevi ().Citation40,Citation41 However, neither subunit could be used as a prognostic marker in patients with localized or metastatic melanoma. Other mechanisms regulating PI3K activity in melanomas include loss of negative regulators such as PTEN or mutational activation ()
Protein kinase B gamma or Akt.
Three isoforms of Akt have been described: Akt1 (PKBα), Akt2 (PKBβ) and Akt3 (PKBγ).Citation46,Citation53 These three variants share > 80% homology and have common and isoform specific functions. Structurally, all three isoforms contain pleckstrin homology (PH), catalytic and regulatory domains.Citation45,Citation46,Citation53–Citation59 Whereas the N-terminal PH domain mediates protein-protein and protein-lipid interactions,Citation60,Citation61 the central catalytic domain (CD) contains a key initiator phosphorylation site (T308) responsible for enzymatic activity.Citation62,Citation63 The carboxy terminal hydrophobic regulatory domain (RD) contains a second phosphorylation site, located on a serine residue (S472), whose phosphorylation completes kinase activation. Although other phosphorylation sites may also regulate the catalytic activity of Akt kinases, their significance remains to be determined.Citation64 In a recent study, E40K mutation in the PH domain has been shown to enhance the enzymatic activity of Akt3 in melanomas.Citation14,Citation65 Splice variants of Akt3 lacking serine 472 have also been identified but the functional significance remains unknown.Citation66,Citation67
Akt is a positive regulator of cell proliferation and survival.Citation7 Aberrant activation of Akt through (1) mutations in PI3K, (2) overexpression, or (3) deletion of PTEN, and (4) mutations in Akt3 has been observed in melanomas.Citation2,Citation3,Citation5,Citation16 Akt activity is also regulated by posttranslational modifications involving phosphorylation or ubiquitination.Citation68 In addition, physical interactions with effector proteins can regulate Akt3 activity in melanomasCitation39,Citation45,Citation46,Citation53–Citation59,Citation69,Citation70. Preferential activation of Akt3 has been reported in melanomasCitation6 but the mechanistic basis for this isoform specific regulation is only beginning to emerge.Citation71
Akt activity is also regulated through dephosphorylation of Thr308 and Ser473 by phosphatases. Phosphatases control the overall phosphorylation status of Akt in melanoma cells.Citation72 Key phosphatases in melanomas include (1) PTEN, (2) the protein phosphatase 2A (PP2A), and (3) PH domain leucine-rich repeat protein phosphatase (PHLPP). Whereas PTEN dephosphorylates PtdIns(3,4,5)P3 to form PtdIns(4,5)P2, PP2A and PHLPP dephosphorylate T308 and S473, respectively, to deactivate Akt kinases.Citation69,Citation73,Citation74 While the role of PTEN in melanomas has been studied extensively, little is known regarding PP2A and PHLPP. One study using mouse melanoma cells showed that PP2A expression is regulated by methylation of a catalytic subunit.Citation72 Inhibition of methylation by okaidic acid then reduced PP2A activity, resulting in increased Akt phosphorylation and proliferation.Citation72 Agents such as chloroethylnitrosourea (CENU) augment methylation of PP2A to reduce cell proliferation and survival.Citation72 Therefore, PP2A promethylating agents are potential candidate melanoma inhibitors. Other mechanisms such as ubiquitination also have been shown to regulate Akt activity in several cancers,Citation70,Citation75 but in melanomas this mechanism of regulation remains to be explored.
Recently, mutations in Akt have been reported in a number of malignancies including melanoma. For example, growth-promoting somatic mutations such as E17K in the PH domain of Akt have been described in a small minority (8%) of cancers of breast, colon (6%), ovary (2%) and skin (1.5%).Citation69,Citation70,Citation76–Citation78 Similarly, E17K mutation in Akt3 protein is reported in melanoma patient tumors and in melanoma cell lines. Analysis of 137 human melanoma specimens and 65 human melanoma cell lines showed expression of mutant E17KAkt3 in two melanoma tumors and in two melanoma cell lines.Citation77 Expression of E17KAkt3 in melanoma cell lines demonstrated the presence of active phosphorylated Akt.Citation77 Although E17KAkt3 found to increase phosphorylated Akt levels, it is unknown whether this could be used as a marker for diagnosis and for measuring the treatment efficacy of pharmacological agents. In addition, very low levels of prevalence of this mutant protein in human melanomas raises concerns whether this is a random event or has any bearing on disease development and drug resistance. Likewise, E49K mutation was reported in a subset of bladder cancer patients,Citation79 but the significance of these mutations remain unknown.
Downstream targets of Akt. Akt substrates can be cytoplasmic or nuclear. The number of proteins found to be regulated by Akt continues to increase as the search for putative substrates bearing Arg-X-Arg-X-X-[Ser/Thr]-Hyd (where X is any amino acid and Hyd is a bulky hydrophobic amino acid) identifies them ().Citation80,Citation81
Activated Akt3 phosphorylates as many as 9,000 substrate proteins, thereby regulating diverse processes such as cell survival, proliferation and migration and impacting chemosensitivity.Citation82 For example, inhibitory phosphorylation of GSK3β promotes cell cycle progression by elevating cyclin D1 levels.Citation83,Citation84 Likewise phosphorylation of PRAS40 at threonine 246 (T246) sequesters PRAS40 thereby prevents its interactions with mTORC1, which increase the nutrient status of various cells.Citation14,Citation85,Citation86 Phosphorylation of PRAS40 has been shown to correlate with Akt activity in melanomas. In another study, phosphorylation of V600EB-Raf by Akt3 decreased its activity to levels promoting rather than inhibiting cell proliferation.Citation65,Citation87 Mechanistically, Akt3 phosphorylates V600EB-Raf on S364 and/or S428, thereby reducing its catalytic activity.Citation65 Ectopic expression of V600EB-Raf in melanocytes induces cellular senescence by elevating the levels of MAPK activity and by upregulating the expression of cyclin-dependent kinase (cdk) inhibitors.Citation88,Citation89 Therefore, genetic changes such as loss of tumor suppressor genes PTEN, p53 or p16INK4A, and upregulation of Akt3 are needed to enable quiescent melanocytic nevi to develop into melanomas.Citation65,Citation90
Other substrates phosphorylated by Akt include apoptotic signal kinase (ASK1), B cell leukemia/lymphoma-2 interacting mediator of cell death (Bim), B cell leukemia/lymphoma-2 associated death agonist (Bad), murine double minute-2 (MDM-2), p21 cyclin dependent kinase inhibitory protein (p21 Cip1), X-linked inhibitor of apoptosis (XIAP) and the forkhead box O3 (Foxo3a) transcription factor.Citation2,Citation7 Akt also phosphorylates IκB kinase (IκK), to enable translocation of NFκB into nucleus. Targeting NFkB and upstream IkKa can inhibit melanoma development.Citation91,Citation92 Akt can also phosphorylate and activate cyclic AMP response element binding protein (CREB) and mammalian target of rapamycin (mTOR), thereby increasing cell survival and drug resistance.Citation2,Citation7
Preclinical Agents Targeting Akt Signaling
Pharmacological agents inhibiting Akt activity have been identified using agent screening and molecular modeling approaches to test their efficacy in vitro and in vivo. Although several agents found effective on cultured cells, only a few showed potency in preclinical trials for inhibiting melanoma tumors growth in mice and causing no noticeable systemic toxicity. Among various agents tested preclinically, only a minority have been evaluated clinically. In the following sections, we provide an overview of the current status of various pharmacological agents inhibiting Akt activity, as well as any potential drawbacks limiting further development.
Lipid based inhibitors of Akt.
Lipid based inhibitors of Akt have exhibited biological activity against melanoma in preclinical and clinical trials.Citation94 Notable examples include (1) alkylphospholipids (ALPs)—perifosine, miltefosine and edelfosine; (2) phosphatidylinositol ether lipid analogs (PIAs); and (3) D-3-deoxy-phosphatidylmyoinositol-1-(R)-2-methoxy-3-octadecyloxyropyl hydrogen phosphate (PX-316)Citation94–Citation96 ().
Alkylphospholipids (APLs). Alkylphospholipids are membrane-permeable ether lipids resistant to degradation by cellular phospholipases.Citation94–Citation96 Accumulation of these synthetic APLs alters denovo phospholipid synthesis as well as translocation of Akt.Citation94–Citation96 Perifosine is a synthetic oral alkylphospholipid inhibitor of Akt that has been tested in several preclinical and clinical trials targeting cancers of the skin, lung, prostate, colon and breast.Citation94–Citation96 Perifosine inhibits translocation of Akt to cell membranes, inhibiting phosphorylation of Akt by PDK1 and PDK2, thereby sensitizing cancer cells to apoptosisCitation94–Citation96 ().
Phosphatidylinositol ether lipid analogs. These agents comprise a class of inhibitors that are structurally similar to PI(3,4)P2 and PI(3,4,5)P3 and interact with PH domain of Akt.Citation97,Citation98 Each is composed of an inositol ring, a linker phosphate or carbonate and an ether lipid side chain.Citation97,Citation98 Docking and modeling studies predicted that these analogs bind to PH domain of Akt in an altered position compared with PtdIns(4,5)P2 and PtdIns (3,4,5)P3s, thereby inhibiting translocation to the plasma membrane.Citation94,Citation97,Citation98 Phosphatidylinositol ether lipids inhibit active Akt without affecting total Akt levels, consequently inducing apoptosis, and decreasing chemo- and radiation-resistance. These inhibitors are being evaluated in clinical trials.
Other inhibitors targeting the PH domain are also being developed and evaluated preclinically as well as clinically. For example, PX-316 (D-3-Deoxy-phosphatidyl-myoinositol-1-(R)-2-methoxy-3-octadecyloxyropyl hydrogen phosphate), a lipid based inhibitor of Akt, has been found to inhibit xenografted breast and colorectal cancers;Citation99 however, its efficacy for killing melanoma cells has not been tested. Furthermore, clinical pharmacokinetic and pharmacodynamic properties have not been established for this agent.
Small molecule inhibitors of Akt.
High-throughput screening approaches have identified selenium containing ISC-4, PBISe, p-XSC; PHT-427, API-1, API-2, API-59CJ-OMe, A443654 and canthine as small molecule pharmacological inhibitors of Akt. These agents inhibited cultured cells and tumors development in animals and are discussed briefly below and in .
ISC-4. Isoselenocyanate-4, a derivative of naturally occurring phenyl butyl isothiocyanate (PBITC), is a selenium-containing cancer therapeutic and chemopreventive agent found effective for inhibiting melanoma and other malignancies in preclinical studies.Citation93,Citation100,Citation101 ISC-4 is synthesized by substituting selenium for sulfur in PBITC.Citation100 Furthermore, preclinical studies have also demonstrated that topical application of ISC-4 compared with PBITC effectively inhibits melanocytic lesions growing in artificial skin reconstructs, a widely used in vitro model for studying melanoma tumors progression in laboratory, and subcutaneous xenografted melanoma tumors ().Citation93,Citation100,Citation101 Intraperitoneal administration of ISC-4 retards the growth of xenografted melanoma tumors with negligible toxicity.Citation93,Citation100,Citation101 Mechanistically, ISC-4 has been shown to inhibit Akt3 signaling in melanomas and induce apoptotic cell death.Citation93,Citation100,Citation101
PBISe. PBISe is an isosteric analog of the iNOS inhibitor, PBIT.Citation102–Citation104 In vitro and in vivo studies demonstrated that topical application of PBISe, but not PBIT inhibited the growth of early melanocytic lesions growing in skin reconstructs and subcutaneous xenografted tumors ().Citation102–Citation104 Intraperitoneal administration of PBISe reduced the growth of xenografted melanoma tumors in mice.Citation102–Citation104 Unlike PBIT, PBISe has inhibitory activity against melanoma. In addition to iNOS inhibition, PBISe blocks Akt signaling and induces pErk1/2 levels in melanomas, resulting in apoptosis and cell cycle arrest.Citation102–Citation104
p-XSC. p-XSC is a selenium containing selenocyanate that has been shown in preclinical studies to have biological activity against melanoma and other malignancies.Citation105,Citation106 Dietary administration of p-XSC inhibited experimental melanoma metastasis in a B16-F10 melanoma model.Citation106 Mice treated with 4, 8 and 15 mg/kg of p-XSC orally developed fewer metastatic tumors compared with control animals ().Citation106 In addition, mice fed with 8–15 mg/kg p-XSC developed significantly smaller tumors compared with control animals.Citation106 Importantly, p-XSC has been shown to induce apoptosis in melanoma tumors without exhibiting similar effects in normal tissues.Citation106
PHT-427. PHT-427 {4-dodecyl-N-(5-(5-(methyl(7-nitrobenzol[c][1,2,5]oxadiazol-4-yl)aminopentyl-1,3,4-thiadiazol-2-yl) benzenesulfonamide}, which binds to the pleckstrin homology (PH) domain, has been shown to inhibit tumors harboring active Akt ().Citation107 Intraperitoneal administration of PHT-427 blocked the growth of xenografted pancreatic tumors ().Citation108 Recently, analogs of PHT-427 with increasing carbon chain length (from C-4 to C-16) have been synthesized and antitumor activity evaluated.Citation108 The C-12 analog was found to exhibit potent Akt and PDPK1 (phosphoinositide-dependent protein kinase-1) inhibitory activity compared with other derivatives.Citation108
API-1. API-1 (NSC-177223) is a pyrido[2,3-d]pyrimidine structurally related to antibiotic sangivamycin ().Citation109 API-1 is a pan-Akt inhibitor that binds to the pleckstrin homology domain of Akt, preventing translocation to cell membranes. In vitro studies demonstrated its selectivity for Akt but not for upstream activators PI3K, PDK1 and PDK2 (mTORC2) or structurally related kinases PKC, SGC, PKA, STAT3, ERK-1/2 or JNK.Citation109 In an animal model, intraperitoneal administration of 10 mg/kg body weight API-1 selectively inhibited xenografted OVCAR3 and PANC1 growth but not OVCAR-5 or COLO357, demonstrating selectivity of this agent for cell lines expressing high levels of Akt activity ().Citation109 Since Akt activity is elevated in most melanoma, API-1 would be predicted to have broad inhibitory activity against melanoma.
API-2. API-2 (triciribine) is a nucleoside analog inhibiting phosphorylation of Akt without affecting the activities of oncogenic kinases PI3K, PDK-1, PKC, PKA, STAT3, Erk1/2 and JNK ().Citation110 API-2 mediated Akt inhibition induced apoptosis and retarded growth of xenografted melanoma, breast, prostate, ovarian and pancreatic tumors.Citation110 Prior to the discovery of its Akt inhibitory activity, triciribine had been shown to inhibit DNA and protein synthesis.Citation111 Although Phase I and Phase II trials have demonstrated clinical efficacy, further study was discontinued due to drug-related toxicities such as hepatotoxicity, hyperglycemia, thrombocytopenia and hypertriglyceridemia, which may have resulted, in part, from the high doses of the agent employed.Citation112,Citation113 Triciribine has been found to accumulate in various tissues, especially liver, gallbladder and pancreas.Citation113 A preclinical study demonstrated anti-tumor activity of API-2 for cell lines expressing high Akt activity when administered at low concentrations.Citation110 For example, intraperitoneal administration of 1 mg/kg body weight of API-2 daily retarded ovarian and pancreatic tumor growth in animals, indicating the relative potency API-2 in reducing tumor development.Citation110 It appears that clinical studies may be warranted to determine whether, in fact, API-2 has antitumor activity in humans at doses associated with acceptable tolerability.
Other Akt inhibitors identified from compound screens include API-59CJ-OMe and KP372.Citation114,Citation115 These agents inhibit Akt and induce apoptosis in various cancer cell lines. Investigators are currently testing preclinical efficacy of these agents in animal models.
BI-69A11. Identified using a virtual docking approach in which 50,000 compounds were screened, BI-69A11 inhibits Akt activity by binding at the ATP catalytic site ().Citation116 BI-69A11 had no effect on the activity of protein kinases, Abl1, p38a, JNK and PI3K. BI-69A11 forms three hydrogen bonds with Lys181, Thr292 and Glu279 in Akt, thereby inhibiting its activity.Citation116 In vitro, BI-69A11 inhibited Akt activity in melanoma cells harboring mutant PTEN and having elevated pAkt levels.Citation116 Furthermore, BI-69A11 reduces pPRAS40, a direct substrate of Akt involved in apoptosis regulation in melanomas.Citation116 In animals, intraperitoneal administration of BI-69A11 retarded xenografted melanoma tumors by inducing apoptosis ().Citation116 Although these preclinical studies have demonstrated efficacy of BI-69A11 for inhibiting Akt activity and melanoma tumor growth, no clinical studies have been reported using this compound.
2-pyrimidyl-5-amidothiophenes. High throughput screening of 2-pyrimidyl-5-amidothiophene derivatives led to discovery of a compound with selectivity for Akt3Citation117 (). Although this ATP-competitive inhibitor was found to target Akt3 at concentrations as low as 3 µM, further validation will be necessary to establish specificity, as structurally similar derivatives showed selectivity to several other kinases.Citation117 For example, a derivative containing a similar backbone inhibited Akt1, 2, and 3 at low nanomolar concentrations in DOV13 ovarian cancer cells.Citation117 In addition, studies using the UACC 903 melanoma cell line have showed that this derivative, compared with other structurally related compounds, had 30-fold greater selectivity toward PKA and other AGC family kinases.Citation117
A-443654 and A-423795. A-443654 is an indazole-pyridine Akt inhibitor that synergizes with chemotherapeutic agents doxorubicin, campothecin and paclitaxelCitation118–Citation120 (). Although A-443654 was identified as an Akt inhibitor, western blot analysis demonstrated elevated pAkt expression following A-443654 treatment, indicating that pAkt level may not be an accurate indicator of inhibitory potency.Citation121 A-423795 is another Akt inhibitor synthesized from the indazole-pyridine compound series.Citation118 This compound inhibits Akt activity in cultured cells and retards growth of xenografted tumorsCitation118 (). Toxicity manifest as lethargy, weight loss and skin irritation at the compound injection site have discouraged investigators from undertaking clinical trials in humans.Citation94
GSK690693. GSK690693 is a novel ATP-competitive pan-Akt inhibitor demonstrated to exhibit anti-tumor activity against xenografted human ovarian (SKOV-3), prostate (LNCaP) and breast (BT474 and HCC-1953) carcinomas.Citation122 Among over 250 kinases that have been screened in vitro, GSK-690693 inhibited Akt1, 2 and 3 with high selectivity at low nanomolar concentrations of 2, 13 and 9 nM respectively for each Akt isoformCitation122 (). However, this inhibitor was less selective for AGC family kinases such as PKA and PKC.Citation122 Interestingly, cell lines sensitive to GSK690693 had high pAkt activity.Citation122 Efficacy of GSK690693 for inhibiting growth of melanoma cell line or melanoma tumor xenografts has not been tested. A recent study evaluating this inhibitor in a panel of in vitro and in vivo xenograft models of the Pediatric Preclinical Testing Program (PPTP) demonstrated that GSK690693, as a single agent, only modestly inhibited growth of solid tumors.Citation123
Hexamethylene bisacetamide (HMBA). HMBA is a hybrid polar compound inhibiting Akt and MAPK signaling cascades.Citation124 In addition, HMBA also represses NFkB and induces transformed cells differentiation.Citation124 Phase I and Phase II clinical trials examining HMBA for the treatment of acute myelogenous leukemia demonstrated significant dose-limiting toxicity, consequently limiting its clinical utility.Citation125 The biological activity of HMBA in melanoma has not yet been evaluated ().
Sertraline. Being a member of the selective serotonin reuptake inhibitor class of anti-depressants, sertraline possesses Akt inhibitory activity and acts as a serotonin transporter antagonist.Citation126 Human melanoma cells treated with sertraline exhibited low pAkt and phosphorylated p70 S6 kinase activities.Citation126 In addition, sertraline treatment induced endoplasmic reticulum stress triggering cellular apoptosis. In animals, intraperitoneal administration of 1 mg/day setraline inhibited xenografted A375 tumor development.Citation126 Although this antidepressant inhibited melanoma tumor development, further investigation in preclinical as well as clinical models are warranted to establish its activity against melanoma, mechanism of action for tumor inhibition and potential toxicity.
Agents targeting mTOR.
mTOR is a 290 kDa cytosolic serine/threonine kinase expressed in most tissues.Citation127 mTOR is one of the downstream members of Akt involved in regulation of the cell cycle, protein synthesis and angiogenesis. Therefore it acts as a nutrient sensor and regulator of translation.Citation127 Upon stimulation, mTOR activates p70S6 kinase and inactivates 4E-BP1 to promote cell proliferation and survival.Citation127 Rapamycin and derivatives of this compound are well-known inhibitors of mTOR ( and ).Citation127,Citation128 These inhibitors bind to the immunophilin FK-506 binding protein, FKBP-12 to inhibit mTOR activity. CCI-779 (Temsirolimus) and RAD001 (Everolimus) are rapamycin derivatives that inhibit the PI3K-mTOR pathway.Citation128 CCI-779 is a water-soluble ester of rapamycinCitation128 that has potent anti-tumor activity for several tumor types including melanoma.Citation128
A possible concern is that, since mTOR can suppress Akt activity through a negative feedback loop, inhibition of mTOR might activate Akt and induce resistance.Citation128,Citation129 This issue should be considered when using mTOR inhibitors. One strategy to circumvent mTOR-mediated resistance is to combine it with an agent targeting Akt. For example, targeting PI3K and mTOR with NVP-BAG956, NVP-BBD130 or NVP-BEZ-235 induced G1 growth arrest mediated by p27 thereby reducing cyclin D1 in melanoma cells.Citation130 However, inhibition of either PI3K or mTOR alone using ZSTK474 or rapamycin, respectively, only marginally reduced cell proliferation.Citation130 Furthermore, oral administration of NVP-BBD130 and NVP-BEZ235 efficiently inhibited B16 melanoma tumor growth in primary tumors and in lymph node metastasis.Citation130 Currently, NVP-BEZ235 is being evaluated in Phase I/II clinical trials for the treatment of metastatic melanoma.Citation130 OSI-027 (Osi Pharmaceuticals) and OXA-01 are novel inhibitors of mTORC1 and mTORC2.Citation131 Preclinical studies using 75 mg/kg OXA-01 demonstrated retardation of xenografted MDA-MB-231 tumor development.Citation132 Efficacy of these agents against melanoma remains to be established.
Hurdles Associated with Moving Preclinical Agents Targeting Akt Signaling into the Clinic
Although targeted inhibition of Akt reduces melanoma development, it is possible that under certain circumstances, it has the potential to induce metastasis, possibly due to differential effects of Akt isoforms on melanoma tumorigenesis or metastasis.Citation32,Citation133 For example, targeted inhibition of Akt2 reduced melanoma metastasis, while downregulation of Akt3 increased metastasis in preclinical animal models.Citation32 Although these preclinical studies highlight some of the potential problems associated with Akt inhibition due to the differential effects of the isoforms in particular cancer cells, the relevance of these observations to the clinical setting remain uncertain but should be considered when planning a clinical trial.
Toxicity associated with PI3K-Akt pathway inhibitors.
Toxicity associated with pharmacological agents targeting PI3-Akt pathway is a major concern hindering the clinical development of these compounds. Since PI3K-Akt signaling is a key pathway regulating many processes including cell survival, glucose metabolism, protein synthesis and cell motility, it is important to consider the effect of targeting this pathway on normal cells and tissues and the resulting consequences for patients. The next section describes a variety of toxicities associated with PI3K-Akt pathway inhibition.
Hyperglycemia. Although Akt is a key regulator of cell survival and proliferation, it is also involved in the regulation of glucose uptake and protein synthesis.Citation140 Therefore, care must be taken while selecting Akt inhibitors for clinical use.Citation141 Preclinical studies have shown that targeting Akt or mTOR can increase hyperglycemia and diabetes due to effects on peripheral tissues and pancreatic β cells.Citation142,Citation143 In addition, Akt-2 null mice exhibited insulin resistance, hyperglycemia, hyperinsulinemia and glucose intolerance.Citation144 Another study has also demonstrated that inhibiting mTOR induced diabetes. Similarly, inhibiting Akt activity using the pan Akt inhibitor GSK690693 induced glucose dysregulation in the form of transient hyperglycemia as well as hyperinsulinemia in animals.Citation142
Gastrointestinal and hematological toxicity. Akt pathway and mTOR inhibitors have been associated with both hematologic and, to a lesser extent, gastrointestinal toxicity.Citation145 In a Phase II study performed in patients with advanced soft tissue sarcoma, administration of oral perifosine in a nine 21-d-cycles treatment induced hematologic and gastrointestinal toxicities.Citation96 Among 16 evaluable patients out of 17 total, hematologic toxicity was mild. However, mild grade 1 or 2 gastrointestinal toxicity in the form of nausea, vomiting and/or diarrhea was frequently observed. No significant biochemical abnormalities were reported using this treatment modality.Citation146 Similarly, a recent Phase I trial showed hematologic toxicity in the form of severe grade 3 to 4 thrombocytopenia when the PKC β and PI3K/Akt pathway inhibitor, enzastaurin, were administered to patients suffering from glioblastoma multiforme.Citation147
mTOR inhibitors have been examined in a number of clinical trials, affording more information about gastrointestinal and hematologic toxicities. Everolimus, an FDA approved agent, has commonly been reported to cause cytopenias, as well as nausea, vomiting, diarrhea, stomatitis or occasionally more serious gastrointestinal toxicities such as pancreatitis.Citation148,Citation149 Temsirolimus, yet another FDA approved agent, is accompanied by similar hematologic and gastrointestinal toxicities. Elevation of liver enzymes and cholangitis has also been observed.Citation150–Citation153 The results of several ongoing clinical trials will provide additional information regarding gastrointestinal and hematologic toxicity.
Clinically Evaluated Agents Targeting Akt Signaling
Few clinical trials in melanoma patients have examined the activity of agents that act primarily or directly through inhibition of PI3K-AKT activity. In contrast, AKT activity has been used as a biomarker of efficacy of a wide range of treatments in the clinical setting.Citation154–Citation156 Indeed, indirectly blocking the AKT pathway may represent an important contribution to an agent's biological activity. Agents being evaluated clinically that are believed to act primarily by inhibition of the AKT pathway—or on immediately downstream effectors—will be summarized in the next section.
Perifosine and miltefosine.
Phase I clinical trials demonstrate gastrointestinal toxicity when perifosine is administered orally.Citation157,Citation158 Studies adjusting the dose and administration regime of perifosine reduced gastrointestinal toxicity, but it still caused nausea, diarrhea, dehydration and fatigue (). Results of Phase II studies using perifosine as a single agent in patients with previously untreated metastatic or locally advanced soft tissue sarcoma; metastatic melanoma or refractory solid tumors have been reported.Citation96,Citation159 These trials demonstrated that oral perifosine did not affect disease progression or overall survival.Citation96 Very few patients exhibited stable disease. For example, a non-randomized, non-blinded multicenter Phase II study assessed the response rate and toxicity of perifosine in 18 patients having unidimensionally measurable and histologically confirmed metastatic melanomas.Citation96 Patients had not received prior chemotherapy, but some had received adjuvant immunotherapy. These patients were given a loading dose of 900 mg perifosine orally on day 1 followed by a maintenance dose of 150 mg on days 2–21 in a 28-d cycle.Citation96 Loading doses for subsequent cycles fixed at 300 mg on day 1. The trial showed no objective response in the 14 evaluable patients. Among these patients, 3 (21%) achieved stable disease whereas the remaining 11 had disease progression.Citation96 Toxicity studies measuring non-hematologic and hematological parameters showed occurrence of grade 3 or 4 non-hematologic toxicities such as diarrhea, arthralgia, nausea and headache and fatigue with perifosine administration.Citation96
One of the major reasons for perifosine failure in clinical trials may relate to suboptimal patient selection.Citation160 In none of the clinical studies were patients selected based on expression or activity of Akt. Recently, a Phase II clinical trial of patients with incurable, recurrent or metastatic squamous cell carcinoma of the head and neck made an attempt to correlate total and pAkt levels with perifosine efficacy.Citation161 However, likely due to a small sample size (19 patients) and lack of pretreatment tumor biopsies, no significant correlation between pAkt and treatment efficacy was observed.Citation161 Currently, perifosine is being tested in Phase I clinical trials in combination with radiation, gemicitabine, docetaxel and paclitaxel.Citation162–Citation164 The combination of perifosine and temozolomide inhibited gliomas more effectively compared with either of the single agents alone in a preclinical study.Citation165 A Phase II trial testing perifosine in combination with dexamethasone in patients with relapsed/refractory multiple myelomas demonstrated partial or minimal response in 38% and stable disease in 47% of patients.Citation95
Clinical studies examining the systemic treatment of melanoma with other lipid based Akt inhibitors have been disappointing with early Phase I and Phase II trials failing to demonstrate significant clinical activity. However, lipid based Akt inhibitor miltefosine has been approved for topical application for patients with cutaneous (nonmelanoma) malignancies.Citation166 It has been shown to be effective against cutaneous lymphomas and breast cancers that have metastasized to skin and has been well tolerated.Citation166
17-AAG.
17-allylamino-17demethoxygeldanamycin (17-AAG), a derivative of the natural compound geldanamycin, blocks hsp90 function and leads to degradation of client proteins, including AKT and BRAFCitation167–Citation169 (). This agent is particularly attractive as a targeted therapeutic in melanoma because it has the potential to inhibit both the AKT and MAP kinase pathways. In a multicenter Phase II trial of 17-AAG in stage III or IV melanoma patients with measurable disease,Citation170 patients were treated once weekly for six weeks. Of 15 evaluable patients, 9 had BRAF mutations and 6 did not.Citation170 No objective clinical responses were observed at the dose and schedule employed.Citation170 The authors concluded that a future study examining a more potent formulation that may be administered more chronically was needed and may be more promising clinically.Citation170
CCI-779 (temsiroliumus).
CCI-779 is an analog of rapamycin and has been shown to have antitumor activity in preclinical models of melanoma.Citation128 Dose-limiting toxicities observed in Phase I trials include myelosuppression, diarrhea, stomatitis, fever, fatigue, and hyperlipidemia (). Skin reactions were also common.Citation171 The California Cancer Consortium conducted a Phase II trial of 23 patients with metastatic melanoma.Citation172 Only one patient experienced a partial response, and the authors concluded that further study of temsiroliums monotherapy was not justified in metastatic melanoma.Citation172 However, the possibility that this agent may be effective in the context of combination drug regimens has not been ruled out.
RAD001 (everolimus).
Another mTOR inhibitor that has received significant attention as a potential treatment for melanoma is RAD001 (everolimus)Citation128 (). A Phase II multicenter trial of 24 patients with metastatic melanoma has been undertaken,Citation173 and the planned interim analysis after 20 patients were enrolled showed no objective clinical responses, though there was significant disease stabilization.Citation173 Fatigue, diarrhea, and anemia were the most common grade 2 toxicities reported; there were no grade 3 toxicities.Citation173 Further enrollment was terminated, but adequate biological activity was felt to warrant further study in combination therapy.Citation173
Lapatinib.
Lapatinib is a small molecule drug that reversibly inhibits ErbB1 and ErbB2 tyrosine kinases and consequently blocks phosphorylation and activation of Akt and Erk1/2.Citation174 Spector and colleagues examined the effect of lapatinib treatment on a variety of biomarkers in 33 patients with assorted nonmelanoma malignancies (predominantly breast cancer) with ErbB1 and ErbB2 overexpression.Citation175 They found that most patients experienced a decrease in tumor pAKT level following treatment, including three of four patients with objective clinical responses.Citation175
Riluzole (2-amino-6-trifluoromethoxybenzothiazole).
Riluzole is a noncompetitive inhibitor of metabotropic glutamate receptor-1 (GRM1) that has been shown to slow the progression of amyotropic lateral sclerosis.Citation176,Citation177 GRM1 is expressed in 60% of human melanomas, and ectopic expression of GRM1 in melanocytes in a murine model resulted in development of melanocytic lesions that were indistinguishable from melanoma.Citation178 Activation of GRM1 results in activation of the MAPK pathway in a BRAF and N-RAS independent manner.Citation179 A Phase 0 trial has been undertaken in which 12 patients with resectable stage III or IV melanoma, treated with 200 mg per day of oral riluzole for 14 d.Citation180 Biopsies were obtained pre-treatment, and lesions were excised following drug administration in order to determine the metabolic response.Citation180 Eleven patients completed the protocol with no significant toxicity or only mild dry mouth (two patients) or dizziness (two patients), and one patient was removed from the study for grade 3 neurologic toxicity (dizziness).Citation180 Six of the eleven evaluable patients had varying degrees of clinical or radiologic tumor regression.Citation180 Three of the six patients had suppression of both the PI3K/AKT and MAPK pathways (and none of seven other patients evaluated for PI3K/AKT activity were found to have suppression).Citation180 The authors concluded that suppression of the MAPK and PI3K/AKT pathways through GRM1 inhibition may be a viable therapeutic strategy for advanced melanoma.Citation180
SR13668.
SR13668 was initially introduced as a chemopreventive agent, designed using computer modeling based on a naturally occurring indole-3-carbinol, and has been shown to inhibit the AKT signaling pathway.Citation138 A clinical trial examining different formulations of SR13668 in healthy donors was performed and is now closed to further enrollment (NCT00896207).
GSK690693.
Safety, tolerability, pharmacokinetics and pharmacodynamics of the pan-AKT inhibitor GSK690693 in patients with lymphomaCitation123 (NCT00493818) has been investigated. Results are not yet available for this study.
GSK2141795.
It is an oral AKT inhibitor currently undergoing Phase I first-in-human testing in patients with solid tumors and lymphoma (NCT00920257); this study was still recruiting patients at the time of manuscript preparation.Citation163 GSK2141795 is also being studied in the setting of solid tumors such as ovarian cancer (NCT01266954) and in conjunction with MEK inhibitor GSK1120212 in cancer (NCT01138085).
Others.
Other AKT pathway inhibitors that are being evaluated in clinical trials in non-melanoma malignancies include therapeutics such as MK2206, triciribine, and mTOR inhibitors.Citation163,Citation181,Citation182 These agents may eventually warrant testing in melanoma.
Combination Therapies
An emerging theme in the development of AKT pathway inhibitors is one that is familiar to the oncology community: poor antineoplastic activity of single-drug regimens. In much the same way that Avastin has been shown to increase survival in colorectal cancer when added to 5-fluorouracil based regimens, it is hoped that agents targeting AKT and other key pathways will enhance the efficacy or synergizing potential with other agents.Citation183
Combination of sorafenib and rapamycin.
To date, most clinical trials of combination therapies have focused on mTOR inhibitors in conjunction with other agents in advanced melanoma.Citation184 For example, targeting MAP kinase pathway with sorafenib and/or the PI3K-Akt-mTOR pathway with mTOR inhibitor rapamycin was found to decrease melanoma growth more effectively than single agents.Citation185 Experimentally, combination of 4 µM sorafenib with 10 nM rapamycin inhibited the growth of six metastatic melanoma cell lines. In addition, this agent combination reduced growth of melanoma tumors developing in organotypic cultures.Citation185 Combination of these two agents enhanced the efficacy by 13.0–27.8% (depended on cell line) compared with either monotherapies.Citation185 Similarly, combining Mek1/2 inhibitor U0126 or PD98059 with rapamycin yielded better cell growth reduction than monotherapies.Citation185 Although, sorafenib combined with rapamycin appears to be a promising strategy for the effective treatment of melanoma, further studies are warranted to evaluate this combination in clinic. For example, it is not known whether this combination could inhibit melanoma tumors growing in mice.
Combination of LY-294002, wortmannin and rapamycin with cisplatin and temozolomide.
In a separate study it was demonstrated that targeting PI3K-Akt pathway using LY-294002, wortmannin and rapamycin in combination with cisplatin and temozolomide inhibited melanoma cell growth in monolayers as well as tumors developing in organotypic cultures.Citation186 Response surface analysis, which establishes relationships between multiple experimental variables and a single response, using combinations of AKT pathway inhibitors and chemotherapeutic agents for inhibiting melanoma cell growth showed that rapamycin and temozolomide synergistically inhibited melanomas compared with either agent alone.Citation186 Similarly, wortmannin and temozolomide synergistically inhibited melanoma cell growth.Citation186 Authors of this study showed that inhibitors targeting PI3K-Akt3 signaling sensitized melanoma cells to chemotherapeutic agents such as cisplatin.Citation186 However, similar effect was not observed when the MAPK pathway was targeted using pharmacological agents.Citation186
Combination of everolimus, paclitaxel and carboplatin.
Several combinations of these agents are currently undergoing clinical evaluation. For example, NCT01014351 is examining combination therapy with everolimus, paclitaxel, and carboplatin in metastatic melanoma. The primary endpoint is progression-free survival, and the planned enrollment is 70 patients. Similarly, the North Central Cancer Treatment Group (NCCTG) has opened a randomized trial (NCT00976573) in which metastatic melanoma patients are treated with paclitaxel, carboplatin, and bevacizumab with or everoliumus; the planned accrual is 148 patients. A third trial (NCT01252251) is being performed at Memorial Sloan Kettering Cancer Center and is studying everolimus in combination with the novel somatostatin antagonist pasirotide in uveal melanoma.
Combination of temsirolimus with AZD6244.
A Phase II study is currently testing mTOR inhibitor temsirolimus in combination with MEK inhibitor AZD6244 in metastatic melanoma. This study (NCT01166126) is being conducted by the Lee Moffitt Cancer Center and is examining temsirolimus in conjunction with MEK inhibitor AZD6244 in stage IV melanoma patients harboring BRAF-mutant tumors. This study is expected to address whether blocking both the BRAF/RAS/MAP kinase and AKT pathways is an effective strategy for treatment of melanoma.
Biomarkers for Agent Selection
In the emerging paradigm of personalized molecular medicine, selection of a particular targeted therapeutic is based, in part, on the particular phenotype of the patient with respect to the target and possibly to up- or downstream proteins regulated by the target. Because of the complexity of signaling pathways, the predicted molecular phenotypes do not always represent useful biomarkers of clinical efficacy. This is complicated by the fact that AKT phosphorylates thousands of proteins, and the extent to which any of these is likely to be surrogates of clinical activity is unknown.Citation2 Nonetheless, perhaps among the best-characterized target of AKT is mTOR.Citation187 Although phosphorylated S473 Akt serves as a biomarker for melanoma tumor progression and development, recent studies have observed elevated pAkt levels when the tumors become resistant to inhibitors targeting MAPK pathway.Citation4,Citation188 Therefore, it is difficult to determine whether elevated pAkt levels are due to elevated expression, or loss of tumor suppressor PTEN or due to induction of drug resistance.Citation2 In addition, in some instances, no direct correlation was found between pAkt status and disease stage. Hence, care must be taken while considering pAkt as a biomarker for selecting more effective therapeutic agents for inhibiting melanoma tumor development.
Reason for Pharmacological Agents Failure in the Clinic
Patient selection and methods for assessing pathway dependence.
To date, small molecule tyrosine kinase inhibitors have had limited clinical activity as monotherapy for the treatment of melanoma. A notable exception is the recently reported Phase I dose-escalation clinical trial that PLX4032 (Vemurafenib, Zelboraf, Roche) exhibited an 80% clinical response rate in patients with V600EBRAF-mutant metastatic melanoma treated at the 960 mg dose.Citation189,Citation190 These results demonstrate proof-of-principle that, in properly selected melanoma patients, targeted therapeutics have the potential to be effective.Citation189,Citation190 As a result of this promising efficacy in V600EBRAF-mutant metastatic melanoma, vemurafenib was recently (in August, 2011) approved by the FDA for this indication. Importantly, none of the patients with tumors harboring wild-type BRAF exhibited clinical responses. This highlights one potential reason for failure of targeted therapeutics, which is absence of a molecular phenotype (e.g., activating mutation or overexpression) rendering the tumor unresponsive to the drug. In fact, studies examining inhibitors of the AKT pathway generally have not restricted enrollment to those patients whose tumors harbor activating mutations or have overexpression of AKT. Hence, patient selection may be an important factor contributing to the limited activity of AKT pathway inhibitors in melanoma.
Secondary mutations canceling the drug effect.
A second potential reason for failure could be the presence of another mutation that renders the target regulatory protein irrelevant. For example, patients with colorectal cancer whose tumors have a mutated form of the downstream regulatory protein K-RAS do not benefit from treatment with the EGFR inhibitor cetuximab, while those patients with wild-type K-RAS may exhibit clinically significant antitumor activity.Citation191 This illustrates that an activating mutation of a downstream regulatory protein can render a targeted therapeutic ineffective. Similarly, gatekeeper mutations in Kit and Abl prevented binding of drugs to these enzymes and thereby induced resistance.Citation191 For the AKT pathway, which has numerous substrates, it is difficult to predict the particular mutations that may have an analogous impact. Nonetheless, this could represent a potential obstacle to development of clinically effective AKT-targeted therapeutics.
Activation of pathways bypassing primary routes.
Bypassing targets inhibited by drugs is a key mechanism leading to drug resistance. A classic example is activation of C-Raf and COT kinases when mutant V600EB-Raf is inhibited by vemurafenib.Citation192,Citation193 Both C-Raf and COT also activate MEK and ERK proteins and thereby induced resistance to drugs targeting B-Raf.Citation192,Citation193 Identification and inhibition of these bypassing mechanisms is also therefore key to the success of various pharmacological agents. Bypass mechanisms circumventing the role of Akt3 signaling in melanomas have yet to be identified.
Studies searching for factors inducing resistance to vemurafenib found that drug resistance was not due to secondary mutations of B-Raf but due to either elevation of other kinase proteins that bypass B-Raf or reactivation of the pathway or upregulation of other unrelated growth factor receptor pathways.Citation192–Citation194 Addressing these concepts, Garraway and coworkers at Dana-Farber Cancer Institute and Harvard Medical School demonstrated that expression of C-Raf and COT, which also stimulate MAPK pathway, in vemurafenib sensitive cells, induced resistance.Citation192,Citation193 Furthermore, analysis of matched pretreatment and vemurafenib treated patient biopsies confirmed expression of COT proteins only after treatment. Another mechanism inducing resistance is activation of growth factor receptors. Roger Lo and colleagues demonstrated this concept experimentally using cell lines and patient biopsies.Citation194 Authors concluded that two mutually exclusive mechanisms, mutations in N-Ras and overexpression of PDGFR, play a key role in the induction of resistance to vemurafenib treatment.Citation194 These and many other studies illustrate the importance of co-targeting multiple proteins either in the same pathway or from other pathways to more effectively inhibit melanomas. Exemplifying this strategy, GSK is currently evaluating a proprietary V600EB-Raf inhibitor in combination with MEK inhibitor in a Phase I clinical trial.
Upregulation of alternate pathways and development of drug resistance.
Recent studies have found that drug resistance could be induced by the upregulation or activation of pathways not related to the one that is inhibited by drug treatment. For example, targeting V600EB-Raf using vemurafenib appeared to activate the PDGFR pathway.Citation194 Similar mechanisms have not yet been reported following Akt inhibition in melanomas. In addition, clinical relevance of pathway activation needs to be explored. Therefore, further studies are warranted to identify pathways activated when Akt pathway is inhibited using pharmacological agents and whether these pathways are inducing resistance in patient tumors.
Mechanisms Leading to the Induction of Drug Resistance
Akt3 mediates resistance to B-Raf inhibitor PLX-4720.
Although it is unclear which pathways are activated when Akt3 is inhibited in melanomas, it is now known that Akt can mediate drug resistance. For example, Akt3 promoted resistance to apoptosis in when B-Raf was targeted in melanomas.Citation188 Using RGP and VGP-like melanoma cells in three-dimensional collagen models, targeting B-Raf using siRNAs or PLX-4720 induced apoptosis mediated by Bim-EL and Bmf.Citation188 However, constitutive activation of Akt3 protected melanoma cells from B-Raf inhibition induced apoptosis.Citation188 This mechanism leading to drug resistance should be taken into account during clinical investigation of pharmacological agents such as PLX-4720.
Akt mediates resistance to MEK1/2 inhibitor AZD6244.
Akt has also been shown to mediate resistance to cell death induced by the potent, highly selective, uncompetitive inhibitor of MEK1/2 AZD6244 in melanoma cells harboring a mutant V600EB-Raf.Citation195 Melanoma cells expressing mutant V600EB-Raf and functional PTEN were found to be sensitive to AZD6244 treatment and showed no elevation in pAkt.Citation195 However, AZD6244 resistant cells, despite expressing normal PTEN showed elevated pAkt treatment indicating that Akt mediates the resistance to AZD6244.Citation195
Strategies to Overcome Drug Resistance
Use of synergistically acting drugs.
Utilizing drug synergism is one of the viable approaches for overcoming drug resistance and associated toxicity. A drug effect is called synergistic only when pharmacological agents targeting two or more proteins yield a greater effect than the sum of targeting each protein individually. Synergism is usually determined by analyzing data using CalcuSyn method.Citation196 In this approach, a drug combination is called synergistic when the combination index (CI) values are less than < 0.9. If the CI value is between 0.9 and 1.1 the combination are additive; and any CI value > 1.1 is antagonistic.Citation196 Proof of principle studies targeting Akt3 and V600EB-Raf demonstrated that inhibiting these two proteins synergistically reduced cell viability and retarded development of xenografted melanomas in mice.Citation136 Furthermore, ceramide-containing liposomes loaded with sorafenib was also found to synergistically inhibit melanoma development.Citation197 However, no clinical trials have been conducted using agents that synergistically inhibit key signaling pathways regulating melanomas in patients. Therefore, studies are warranted to identify targets and targeted agents that synergistically inhibit melanoma development.
Agents targeting PI3K and mTORC1 and mTORC2.
Even though targeting Akt3 alone has been shown to inhibit melanoma development by triggering apoptosis, complete tumor inhibition has been reported neither in preclinical studies nor in clinical trials, indicating the need for identification of additional targets in this pathway and development of compounds for simultaneously targeting key members of this signaling cascade.
PI-103 or GDC-0941 and rapamycin. A recent preclinical study showed that vertical inhibition (inhibition of activity of members of the same regulatory pathway) of PI3K and mTORC1 and mTORC2 synergistically reduced melanoma cell growth in vitro and in vivoCitation50 (). Co-targeting class-I PI3K using the specific inhibitor PI-103 or GDC-0941 and mTORC1 inhibitor rapamycin reduced cultured melanoma cells viability more effectively than either agent alone.Citation50 Furthermore, co-administering 20 mg/kg PI-103 and 1 mg/kg rapamycin reduced xenografted melanoma tumors by 50%.Citation50 However, at these concentrations neither compound had significant effects on tumor growth.Citation50 Analogous studies are needed to demonstrate the effect of co-targeting Akt and its downstream members ().
Agents targeting PI3K and mTOR using dual inhibitors.
NVP-BEZ235, an imidazoquinoline derivative inhibiting both PI3K and mTOR by binding to the ATP binding site, has been tested in preclinical models of melanoma.Citation44,Citation130 In preclinical studies, blocking PI3K with ZSTK474 or blocking mTOR with rapamycin yielded promising results.Citation44,Citation130 It is promising that NVP-BEZ235 was found to be more potent than the combination of ZSTK474 and rapamycin with respect to inhibition of melanoma cell proliferation. Currently this inhibitor is under Phase I/II clinical trial evaluationCitation198
Agents targeting upstream growth factor receptors in combination with expression of tumor suppressor proteins.
Several research groups are currently testing the efficacy of chemotherapeutic agents targeting an upstream growth factor receptor and expressing a tumor suppressor in preclinical and clinical trials. For example, PTEN loss confers resistance to B-Raf inhibitors, and targeting V600EB-Raf in combination with PTEN expression has potential to inhibit melanoma development.Citation28,Citation31 Mechanistically, expression of PTEN in advanced melanomas lacking PTEN inhibits Akt3 activity and thereby reduced cell survival, induced apoptosis and sensitized cells to therapeutic agents.Citation5,Citation6,Citation14 Furthermore targeting V600EB-Raf using siRNA in combination with Akt3 inhibition reduced metastatic melanoma viability, by inducing apoptotic cell death.Citation65,Citation136 Although various preclinical, proof-of-concept studies have demonstrated efficacy of targeting MAPK pathway in conjunction with PTEN expression or Akt3 inhibition for melanoma inhibition, it is currently unknown whether this combination could inhibit melanoma tumor development in patients. Therefore, clinical trials are warranted to test this combination in patients. In addition, preclinical studies should identify pharmacological agents that can induce PTEN expression specifically in melanoma cells. For example, agents that promote PTEN expression by controlling various epigenetic mechanisms are attractive candidate drugs. In this line, in vitro studies showed the antitumor activity of demethylating agent 5-Azacytidine, and histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA).
Expression of Interleukin-24 (IL-24), a novel tumor suppressor/cytokine, inhibited proliferation and induced apoptosis. Expression of this protein in combination with erlotinib reduced melanoma growth more effectively than each alone.Citation199 Several studies have demonstrated that IL-24 is expressed predominantly in normal human melanocytes, whereas only very low levels were observed in metastatic melanomas.Citation200 Likewise, erlotinib, a small-molecule epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, reduced melanoma viability in in vitro studies.Citation199 Combination of IL-24 and erlotinib inhibited melanoma viability by inducing caspase-3 and -9 cleavage, and downregulating phosphorylated EGFR and pAkt.Citation199 Although these data demonstrate efficacy of this combination in preclinical studies, the activity of this combination has not yet been evaluated clinically.
What Does the Future Hold for Targeting Akt or the PI3K Pathways in Melanoma?
It is now well-established that targeting Akt3 inhibits melanoma tumor development, sensitizes cells to chemotherapeutic agents and decreases drug resistance induced by pharmacological agents targeting MAPK. However, the cellular roles of other Akt isoforms remain unknown. A recent study demonstrated that Akt2 mediates melanoma cells metastasis.Citation32 Functional significance of Akt1 is yet to be established in melanomas. Therefore, it is important to establish the role of each Akt isoform before testing a pharmacological agent in the clinic, particularly since any of the three isoforms has the theoretical potential to promote metastasis. Consequently, one focus of future drug discovery and development research should be the synthesis of highly selective, isoform specific inhibitors for targeting the specific Akt isoforms in melanomas.
Key Findings and Weaknesses
Although several preclinical studies have demonstrated the therapeutic potential of targeting Akt3 signaling in melanomas, not much is known regarding the effect of inhibiting this target in the clinic. Unavailability of clinically viable pharmacological agents specifically targeting particular isoforms limits these studies. Furthermore, clinical trials investigating Akt inhibitors such as perifosine were conducted in patients without selection for those harboring activated Akt. Therefore, suboptimal patient selection or stratification may be adversely affect outcomes of clinical trials, and should be considered when interpreting clinical results. Furthermore, expression and activity of positive and negative regulators that influence the activity of this pathway also should be considered as aberrant expression patterns of these proteins might affect the downstream target proteins.
Goals Achieved and Yet to Achieve
Further study is necessary before AKT inhibitors are approved by the FDA for clinical use in melanoma. The most important step is identification of lead compounds with clinical activity and an acceptable toxicity profile. It may be that the optimal use of AKT inhibitors in melanoma is to employ them in combination regimens along with cytotoxic drugs and/or other targeted molecular agents. Additionally, some targeted therapeutics, such as vemurafenib, may be effective only in patients with mutated (and/or overexpressed) targets. Hence, if initial studies fail to show significant clinical benefit in patients, it may require proper patient selection based on activity of the Akt pathway to determine the optimal efficacy and clinical role for these therapeutic agents.
Areas of Research that Might be Interesting to Pursue
Combination of low dose perifosine and ipilimumab.
While targeted agents are unlikely to offer a cure for stage IV melanoma in the near future, there have been exciting advancements. Ipilimumab, an anti-CTLA4 monoclonal antibody that activates cytotoxic T cells, was recently shown in a randomized, prospective multi-center trial to improve survival by four months compared with the control group.Citation201 During preparation of this manuscript, ipilimumab was approved by the FDA for the treatment of metastatic melanoma. At this point, ipilimumab appears to be among the most promising targeted agents available for the treatment of this disease. Therefore it is interesting to evaluate the clinical efficacy of combining Akt inhibitor perifosine or its derivatives such as miltefosine and ipilimumab.
Combination of low dose perifosine and vemurafenib.
Early Phase I dose-escalation trial results using V600EB-RAF inhibitor vemurafenib showed high response rate in patients with BRAF-mutant metastatic melanoma.Citation190 Among 32 patients harboring V600EB-Raf mutation, 24 individuals showed a partial response and 2 exhibited complete responses to vemurafenib treatment. Vemurafenib was also approved by the FDA during preparation of this manuscript and represents another exciting addition to the arsenal of therapies for stage IV melanoma. Among the compelling questions at this point is whether the combination of low dose perifosine and vemurafenib might further enhance efficacy in metastatic melanoma.
Figures and Tables
Figure 1 Schematic representation of key therapeutic targets in the PI3K/Akt and MAPK pathways together with pharmacological agents inhibiting members of these signaling cascades. Shown are the key therapeutic targets regulating melanoma development. Whereas PI3K/Akt3 pathway involved in cell survival and apoptosis regulation, MAPK pathway is implicated in cell proliferation. Targeting members of these signaling cascades using pharmacological agents has been shown to inhibit melanoma tumor development. Pharmacological agents inhibiting PI3K/Akt signaling; perifosine, 17-AAG, CCI-779 and RAD-001 have been evaluated in clinical trials. Vemurafenib and sorafenib targeting MAPK are also being evaluated in various clinical trials for inhibiting melanomas.
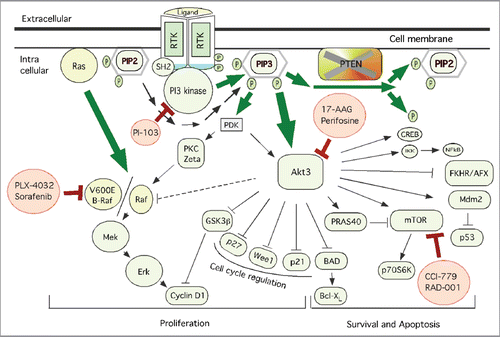
Figure 2 Mechanisms regulating PI3K pathway in melanoma. PI3K is one of the key proteins involved in the development of melanoma. PI3K expression and activities are regulated by mutational activation, elevated protein expression and loss of negative regulators such as PTEN. Activated PI3K converts PIP2s in to PIP3s, which in turn binds to plextrin homology domains of various kinases such as Akt thereby helps in the translocation of these proteins to cell membranes.
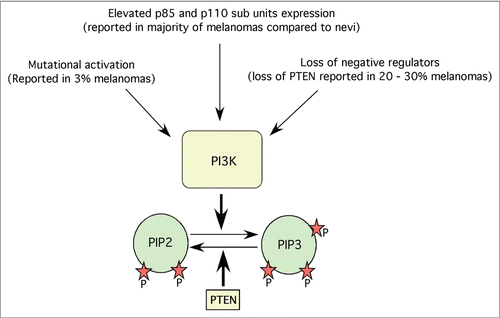
Table 1 Pharmacological agents that are under clinical evaluation for inhibiting PI3K/Akt3 pathway in melanomas
Table 2 Preclinically evaluated pharmacological agents targeting members of PI3K/Akt3 signaling
Table 3 Potential pharmacological agent combinations inhibiting key signaling proteins in PI3K/Akt3 pathway in melanomas
Acknowledgments
NIH CA-127892-01A and The Foreman Foundation for Melanoma Research (GPR). Melanoma Research Foundation (SVM).
Related Research Data
References
- Madhunapantula SV, Robertson GP. Is B-Raf a good therapeutic target for melanoma and other malignancies?. Cancer Res 2008; 68:5 - 8; PMID: 18172288; http://dx.doi.org/10.1158/0008-5472.CAN-07-2038
- Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res 2009; 22:400 - 419; PMID: 19493313; http://dx.doi.org/10.1111/j.1755-148X.2009.00585.x
- Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev 2005; 24:273 - 285; PMID: 15986137; http://dx.doi.org/10.1007/s10555-005-1577-9
- Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol 2010; 80:624 - 637; PMID: 20450891; http://dx.doi.org/10.1016/j.bcp.2010.04.029
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res 2003; 63:2881 - 2890; PMID: 12782594
- Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res 2004; 64:7002 - 7010; PMID: 15466193; http://dx.doi.org/10.1158/0008-5472.CAN-04-1399
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261 - 1274; PMID: 17604717; http://dx.doi.org/10.1016/j.cell.2007.06.009
- Bunney TD, Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer 2010; 10:342 - 352; PMID: 20414202; http://dx.doi.org/10.1038/nrc2842
- Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des 2010; 16:34 - 44; PMID: 20214616; http://dx.doi.org/10.2174/138161210789941865
- Lazo JS, Wipf P. Phosphatases as targets for cancer treatment. Curr Opin Investig Drugs 2009; 10:1297 - 1304; PMID: 19943201
- Smalley KS, Herlyn M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann N Y Acad Sci 2005; 1059:16 - 25; PMID: 16382039; http://dx.doi.org/10.1196/annals.1339.005
- Hsu MY, Meier F, Herlyn M. Melanoma development and progression: a conspiracy between tumor and host. Differentiation 2002; 70:522 - 536; PMID: 12492494; http://dx.doi.org/10.1046/j.1432-0436.2002.700906.x
- Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest 2005; 115:813 - 824; PMID: 15841168
- Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res 2007; 67:3626 - 3636; PMID: 17440074; http://dx.doi.org/10.1158/0008-5472.CAN-06-4234
- Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol 2005; 23:1473 - 1482; PMID: 15735123; http://dx.doi.org/10.1200/JCO.2005.07.168
- Aziz SA, Davies M, Pick E, Zito C, Jilaveanu L, Camp RL, et al. Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin Cancer Res 2009; 15:3029 - 3036; PMID: 19383818; http://dx.doi.org/10.1158/1078-0432.CCR-08-2768
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550 - 562; PMID: 19629070; http://dx.doi.org/10.1038/nrc2664
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 2000; 346:561 - 576; PMID: 10698680; http://dx.doi.org/10.1042/0264-6021:3460561
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009; 37:217 - 222; PMID: 19143635; http://dx.doi.org/10.1042/BST0370217
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol 2009; 7:e38; PMID: 19209957; http://dx.doi.org/10.1371/journal.pbio.1000038
- Zheng Y, Peng M, Wang Z, Asara JM, Tyner AL. Protein tyrosine kinase 6 directly phosphorylates AKT and promotes AKT activation in response to epidermal growth factor. Mol Cell Biol 2010; 30:4280 - 4292; PMID: 20606012; http://dx.doi.org/10.1128/MCB.00024-10
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999; 99:323 - 334; PMID: 10555148; http://dx.doi.org/10.1016/S0092-8674(00)81663-3
- Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 1999; 9:125 - 128; PMID: 10203785; http://dx.doi.org/10.1016/S0962-8924(99)01519-6
- Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet 2002; 70:829 - 844; PMID: 11875759; http://dx.doi.org/10.1086/340026
- Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene 2006; 374:1 - 9; PMID: 16675164; http://dx.doi.org/10.1016/j.gene.2006.02.024
- Guldberg P, thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res 1997; 57:3660 - 3663; PMID: 9288767
- Tsao H, Zhang X, Benoit E, Haluska FG. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 1998; 16:3397 - 3402; PMID: 9692547; http://dx.doi.org/10.1038/sj.onc.1201881
- Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011; 71:2750 - 2760; PMID: 21317224; http://dx.doi.org/10.1158/0008-5472.CAN-10-2954
- Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 Activity Promotes Development of Malignant Melanoma. Cancer Res 2004; In press PMID: 15466193; http://dx.doi.org/10.1158/0008-5472.CAN-04-1399
- Mikhail M, Velazquez E, Shapiro R, Berman R, Pavlick A, Sorhaindo L, et al. PTEN expression in melanoma: relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res 2005; 11:5153 - 5157; PMID: 16033830; http://dx.doi.org/10.1158/1078-0432.CCR-05-0397
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544 - 552; PMID: 19282848; http://dx.doi.org/10.1038/ng.356
- Nogueira C, Kim KH, Sung H, Paraiso KH, Dannenberg JH, Bosenberg M, et al. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010; 29:6222 - 6232; PMID: 20711233; http://dx.doi.org/10.1038/onc.2010.349
- Mounir Z, Krishnamoorthy JL, Robertson GP, Scheuner D, Kaufman RJ, Georgescu MM, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal 2009; 2:ra85; PMID: 20029030; http://dx.doi.org/10.1126/scisignal.2000389
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 1999; 96:4240 - 4245; PMID: 10200246; http://dx.doi.org/10.1073/pnas.96.8.4240
- Li L, Ross AH. Why is PTEN an important tumor suppressor?. J Cell Biochem 2007; 102:1368 - 1374; PMID: 17972252; http://dx.doi.org/10.1002/jcb.21593
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA 1999; 96:2110 - 2115; PMID: 10051603; http://dx.doi.org/10.1073/pnas.96.5.2110
- Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene 2003; 22:3113 - 3122; PMID: 12789288; http://dx.doi.org/10.1038/sj.onc.1206451
- Huang W, Chang HY, Fei T, Wu H, Chen YG. GSK3 beta mediates suppression of cyclin D2 expression by tumor suppressor PTEN. Oncogene 2007; 26:2471 - 2482; PMID: 17043650; http://dx.doi.org/10.1038/sj.onc.1210033
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4:988 - 1004; PMID: 16341064; http://dx.doi.org/10.1038/nrd1902
- Franke TF. PI3K/Akt: getting it right matters. Oncogene 2008; 27:6473 - 6488; PMID: 18955974; http://dx.doi.org/10.1038/onc.2008.313
- Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 2008; 1784:159 - 185; PMID: 17997386
- Fruman DA. Regulatory subunits of class IA PI3K. Curr Top Microbiol Immunol 2010; 346:225 - 244; PMID: 20563711; http://dx.doi.org/10.1007/82_2010_39
- Vogt PK, Gymnopoulos M, Hart PI Jr. 3-kinase and cancer: changing accents. Curr Opin Genet Dev 2009; 19:12 - 17; PMID: 19185485; http://dx.doi.org/10.1016/j.gde.2008.11.011
- Aziz SA, Jilaveanu LB, Zito C, Camp RL, Rimm DL, Conrad P, et al. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res 2010; 16:6029 - 6039; PMID: 21169255; http://dx.doi.org/10.1158/1078-0432.CCR-10-1490
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA 2001; 98:10983 - 10985; PMID: 11572954; http://dx.doi.org/10.1073/pnas.211430998
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14:381 - 395; PMID: 11882383; http://dx.doi.org/10.1016/S0898-6568(01)00271-6
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 1997; 89:457 - 467; PMID: 9150145; http://dx.doi.org/10.1016/S0092-8674(00)80226-3
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI (3)K and PKB. Nature 1997; 385:544 - 548; PMID: 9020362; http://dx.doi.org/10.1038/385544a0
- Polsky D, Cordon-Cardo C. Oncogenes in melanoma. Oncogene 2003; 22:3087 - 3091; PMID: 12789285; http://dx.doi.org/10.1038/sj.onc.1206449
- Werzowa J, Koehrer S, Strommer S, Cejka D, Fuereder T, Zebedin E, et al. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. J Invest Dermatol 2011; 131:495 - 503; PMID: 21048785; http://dx.doi.org/10.1038/jid.2010.327
- Tachiiri S, Sasai K, Oya N, Hiraoka M. Enhanced cell killing by overexpression of dominant-negative phosphatidylinositol 3-kinase subunit, Deltap85, following genotoxic stresses. Jpn J Cancer Res 2000; 91:1314 - 1318; PMID: 11123431; http://dx.doi.org/10.1111/j.1349-7006.2000.tb00919.x
- Harfouche R, Basu S, Soni S, Hentschel DM, Mashelkar RA, Sengupta S. Nanoparticle-mediated targeting of phosphatidylinositol-3-kinase signaling inhibits angiogenesis. Angiogenesis 2009; 12:325 - 338; PMID: 19685150; http://dx.doi.org/10.1007/s10456-009-9154-4
- Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell 2002; 111:293 - 303; PMID: 12419241; http://dx.doi.org/10.1016/S0092-8674(02)01083-8
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2001; 2:760 - 768; PMID: 11584303; http://dx.doi.org/10.1038/35096067
- Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett 2003; 546:108 - 112; PMID: 12829245; http://dx.doi.org/10.1016/S0014-5793(03)00562-3
- Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 2004; 3:268 - 275; PMID: 15034304; http://dx.doi.org/10.4161/cbt.3.3.703
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29:233 - 242; PMID: 15130559; http://dx.doi.org/10.1016/j.tibs.2004.03.006
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 2001; 26:657 - 664; PMID: 11701324; http://dx.doi.org/10.1016/S0968-0004(01)01958-2
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999; 13:2905 - 2927; PMID: 10579998; http://dx.doi.org/10.1101/gad.13.22.2905
- Lietzke SE, Bose S, Cronin T, Klarlund J, Chawla A, Czech MP, et al. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol Cell 2000; 6:385 - 394; PMID: 10983985; http://dx.doi.org/10.1016/S1097-2765(00)00038-1
- Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, et al. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell 2000; 6:373 - 384; PMID: 10983984; http://dx.doi.org/10.1016/S1097-2765(00)00037-X
- Jones PF, Jakubowicz T, Hemmings BA. Molecular cloning of a second form of rac protein kinase. Cell Regul 1991; 2:1001 - 1009; PMID: 1801921
- Andjelković M, Jones PF, Grossniklaus U, Cron P, Schier AF, Dick M, et al. Developmental regulation of expression and activity of multiple forms of the Drosophila RAC protein kinase. J Biol Chem 1995; 270:4066 - 4075; PMID: 7876156; http://dx.doi.org/10.1074/jbc.270.8.4066
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996; 15:6541 - 6551; PMID: 8978681
- Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res 2008; 68:3429 - 3439; PMID: 18451171; http://dx.doi.org/10.1158/0008-5472.CAN-07-5867
- Konishi H, Kuroda S, Tanaka M, Matsuzaki H, Ono Y, Kameyama K, et al. Molecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem Biophys Res Commun 1995; 216:526 - 534; PMID: 7488143; http://dx.doi.org/10.1006/bbrc.1995.2654
- Brodbeck D, Hill MM, Hemmings BA. Two splice variants of protein kinase B gamma have different regulatory capacity depending on the presence or absence of the regulatory phosphorylation site serine 472 in the carboxyl-terminal hydrophobic domain. J Biol Chem 2001; 276:29550 - 29558; PMID: 11387345; http://dx.doi.org/10.1074/jbc.M104633200
- Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol 2010; 346:31 - 56; PMID: 20517722; http://dx.doi.org/10.1007/82_2010_58
- Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res 2010; 2:19 - 42; PMID: 20182580
- Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle 2010; 9:487 - 497; PMID: 20081374; http://dx.doi.org/10.4161/cc.9.3.10508
- Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 2009; 8:2502 - 2508; PMID: 19597332; http://dx.doi.org/10.4161/cc.8.16.9335
- Guénin S, Schwartz L, Morvan D, Steyaert JM, Poignet A, Madelmont JC, et al. PP2A activity is controlled by methylation and regulates oncoprotein expression in melanoma cells: a mechanism which participates in growth inhibition induced by chloroethylnitrosourea treatment. Int J Oncol 2008; 32:49 - 57; PMID: 18097542
- Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci USA 2007; 104:19011 - 19016; PMID: 18006659; http://dx.doi.org/10.1073/pnas.0706696104
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005; 18:13 - 24; PMID: 15808505; http://dx.doi.org/10.1016/j.molcel.2005.03.008
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009; 325:1134 - 1138; PMID: 19713527; http://dx.doi.org/10.1126/science.1175065
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007; 448:439 - 444; PMID: 17611497; http://dx.doi.org/10.1038/nature05933
- Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer 2008; 99:1265 - 1268; PMID: 18813315; http://dx.doi.org/10.1038/sj.bjc.6604637
- Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell 2007; 12:104 - 107; PMID: 17692802; http://dx.doi.org/10.1016/j.ccr.2007.07.014
- Askham JM, Platt F, Chambers PA, Snowden H, Taylor CF, Knowles MA. AKT1 mutations in bladder cancer: identification of a novel oncogenic mutation that can co-operate with E17K. Oncogene 2010; 29:150 - 155; PMID: 19802009; http://dx.doi.org/10.1038/onc.2009.315
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem 1995; 270:27489 - 27494; PMID: 7499206; http://dx.doi.org/10.1074/jbc.270.46.27489
- Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, et al. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem 2000; 275:36108 - 36115; PMID: 10945990; http://dx.doi.org/10.1074/jbc.M005497200
- Rosner M, Hanneder M, Freilinger A, Hengstschlager M. Nuclear/cytoplasmic localization of Akt activity in the cell cycle. Amino Acids 2007; 32:341 - 345; PMID: 17357828; http://dx.doi.org/10.1007/s00726-007-0509-0
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol 2005; 17:150 - 157; PMID: 15780591; http://dx.doi.org/10.1016/j.ceb.2005.02.010
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 2004; 30:193 - 204; PMID: 15023437; http://dx.doi.org/10.1016/j.ctrv.2003.07.007
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 2003; 278:10189 - 10194; PMID: 12524439; http://dx.doi.org/10.1074/jbc.M210837200
- Wang L, Harris TE, Roth RA, Lawrence JC Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem 2007; 282:20036 - 20044; PMID: 17510057; http://dx.doi.org/10.1074/jbc.M702376200
- Ikenoue T, Kanai F, Hikiba Y, Tanaka Y, Imamura J, Ohta M, et al. Functional consequences of mutations in a putative Akt phosphorylation motif of B-raf in human cancers. Mol Carcinog 2005; 43:59 - 63; PMID: 15791648; http://dx.doi.org/10.1002/mc.20102
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008; 132:363 - 374; PMID: 18267069; http://dx.doi.org/10.1016/j.cell.2007.12.032
- Houben R, Ortmann S, Drasche A, Troppmair J, Herold MJ, Becker JC. Proliferation arrest in B-Raf mutant melanoma cell lines upon MAPK pathway activation. J Invest Dermatol 2009; 129:406 - 414; PMID: 18650848; http://dx.doi.org/10.1038/jid.2008.214
- Goel VK, Ibrahim N, Jiang G, Singhal M, Fee S, Flotte T, et al. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009; 28:2289 - 2298; PMID: 19398955; http://dx.doi.org/10.1038/onc.2009.95
- Yang J, Splittgerber R, Yull FE, Kantrow S, Ayers GD, Karin M, et al. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest 2010; 120:2563 - 2574; PMID: 20530876; http://dx.doi.org/10.1172/JCI42358
- Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009; 125:2863 - 2870; PMID: 19609947; http://dx.doi.org/10.1002/ijc.24748
- Sharma A, Sharma AK, Madhunapantula SV, Desai D, Huh SJ, Mosca P, et al. Targeting Akt3 Signaling in Malignant Melanoma Using Isoselenocyanates. Clin Cancer Res 2009; 15:1674 - 1685; PMID: 19208796; http://dx.doi.org/10.1158/1078-0432.CCR-08-2214
- LoPiccolo J, Granville CA, Gills JJ, Dennis PA. Targeting Akt in cancer therapy. Anticancer Drugs 2007; 18:861 - 874; PMID: 17667591
- Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep 2009; 11:102 - 110; PMID: 19216841; http://dx.doi.org/10.1007/s11912-009-0016-4
- Ernst DS, Eisenhauer E, Wainman N, Davis M, Lohmann R, Baetz T, et al. Phase II study of perifosine in previously untreated patients with metastatic melanoma. Invest New Drugs 2005; 23:569 - 575; PMID: 16034524; http://dx.doi.org/10.1007/s10637-005-1157-4
- Castillo SS, Brognard J, Petukhov PA, Zhang C, Tsurutani J, Granville CA, et al. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res 2004; 64:2782 - 2792; PMID: 15087394; http://dx.doi.org/10.1158/0008-5472.CAN-03-1530
- Gills JJ, Dennis PA. The development of phosphatidylinositol ether lipid analogues as inhibitors of the serine/threonine kinase, Akt. Expert Opin Investig Drugs 2004; 13:787 - 797; PMID: 15212619; http://dx.doi.org/10.1517/13543784.13.7.787
- Meuillet EJ, Ihle N, Baker AF, Gard JM, Stamper C, Will'iams R, et al. In vivo molecular pharmacology and antitumor activity of the targeted Akt inhibitor PX-316. Oncol Res 2004; 14:513 - 527; PMID: 15559765
- Sharma AK, Sharma A, Desai D, Madhunapantula SV, Huh SJ, Robertson GP, et al. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. J Med Chem 2008; 51:7820 - 7826; PMID: 19053750; http://dx.doi.org/10.1021/jm800993r
- Nguyen N, Sharma A, Nguyen N, Sharma AK, Desai D, Huh SJ, et al. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev Res (Phila) 2011; 4:248 - 258; PMID: 21097713; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0106
- Madhunapantula SV, Desai D, Sharma A, Huh S, Amin S, Robertson GP. PBIse, a novel selenium containing drug for the treatment of malignant melanoma. Mol Cancer Ther 2008; 7:1297 - 1308; PMID: 18483317; http://dx.doi.org/10.1158/1535-7163.MCT-07-2267
- Desai D, Madhunapantula SV, Gowdahalli K, Sharma A, Chandagaludoreswamy R, El-Bayoumy K, et al. Synthesis and characterization of a novel iNOS/Akt inhibitor Se,Se'-1,4-phenylenebis(1,2-ethanediyl) bisisoselenourea (PBISe)-against colon cancer. Bioorg Med Chem Lett 2010; 20:2038 - 2043; PMID: 20153642; http://dx.doi.org/10.1016/j.bmcl.2009.09.071
- Chung CY, Madhunapantula SV, Desai D, Amin S, Robertson GP. Melanoma Prevention Using Topical PBISe. Cancer Prev Res (Phila) 2011;
- Reddy BS, Rivenson A, Kulkarni N, Upadhyaya P, el-Bayoumy K. Chemoprevention of colon carcinogenesis by the synthetic organoselenium compound 1,4-phenylenebis(methylene)selenocyanate. Cancer Res 1992; 52:5635 - 5640; PMID: 1394188
- Tanaka T, Kohno H, Murakami M, Kagami S, El-Bayoumy K. Suppressing effects of dietary supplementation of the organoselenium 1,4-phenylenebis (methylene)selenocyanate and the Citrus antioxidant auraptene on lung metastasis of melanoma cells in mice. Cancer Res 2000; 60:3713 - 3716; PMID: 10919638
- Moses SA, Ali MA, Zuohe S, Du-Cuny L, Zhou LL, Lemos R, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res 2009; 69:5073 - 5081; PMID: 19491272; http://dx.doi.org/10.1158/0008-5472.CAN-08-3839
- Meuillet EJ, Zuohe S, Lemos R, Ihle N, Kingston J, Watkins R, et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther 2010; 9:706 - 717; PMID: 20197390; http://dx.doi.org/10.1158/1535-7163.MCT-09-0985
- Kim D, Sun M, He L, Zhou QH, Chen J, Sun XM, et al. A small molecule inhibits Akt through direct binding to Akt and preventing Akt membrane translocation. J Biol Chem 2010; 285:8383 - 8394; PMID: 20068047; http://dx.doi.org/10.1074/jbc.M109.094060
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 2004; 64:4394 - 4399; PMID: 15231645; http://dx.doi.org/10.1158/0008-5472.CAN-04-0343
- Schweinsberg PD, Smith RG, Loo TL. Identification of the metabolites of an antitumor tricyclic nucleoside (NSC-154020). Biochem Pharmacol 1981; 30:2521 - 2526; PMID: 7306204; http://dx.doi.org/10.1016/0006-2952(81)90577-3
- Feun LG, Blessing JA, Barrett RJ, Hanjani P. A phase II trial of tricyclic nucleoside phosphate in patients with advanced squamous cell carcinoma of the cervix. A Gynecologic Oncology Group Study. Am J Clin Oncol 1993; 16:506 - 508; PMID: 8256767; http://dx.doi.org/10.1097/00000421-199312000-00010
- Feun LG, Savaraj N, Bodey GP, Lu K, Yap BS, Ajani JA, et al. Phase I study of tricyclic nucleoside phosphate using a five-day continuous infusion schedule. Cancer Res 1984; 44:3608 - 3612; PMID: 6744283
- Tang HJ, Jin X, Wang S, Yang D, Cao Y, Chen J, et al. A small molecule compound inhibits AKT pathway in ovarian cancer cell lines. Gynecol Oncol 2006; 100:308 - 317; PMID: 16209885; http://dx.doi.org/10.1016/j.ygyno.2005.08.044
- Koul D, Shen R, Bergh S, Sheng X, Shishodia S, Lafortune TA, et al. Inhibition of Akt survival pathway by a small-molecule inhibitor in human glioblastoma. Mol Cancer Ther 2006; 5:637 - 644; PMID: 16546978; http://dx.doi.org/10.1158/1535-7163.MCT-05-0453
- Gaitonde S, De SK, Tcherpakov M, Dewing A, Yuan H, Riel-Mehan M, et al. BI-69A11-mediated inhibition of AKT leads to effective regression of xenograft melanoma. Pigment Cell Melanoma Res 2009; 22:187 - 195; PMID: 19175524; http://dx.doi.org/10.1111/j.1755-148X.2009.00544.x
- Lin X, Murray JM, Rico AC, Wang MX, Chu DT, Zhou Y, et al. Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for AKT: synthesis and SAR studies. Bioorg Med Chem Lett 2006; 16:4163 - 4168; PMID: 16765046; http://dx.doi.org/10.1016/j.bmcl.2006.05.092
- Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, de Jong R, et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther 2005; 4:977 - 986; PMID: 15956255; http://dx.doi.org/10.1158/1535-7163.MCT-05-0005
- Morgan-Lappe S, Woods KW, Li Q, Anderson MG, Schurdak ME, Luo Y, et al. RNAi-based screening of the human kinome identifies Akt-cooperating kinases: a new approach to designing efficacious multitargeted kinase inhibitors. Oncogene 2006; 25:1340 - 1348; PMID: 16247451; http://dx.doi.org/10.1038/sj.onc.1209169
- Shi Y, Liu X, Han EK, Guan R, Shoemaker AR, Oleksijew A, et al. Optimal classes of chemotherapeutic agents sensitized by specific small-molecule inhibitors of akt in vitro and in vivo. Neoplasia 2005; 7:992 - 1000; PMID: 16331885; http://dx.doi.org/10.1593/neo.05355
- Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T, et al. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene 2007; 26:5655 - 5661; PMID: 17334390; http://dx.doi.org/10.1038/sj.onc.1210343
- Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 2008; 68:2366 - 2374; PMID: 18381444; http://dx.doi.org/10.1158/0008-5472.CAN-07-5783
- Carol H, Morton CL, Gorlick R, Kolb EA, Keir ST, Reynolds CP, et al. Initial testing (stage 1) of the Akt inhibitor GSK690693 by the pediatric preclinical testing program. Pediatr Blood Cancer 2010; 55:1329 - 1337; PMID: 20740623; http://dx.doi.org/10.1002/pbc.22710
- Dey A, Wong E, Kua N, Teo HL, Tergaonkar V, Lane D. Hexamethylene bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NF kappaB activity: implications for cancer therapy. Cell Cycle 2008; 7:3759 - 3767; PMID: 19029824; http://dx.doi.org/10.4161/cc.7.23.7213
- Andreeff M, Stone R, Michaeli J, Young CW, Tong WP, Sogoloff H, et al. Hexamethylene bisacetamide in myelodysplastic syndrome and acute myelogenous leukemia: a phase II clinical trial with a differentiation-inducing agent. Blood 1992; 80:2604 - 2609; PMID: 1421378
- Reddy KK, Lefkove B, Chen LB, Govindarajan B, Carracedo A, Velasco G, et al. The antidepressant sertraline downregulates Akt and has activity against melanoma cells. Pigment Cell Melanoma Res 2008; 21:451 - 456; PMID: 18710373; http://dx.doi.org/10.1111/j.1755-148X.2008.00481.x
- Proud CG. mTOR Signalling in Health and Disease. Biochem Soc Trans 2011; 39:431 - 436; PMID: 21428914; http://dx.doi.org/10.1042/BST0390431
- Roychowdhury A, Sharma R, Kumar S. Recent advances in the discovery of small molecule mTOR inhibitors. Future Med Chem 2010; 2:1577 - 1589; PMID: 21426150; http://dx.doi.org/10.4155/fmc.10.233
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26:1932 - 1940; PMID: 17001314; http://dx.doi.org/10.1038/sj.onc.1209990
- Marone R, Erhart D, Mertz AC, Bohnacker T, Schnell C, Cmiljanovic V, et al. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res 2009; 7:601 - 613; PMID: 19372588; http://dx.doi.org/10.1158/1541-7786.MCR-08-0366
- Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, et al. Reduced VEGF Production, Angiogenesis, and Vascular Regrowth Contribute to the Antitumor Properties of Dual mTORC1/mTORC2 Inhibitors. Cancer Res 2011; 71:1573 - 1583; PMID: 21363918; http://dx.doi.org/10.1158/0008-5472.CAN-10-3126
- Maira SM, Furet P, Stauffer F. Discovery of novel anticancer therapeutics targeting the PI3K/Akt/mTOR pathway. Future Med Chem 2009; 1:137 - 155; PMID: 21426073; http://dx.doi.org/10.4155/fmc.09.5
- Heron-Milhavet L, Khouya N, Fernandez A, Lamb NJ. Akt1 and Akt2: Differentiating the aktion. Histol Histopathol 2011; 26:651 - 662; PMID: 21432781
- Yoeli-Lerner M, Chin YR, Hansen CK, Toker A. Akt/protein kinase b and glycogen synthase kinase-3beta signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol Cancer Res 2009; 7:425 - 432; PMID: 19258413; http://dx.doi.org/10.1158/1541-7786.MCR-08-0342
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene 2003; 22:3138 - 3151; PMID: 12789290; http://dx.doi.org/10.1038/sj.onc.1206454
- Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, et al. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res 2008; 68:7638 - 7649; PMID: 18794153; http://dx.doi.org/10.1158/0008-5472.CAN-07-6614
- Gaitonde S, De SK, Tcherpakov M, et al. BI-69A11-mediated inhibition of AKT leads to effective regression of xenograft melanoma. Pigment Cell Melanoma Res 2009;
- Kapetanovic IM, Muzzio M, Hu SC, Crowell JA, Rajewski RA, Haslam JL, et al. Pharmacokinetics and enhanced bioavailability of candidate cancer preventative agent, SR13668 in dogs and monkeys. Cancer Chemother Pharmacol 2010; 65:1109 - 1116; PMID: 19756605; http://dx.doi.org/10.1007/s00280-009-1116-4
- Chao WR, Yean D, Amin K, Green C, Jong L. Computer-aided rational drug design: a novel agent (SR13668) designed to mimic the unique anticancer mechanisms of dietary indole-3-carbinol to block Akt signaling. J Med Chem 2007; 50:3412 - 3415; PMID: 17602463; http://dx.doi.org/10.1021/jm070040e
- Vasudevan KM, Garraway LA. AKT signaling in physiology and disease. Curr Top Microbiol Immunol 2010; 347:105 - 133; PMID: 20549472; http://dx.doi.org/10.1007/82_2010_66
- Cherrin C, Haskell K, Howell B, Jones R, Leander K, Robinson R, et al. An allosteric Akt inhibitor effectively blocks Akt signaling and tumor growth with only transient effects on glucose and insulin levels in vivo. Cancer Biol Ther 2010; 9:493 - 503; PMID: 20139722; http://dx.doi.org/10.4161/cbt.9.7.11100
- Crouthamel MC, Kahana JA, Korenchuk S, Zhang SY, Sundaresan G, Eberwein DJ, et al. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin Cancer Res 2009; 15:217 - 225; PMID: 19118049; http://dx.doi.org/10.1158/1078-0432.CCR-08-1253
- Sudarsanam S, Johnson DE. Functional consequences of mTOR inhibition. Curr Opin Drug Discov Devel 2010; 13:31 - 40; PMID: 20047144
- Tan K, Kimber WA, Luan J, et al. Analysis of genetic variation in Akt2/PKB-beta in severe insulin resistance, lipodystrophy, type 2 diabetes, and related metabolic phenotypes. Diabetes 2007; 56:714 - 719; PMID: 17327441; http://dx.doi.org/10.2337/db06-0921
- Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf 2011; 34:97 - 115; PMID: 21247219; http://dx.doi.org/10.2165/11585040-000000000-00000
- Knowling M, Blackstein M, Tozer R, Bramwell V, Dancey J, Dore N, et al. A phase II study of perifosine (D-21226) in patients with previously untreated metastatic or locally advanced soft tissue sarcoma: A National Cancer Institute of Canada Clinical Trials Group trial. Invest New Drugs 2006; 24:435 - 439; PMID: 16528479; http://dx.doi.org/10.1007/s10637-006-6406-7
- Butowski N, Chang SM, Lamborn KR, Polley MY, Parvataneni R, Hristova-Kazmierski M, et al. Enzastaurin plus temozolomide with radiation therapy in glioblastoma multiforme: a phase I study. Neurooncol 2010; 12:608 - 613; PMID: 20156802; http://dx.doi.org/10.1093/neuonc/nop070
- Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, et al. Phase II trial of the oral mammalian target of rapamycin inhibitor everolimus in relapsed or refractory Waldenstrom macroglobulinemia. J Clin Oncol 2010; 28:1408 - 1414; PMID: 20142598; http://dx.doi.org/10.1200/JCO.2009.24.0994
- Soria JC, Shepherd FA, Douillard JY, Wolf J, Giaccone G, Crino L, et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann Oncol 2009; 20:1674 - 1681; PMID: 19549709; http://dx.doi.org/10.1093/annonc/mdp060
- Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer 2010; 10:368; PMID: 20630061; http://dx.doi.org/10.1186/1471-2407-10-368
- Temkin SM, Yamada SD, Fleming GF. A phase I study of weekly temsirolimus and topotecan in the treatment of advanced and/or recurrent gynecologic malignancies. Gynecol Oncol 2010; 117:473 - 476; PMID: 20347480; http://dx.doi.org/10.1016/j.ygyno.2010.02.022
- Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol 2005; 23:5294 - 5304; PMID: 15998902; http://dx.doi.org/10.1200/JCO.2005.23.622
- Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 2005; 23:5314 - 5322; PMID: 15955899; http://dx.doi.org/10.1200/JCO.2005.66.130
- Yang SX, Costantino JP, Kim C, Mamounas EP, Nguyen D, Jeong JH, et al. Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node-positive breast cancer. J Clin Oncol 2010; 28:2974 - 2981; PMID: 20479407; http://dx.doi.org/10.1200/JCO.2009.26.1602
- Andre F, Nahta R, Conforti R, Boulet T, Aziz M, Yuan LX, et al. Expression patterns and predictive value of phosphorylated AKT in early-stage breast cancer. Ann Oncol 2008; 19:315 - 320; PMID: 17804473; http://dx.doi.org/10.1093/annonc/mdm429
- Tokunaga E, Kataoka A, Kimura Y, Oki E, Mashino K, Nishida K, et al. The association between Akt activation and resistance to hormone therapy in metastatic breast cancer. Eur J Cancer 2006; 42:629 - 635; PMID: 16464571; http://dx.doi.org/10.1016/j.ejca.2005.11.025
- Crul M, Rosing H, de Klerk GJ, Dubbelman R, Traiser M, Reichert S, et al. Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur J Cancer 2002; 38:1615 - 1621; PMID: 12142051; http://dx.doi.org/10.1016/S0959-8049(02)00127-2
- Van Ummersen L, Binger K, Volkman J, Marnocha R, Tutsch K, Kolesar J, et al. A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res 2004; 10:7450 - 7456; PMID: 15569974; http://dx.doi.org/10.1158/1078-0432.CCR-03-0406
- Bailey HH, Mahoney MR, Ettinger DS, Maples WJ, Fracasso PM, Traynor AM, et al. Phase II study of daily oral perifosine in patients with advanced soft tissue sarcoma. Cancer 2006; 107:2462 - 2467; PMID: 17058289; http://dx.doi.org/10.1002/cncr.22308
- Hennessy BT, Lu Y, Poradosu E, Yu Q, Yu S, Hall H, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res 2007; 13:7421 - 7431; PMID: 18094426; http://dx.doi.org/10.1158/1078-0432.CCR-07-0760
- Argiris A, Cohen E, Karrison T, Esparaz B, Mauer A, Ansari R, et al. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol Ther 2006; 5:766 - 770; PMID: 16760642; http://dx.doi.org/10.4161/cbt.5.7.2874
- Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res 2010; 70:2548 - 2557; PMID: 20197468; http://dx.doi.org/10.1158/0008-5472.CAN-09-2503
- Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 2010; 19:1355 - 1366; PMID: 20846000; http://dx.doi.org/10.1517/13543784.2010.520701
- Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, Mitsiades C, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood 2006; 107:4053 - 4062; PMID: 16418332; http://dx.doi.org/10.1182/blood-2005-08-3434
- Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res 2005; 65:7429 - 7435; PMID: 16103096; http://dx.doi.org/10.1158/0008-5472.CAN-05-1042
- Terwogt JM, Mandjes IA, Sindermann H, Beijnen JH, ten Bokkel Huinink WW. Phase II trial of topically applied miltefosine solution in patients with skin-metastasized breast cancer. Br J Cancer 1999; 79:1158 - 1161; PMID: 10098751; http://dx.doi.org/10.1038/sj.bjc.6690184
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 1997; 89:239 - 250; PMID: 9108479; http://dx.doi.org/10.1016/S0092-8674(00)80203-2
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 2000; 97:10832 - 10837; PMID: 10995457; http://dx.doi.org/10.1073/pnas.170276797
- Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci USA 2006; 103:57 - 62; PMID: 16371460; http://dx.doi.org/10.1073/pnas.0609973103
- Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res 2008; 14:8302 - 8307; PMID: 19088048; http://dx.doi.org/10.1158/1078-0432.CCR-08-1002
- Peralba JM, DeGraffenried L, Friedrichs W, Fulcher L, Grünwald V, Weiss G, et al. Pharmacodynamic Evaluation of CCI-779, an Inhibitor of mTOR, in Cancer Patients. Clin Cancer Res 2003; 9:2887 - 2892; PMID: 12912932
- Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, et al. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer 2005; 104:1045 - 1048; PMID: 16007689; http://dx.doi.org/10.1002/cncr.21265
- Markovic SN, Suman VJ, Ingle JN, Kaur JS, Pitot HC, Loprinzi CL, et al. Peptide vaccination of patients with metastatic melanoma: improved clinical outcome in patients demonstrating effective immunization. Am J Clin Oncol 2006; 29:352 - 360; PMID: 16891861; http://dx.doi.org/10.1097/01.coc.0000217877.78473.a4
- Nelson MH, Dolder CR. Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Ann Pharmacother 2006; 40:261 - 269; PMID: 16418322; http://dx.doi.org/10.1345/aph.1G387
- Spector NL, Xia W, Burris H 3rd, Hurwitz H, Dees EC, Dowlati A, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005; 23:2502 - 2512; PMID: 15684311; http://dx.doi.org/10.1200/JCO.2005.12.157
- Swash M. New ideas for therapy in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2005; 6:3 - 4; PMID: 16036419; http://dx.doi.org/10.1080/14660820510035379
- Khan AJ, Wall B, Ahlawat S, Green C, Schiff D, Mehnert JM, et al. Riluzole enhances ionizing radiation-induced cytotoxicity in human melanoma cells that ectopically express metabotropic glutamate receptor 1 in vitro and in vivo. Clin Cancer Res 2011; 17:1807 - 1814; PMID: 21325066; http://dx.doi.org/10.1158/1078-0432.CCR-10-1276
- Marín YE, Namkoong J, Cohen-Solal K, Shin SS, Martino JJ, Oka M, et al. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal 2006; 18:1279 - 1286; PMID: 16305822; http://dx.doi.org/10.1016/j.cellsig.2005.10.012
- Namkoong J, Shin SS, Lee HJ, Marín YE, Wall BA, Goydos JS, et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res 2007; 67:2298 - 2305; PMID: 17332361; http://dx.doi.org/10.1158/0008-5472.CAN-06-3665
- Yip D, Le MN, Chan JL, Lee JH, Mehnert JA, Yudd A, et al. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin Cancer Res 2009; 15:3896 - 3902; PMID: 19458050; http://dx.doi.org/10.1158/1078-0432.CCR-08-3303
- Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, et al. eEF-2 Kinase Dictates Cross-Talk between Autophagy and Apoptosis Induced by Akt Inhibition, Thereby Modulating Cytotoxicity of Novel Akt Inhibitor MK-2206. Cancer Res 2011; 71:2654 - 2663; PMID: 21307130; http://dx.doi.org/10.1158/0008-5472.CAN-10-2889
- Garrett CR, Coppola D, Wenham RM, Cubitt CL, Neuger AM, Frost TJ, et al. Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest New Drugs 2011; 29:1381 - 1389; PMID: 20644979; http://dx.doi.org/10.1007/s10637-010-9479-2
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335 - 2342; PMID: 15175435; http://dx.doi.org/10.1056/NEJMoa032691
- López-Fauqued M, Gil R, Grueso J, Hernandez-Losa J, Pujol A, et al. The dual PI3K/mTOR inhibitor PI-103 promotes immunosuppression, in vivo tumor growth and increases survival of sorafenib-treated melanoma cells. Int J Cancer 2010; 126:1549 - 1561; PMID: 19810100
- Lasithiotakis KG, Sinnberg TW, Schittek B, Flaherty KT, Kulms D, Maczey E, et al. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J Invest Dermatol 2008; 128:2013 - 2023; PMID: 18323781; http://dx.doi.org/10.1038/jid.2008.44
- Sinnberg T, Lasithiotakis K, Niessner H, et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J Invest Dermatol 2009; 129:1500 - 1515; PMID: 19078992; http://dx.doi.org/10.1038/jid.2008.379
- Pópulo H, Soares P, Rocha AS, Silva P, Lopes JM. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res 2010; 20:107 - 117; PMID: 20173664; http://dx.doi.org/10.1097/CMR.0b013e32832ccd09
- Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res 2010; 70:6670 - 6681; PMID: 20647317; http://dx.doi.org/10.1158/0008-5472.CAN-09-4471
- Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010; 467:596 - 599; PMID: 20823850; http://dx.doi.org/10.1038/nature09454
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 819; PMID: 20818844; http://dx.doi.org/10.1056/NEJMoa1002011
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757 - 1765; PMID: 18946061; http://dx.doi.org/10.1056/NEJMoa0804385
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464:427 - 430; PMID: 20179705; http://dx.doi.org/10.1038/nature08902
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468:968 - 972; PMID: 21107320; http://dx.doi.org/10.1038/nature09627
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468:973 - 977; PMID: 21107323; http://dx.doi.org/10.1038/nature09626
- Gopal YN, Deng W, Woodman SE, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res 2010; 70:8736 - 8747; PMID: 20959481; http://dx.doi.org/10.1158/0008-5472.CAN-10-0902
- Chang TT, Chou TC. Rational approach to the clinical protocol design for drug combinations: a review. Acta Paediatr Taiwan 2000; 41:294 - 302; PMID: 11198934
- Tran MA, Smith CD, Kester M, Robertson GP. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res 2008; 14:3571 - 3581; PMID: 18519791; http://dx.doi.org/10.1158/1078-0432.CCR-07-4881
- Manara MC, Nicoletti G, Zambelli D, Ventura S, Guerzoni C, Landuzzi L, et al. NVP-BEZ235 as a new therapeutic option for sarcomas. Clin Cancer Res 2010; 16:530 - 540; PMID: 20068094; http://dx.doi.org/10.1158/1078-0432.CCR-09-0816
- Deng WG, Kwon J, Ekmekcioglu S, Poindexter NJ, Grimm EA. IL-24 gene transfer sensitizes melanoma cells to erlotinib through modulation of the Apaf-1 and Akt signaling pathways. Melanoma Res 2010; In press PMID: 20216471
- Fisher PB, Sarkar D, Lebedeva IV, Emdad L, Gupta P, Sauane M, et al. Melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24): novel gene therapeutic for metastatic melanoma. Toxicol Appl Pharmacol 2007; 224:300 - 307; PMID: 17208263; http://dx.doi.org/10.1016/j.taap.2006.11.021
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711 - 723; PMID: 20525992; http://dx.doi.org/10.1056/NEJMoa1003466