Abstract
Glutamine metabolism is crucial for cancer cell growth via the generation of intermediate molecules in the tricarboxylic acid (TCA) cycle, antioxidants and ammonia. The goal of the current study was to evaluate the effects of glutamine on metabolism in the breast cancer tumor microenvironment, with a focus on autophagy and cell death in both epithelial and stromal compartments. For this purpose, MCF7 breast cancer cells were cultured alone or co-cultured with non-transformed fibroblasts in media containing high glutamine and low glucose (glutamine +) or under control conditions, with no glutamine and high glucose (glutamine –). Here, we show that MCF7 cells maintained in co-culture with glutamine display increased mitochondrial mass, as compared with control conditions. Importantly, treatment with the autophagy inhibitor chloroquine abolishes the glutamine-induced augmentation of mitochondrial mass. It is known that loss of caveolin-1 (Cav-1) expression in fibroblasts is associated with increased autophagy and an aggressive tumor microenvironment. Here, we show that Cav-1 downregulation which occurs in fibroblasts maintained in co-culture specifically requires glutamine. Interestingly, glutamine increases the expression of autophagy markers in fibroblasts, but decreases expression of autophagy markers in MCF7 cells, indicating that glutamine regulates the autophagy program in a compartment-specific manner. Functionally, glutamine protects MCF7 cells against apoptosis, via the upregulation of the anti-apoptotic and anti-autophagic protein TIGAR. Also, we show that glutamine cooperates with stromal fibroblasts to confer tamoxifen-resistance in MCF7 cancer cells. Finally, we provide evidence that co-culture with fibroblasts (1) promotes glutamine catabolism, and (2) decreases glutamine synthesis in MCF7 cancer cells. Taken together, our findings suggest that autophagic fibroblasts may serve as a key source of energy-rich glutamine to fuel cancer cell mitochondrial activity, driving a vicious cycle of catabolism in the tumor stroma and anabolic tumor cell expansion.
Introduction
Glutamine is a crucial amino acid in cancer cell metabolism. It is the most abundant free amino acid in plasma and cancer patients have increased glutamine plasma levels.Citation1 Also, cancer cells show high glutamine uptakeCitation2 and they metabolize glutamine at a much higher rate than any other aminoacid.Citation3,Citation4 The mechanism(s) by which glutamine promotes cancer growth are poorly understood. However, it is known that glutamine plays an important role in replenishing catabolic and anabolic intermediate metabolites, in generating antioxidants and modulating autophagy. Rapidly proliferating cells require glutamine and its byproduct α-ketoglutarate to replenish tricarboxylic acid (TCA) cycle intermediates during cell growth. For example, in proliferating glioblastoma cells, the TCA cycle intermediate, oxaloacetate, is derived predominantly from glutamine.Citation5 Glutamine is also essential in catabolic reactions generating ATP, in anabolic reactions for nucleotide and fatty acid synthesis and in generating the antioxidants NADH and glutathione.Citation1 Also, ammonia derived from glutamine is an important diffusible stimulator of autophagy, which could possibly promote cancer cell survival and increase resistance to anticancer drugs.Citation6
Glutamine may also be important in tumor progression because it allows metabolic-coupling between different organ systems and perhaps between different compartments within a tumor. Despite having high energetic requirements, tumors excrete large amounts of energy-rich metabolites, such as glutamine, alanine and lactate. This may seem metabolically inefficient, but it is now understood that the release and uptake of these metabolites and their byproducts between different organs and/or tumor compartments is energetically efficient and promotes tumor growth.Citation7 For example, lactate and alanine secreted by tumors are converted into glucose in the liver by gluconeogenesis (Cori cycle) and subsequently taken up by cancer cells.Citation8,Citation9 Metabolite transfer and metabolic-coupling can also occur within the tumor itself.Citation10–Citation13 Cancer-associated fibroblasts or hypoxic epithelial cancer cells with impaired oxidative phosphorylation secrete lactate that is then absorbed by epithelial cancer cells with functional mitochondria to sustain their oxidative phosphorylation.Citation10,Citation14
We and others have found that the loss of stromal Cav-1 is associated with poor clinical outcome in breast cancer and prostate cancer.Citation14–Citation18 Loss of stromal Cav-1 leads to metabolic-coupling between the epithelial and stromal tumor compartments, with high secretion of glutamine from the stroma.Citation14,Citation19 Some of the mechanisms by which a loss of stromal Cav-1 induces metabolic-coupling and promotes tumor growth have recently been elucidated. Via the generation of reactive oxygen species (ROS), epithelial cancer cells induce a loss of Cav-1 in fibroblasts. Fibroblasts with a loss of Cav-1 display catabolic metabolism with increased autophagy and mitophagy, impaired mitochondrial function and release high-energy metabolites (such as lactate) and macromolecules (such as glutamine and other aminoacids). Conversely, epithelial cancer cells in proximity to these catabolic fibroblasts have increased uptake of these high-energy metabolites to sustain their oxidative metabolism and growth.Citation11,Citation12,Citation19–Citation21
We have previously shown that loss of Cav-1 in the stroma greatly increases autophagy in fibroblasts with increased secretion of glutamine and other aminoacids, and increases mitochondrial mass in adjacent epithelial cancer cells.Citation11,Citation12,Citation19 Since glutamine is essential for cancer metabolism and glutamine-derived ammonia is a stimulator of autophagy, we aimed to investigate the effects of glutamine on fibroblast and epithelial cancer cell autophagy, cell survival and bioenergetics in a well-characterized epithelial-stromal model of breast cancer.
Here, we demonstrate that glutamine exerts different metabolic effects on the epithelial and stromal tumor compartments. We show that glutamine generates aggressive tumor cells with higher mitochondrial mass and decreased autophagy, and transforms the stroma into a tumor-promoting state, with a loss of Cav-1 and increased autophagy. Therefore, glutamine-induced autophagy in fibroblasts promotes mitochondrial biogenesis in epithelial cancer cells. Autophagy inhibitors such as chloroquine are a promising treatment strategy for tumors with high glutamine uptake and high stromal autophagy, since they would metabolically uncouple cancer cells and fibroblasts, and reduce mitochondrial oxidative activity in the epithelial cancer cells.
Results
In a coculture model, glutamine increases mitochondrial mass in MCF7 cells and decreases Cav-1 expression in fibroblasts.
To evaluate the effects of glutamine in the tumor microenvironment, we used a coculture model system comprised of (1) human fibroblasts immortalized with the telomerase catalytic domain (hTERT-BJ1 fibroblasts), and (2) human breast cancer cells (MCF7). We are able to distinguish the two cell populations by using antibodies against cytokeratins 8/18 (K8/18) which are only expressed in MCF7 cells. Cells were maintained under two different culture conditions: (1) high glutamine with low glucose media [called thereafter glutamine (+) or high glutamine media, containing 10 mM glutamine and 5 mM glucose], and (2) high glucose with no glutamine media [called hereafter glutamine (−) or high glucose media, containing 0 mM glutamine and 25 mM glucose].
To visualize mitochondrial mass, we employed an antibody that stains the intact mitochondrial membrane and whose expression correlates with mitochondrial activity.Citation22 shows that the mitochondrial mass is specifically increased in MCF7 cells in coculture maintained in high glutamine media, as compared with no glutamine media. Importantly, the mitochondrial mass is augmented specifically in the epithelial cancer cells, but not in the fibroblast compartment. Therefore, mitochondrial biogenesis appears to be regulated by glutamine in a compartment-specific manner. As critical negative controls, homotypic cultures of fibroblasts and MCF7 cells were maintained in parallel in high glutamine media and stained with the intact mitochondria membrane antibody. shows that under these conditions, the mitochondrial mass is slightly higher in fibroblasts than in MCF7 cells, suggesting that the glutamine-induced augmentation of MCF7 cell mitochondria mass requires the presence of fibroblasts.
We next assessed if glutamine modulates Cav-1 expression in this fibroblast-MCF7 cell coculture system. shows that Cav-1 expression is decreased in the fibroblasts of cocultures maintained in a high glutamine media, as compared with the fibroblasts of cocultures maintained in media without glutamine. It has previously been shown that a loss of stromal Cav-1 increases (1) the secretion of glutamine,Citation19 and (2) the mitochondrial mass and activity of adjacent epithelial cancer cells.Citation11 Thus, our results suggest that fibroblast-derived glutamine may directly increase the mitochondrial mass of epithelial cancer cells.
Coculture with fibroblasts promotes glutamine catabolism, but decreases glutamine synthesis in MCF7 cancer cells.
Our data indicate that fibroblasts may generate glutamine that is absorbed by adjacent cancer cells to increase their mitochondrial mass. We reasoned that if fibroblasts are a source of glutamine, then the presence of fibroblasts may be sufficient to reduce glutamine neogenesis in adjacent cancer cells. To this end, we evaluated the expression of glutamate-ammonia ligase (GLUL) in MCF7 cells cultured alone or in coculture in high glutamine media. GLUL is the key enzyme in glutamine neogenesis, catalyzing the synthesis of glutamine from glutamate and ammonia. shows that MCF7 cells cultured alone have high GLUL expression. However, when cocultured with fibroblasts, MCF7 cells display a significant decrease in GLUL expression, suggesting that fibroblasts induce the downregulation of GLUL in MCF7 cells. As fibroblasts lacking Cav-1 display increased glutamine secretion, the decreased glutamine synthesis in cocultured MCF7 cells suggests the transfer of glutamine from fibroblasts to MCF7 cells.
We then investigated if the presence of fibroblasts stimulates glutamine catabolism in adjacent cancer cells. To this end, the expression of glutaminase (GLS) and glutamate dehydrogenase 1 (GLUD1) was assessed in MCF7 cells cultured alone or in coculture in high glutamine media. GLS and GLUD1 are mitochondrial enzymes that catalyze the first and second step of glutamine catabolism, respectively. GLS catalyzes the conversion of glutamine to glutamate, while GLUD1 catalyzes the deamination of glutamate to α-ketoglutarate and ammonia. As α-ketoglutarate is an intermediate of the TCA cycle, glutamine catabolism directly fuels mitochondrial activity. and C shows that GLS and GLUD1 are both greatly upregulated in cocultured MCF7 cells as compared with MCF7 cells alone, suggesting that fibroblasts promote the catabolism of glutamine in cancer cells.
We also evaluated if the presence of fibroblasts modulates the expression of key glutamine transporters. To this end, we determined the expression of SLC6A14 and SLC7A5 in MCF7 cells cultured alone or in coculture in high glutamine media. SLC6A14 mediates the uptake of glutamine, while SLC7A5 mediates the efflux of glutamine to the extracellular space. Interestingly, shows that SLC6A14 is greatly upregulated in cocultured MCF7 cells as compared with MCF7 cells alone, suggesting that the presence of fibroblasts stimulates the uptake of glutamine in cancer cells. Conversely, shows that SLC7A5 is downregulated in cocultured MCF7 cells as compared with MCF7 cells alone, suggesting that the presence of fibroblasts inhibits the cellular efflux of glutamine in cancer cells.
The autophagy inhibitor chloroquine blocks glutamine-induced mitochondrial biogenesis in MCF7 cells.
We next investigated if autophagy plays a role in the glutamine-induced augmentation of MCF7 cell mitochondrial mass. Previous studies have shown that a loss of stromal Cav-1 induces autophagy and glutamine production,Citation19 and glutamine is a stimulator of autophagy.Citation6 To disrupt this feed-forward mechanism, fibroblast-MCF7 cell cocultures were maintained in high glutamine media and were treated with the autophagy inhibitor chloroquine. To visualize the mitochondrial mass in the epithelial and stromal compartments, samples were immunostained with an antibody against the intact mitochondrial membrane. shows that treatment with chloroquine strongly reduces glutamine-induced mitochondrial biogenesis in epithelial cancer cells in coculture. No significant changes were observed in the mitochondrial mass of fibroblasts in coculture upon chloroquine treatment. These results suggest that glutamine-induced mitochondrial biogenesis in epithelial cancer cells in coculture is due to elevated autophagy in the fibroblasts.
To evaluate if increased autophagy of the stromal cells confers a growth advantage to cancer cells, MCF7 cells were cultured alone or cocultured with fibroblasts in high glutamine media in the presence of chloroquine or vehicle alone (CTRL). Apoptosis was then measured with propidium iodide (PI) staining. shows that chloroquine does not induce apoptosis in MCF7 cells cultured alone, but induces a 1.8-fold increase in the apoptosis of MCF7 cells cocultured with fibroblasts. These results suggest that increased autophagy in stromal cells normally protects cancer cells against apoptosis.
Glutamine increases autophagy in fibroblasts, but decreases autophagy in cancer cells.
In order to directly assess if glutamine induces autophagy in a compartment-specific manner, cultures of fibroblasts alone or MCF7 cells alone were incubated in media containing high glutamine or no glutamine. Cells were then analyzed by immuno-blot analysis using a panel of autophagy markers. shows that high glutamine media strongly increases the expression of autophagy markers (Lamp-1 and cathepsin B) in fibroblasts. However, MCF7 cells cultured in high glutamine media show just the opposite results, with decreased expression of autophagy markers, Lamp-1 and Beclin-1 (). These results clearly indicate that glutamine differentially modulates autophagy in a cell-type and compartment-specific manner.
Glutamine protects MCF7 cells from apoptosis, with increased TIGAR expression.
To determine if high glutamine confers a functional advantage in epithelial cancer cells, we next evaluated the apoptotic rates of MCF7 cells alone, maintained in high glutamine or high glucose media. To this end, the percentage of MCF7 cells that are positive for Annexin V (a marker of early and late apoptosis) and PI (a marker of late apoptotic and dead cells) was measured under these conditions. Interestingly, MCF7 cells cultured with high glutamine display a 2-fold decrease in apoptosis, as measured by the percentage of Annexin V positive cells, compared with cells cultured with high glucose (, left). Similarly, MCF7 cells with high glutamine show a 1.7-fold decrease in total cell death as measured by the percentage of cells that are Annexin V positive and/or uptake PI (, right). Thus, glutamine protects MCF7 cells against apoptosis. These findings are consistent with our previous studies showing that fibroblasts with Cav-1 downregulation secrete more glutamine and also protect MCF7 cells from apoptosis.Citation23
To investigate the mechanism(s) by which glutamine protects MCF7 cells from apoptosis, we assessed the expression of the protein TIGAR in MCF7 cells cultured with high glutamine media or high glucose media. TIGAR is a multi-functional protein that inhibits glycolysis, apoptosis and autophagy. Western blot analysis shows that TIGAR expression is greatly increased in MCF7 cells cultured with glutamine, compared with cells cultured without glutamine (). These results indicate that upregulation of TIGAR may be one of the mechanism by which glutamine protects epithelial cancer cells against apoptosis.
Glutamine and fibroblasts co-operate to protect MCF7 cells against tamoxifen-induced cell death.
We have previously shown that fibroblasts confer tamoxifen-resistance in MCF7 cells.Citation24 We next evaluated if glutamine and fibroblasts cooperate to protect MCF7 cells against tamoxifen-induced cell death. To this end, MCF7 cells in homotypic culture or cocultured with fibroblasts were maintained under either high glutamine or no glutamine conditions for 4 d. Then, the cells were treated with tamoxifen (12 µM) or vehicle alone (CTR) for 24 h. Apoptosis was measured with annexin V and PI staining. and B show the percentage of apoptotic or dead MCF7 cells (Annexin V positive and/or PI positive). Note that for simplicity of explanation, the same graph is shown twice in panel and B.
shows that MCF7 cells cultured alone with high glutamine display a 3.3-fold decrease in apoptosis compared with cells cultured with no glutamine (first and second bars). Interestingly, MCF7 cell tamoxifen-induced apoptosis is decreased by 1.5-fold under high glutamine conditions, as compared with no glutamine (third and fourth bars), suggesting that high glutamine protects MCF7 cells from tamoxifen-induced apoptosis.
We then evaluated the combined effects of glutamine and fibroblasts. Interestingly, under untreated conditions (CTR), cocultured MCF7 cells with high glutamine show a 1.9-fold decrease in apoptosis compared with cells cultured with no glutamine (fifth and sixth bar). Moreover, high glutamine greatly protects cocultured MCF7 cells against tamoxifen-induced apoptosis. Note that upon tamoxifen treatment, cocultured MCF7 cells in high glutamine show a 3.4-fold decrease in apoptosis compared with cells cultured with no glutamine (seventh and eighth bars). These results indicate that high glutamine protects MCF7 cells from tamoxifen-induced apoptosis, and that glutamine and fibroblasts co-operate to induce tamoxifen resistance.
further illustrates that fibroblasts cooperate with glutamine to confer tamoxifen-resistance. In untreated control conditions (CTR), cocultured MCF7 cells maintained with glutamine show a 1.5-fold reduction in apoptosis compared with MCF7 cells alone (second and sixth bars). However, upon tamoxifen treatment in the presence of glutamine, fibroblasts induce a 4.3-fold decrease in MCF7 cell apoptosis (fourth and eighth bars). Taken together, these results clearly demonstrate that metabolic re-arrangements in the tumor microenvironment may drive tamoxifen-resistance.
Most remarkably, in the absence of glutamine, > 85% of the MCF7 cells undergo apoptosis in response to tamoxifen treatment (, panels A and B). In contrast, in the presence of glutamine and stromal fibroblasts, > 85% of the MCF7 cells are protected against tamoxifen, and fail to undergo apoptosis (compare the third and eighth bars in ). Thus, tamoxifen-resistance may be a metabloic/stromal phenomenon, conferred by glutamine and autophagic host fibroblasts.
Discussion
The current study shows that the effects of glutamine in the tumor microenvironment are compartment-specific. In the tumor epithelial compartment, glutamine augments mitochondrial biogenesis, decreases autophagy and protects cancer cells from apoptotic cell death, with the upregulation of TIGAR expression. However, glutamine has opposite effects in the stromal compartment, by decreasing Cav-1 expression and increasing stromal autophagy.
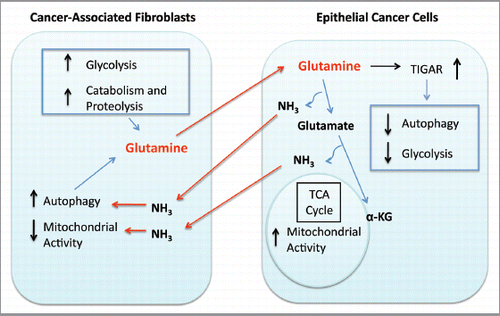
Based on these and other supporting data, we propose a working model in which metabolic-coupling occurs between epithelial cancer cells and the tumor stroma. This working model () indicates that cancer-associated fibroblasts (CAFs) undergo an autophagic program, leading to the generation and secretion of high glutamine levels into the tumor microenvironment. Cancer cells then take up glutamine and convert it to glutamate and ammonia. Glutamate is further catabolized to α-ketoglutarate, which then enters the TCA cycle and increases the mitochondrial activity of epithelial cancer cells. Epithelial cancer cells are protected from autophagy and apoptosis by the upregulation of the autophagy inhibitor TIGAR. The other byproduct, ammonia, freely diffuses into the microenvironment, and stimulates autophagy and glutamine production in CAFs. Finally, the use of autophagy inhibitors such as chloroquine can uncouple the epithelial and stromal compartments, driving a decrease in epithelial mitochondrial activity.
Interestingly, our newly proposed model of metabolic-coupling between epithelial and stromal cancer cells dramatically resembles a well-accepted physiological process, known as the glutamate-glutamine cycle in the brain.Citation25–Citation29 In the central nervous system, neurons are metabolically coupled to astrocytes. Astrocytes are responsible to provide neurons with the glutamate-precursor glutamine and with lactate, which is used by neuronal cells as preferred energy substrates. The export of glutamine from astrocytes and the uptake of glutamine by neurons are integral steps of the glutamate-glutamine cycle. Thus, astrocytes avidly accumulate glutamate by the high affinity transporter GLT1, and glutamine synthase (GLUL) activity is confined almost exclusively in astrocytes.Citation25 Conversely, neurons take up glutamine and express glutaminase (GLS) that catalyzes the hydrolysis of glutamine to glutamate.Citation25 Thus, cancer cells may be able to imitate and exploit a known physiological process to sustain their growth.
Our results are also consistent with observations made in cancer patients with cachexia. Autophagy in the skeletal muscle of cancer patients with cachexia is well described, and is believed to promote tumor growth. In fact, more than 90% of the body's glutamine is stored in the muscle and glutamine is the main aminoacid excreted from muscle under catabolic stress conditions. The presence of tumors leads to increased glutamine excretion from muscle.Citation30,Citation31 The glutamine-generating enzyme glutamate ammonia ligase (GLUL) is also increased in patients with cachexia.Citation1 Therefore, there is a strong suggestion that in patients with cancer-induced cachexia, metabolic coupling occurs systemically between muscle and epithelial cancer cells. Glutamine secretion is increased in the muscle, and conversely, high glutamine uptake occurs in cancer cells, which then generate ammonia. Ammonia diffuses out of cancer cells, and then promotes an autophagic microenvironment, further fueling catabolism and cachexia.
Our results indicate that the glutamine neosynthesis is decreased (as judged by GLUL staining), while glutamine catabolism is increased (as judged by GLS and GLUD1 staining) in epithelial cancer cells cocultured with fibroblasts, suggesting that stromal cells are a major source of glutamine in the tumor microenvironment. Consistent with this proposal, the expression of the transporter for the glutamine uptake, SLC6A14, is largely confined to epithelial cancer cells under coculture conditions. Future studies are warranted to assess glutamine metabolism in the epithelial and stromal compartments of patients with cancer-induced cachexia.
Not only are glutamine levels high in cancer patients, but cancer cells also exhibit high glutaminase (GLS) activityCitation32 and GLS levels correlate with tumor proliferation.Citation33,Citation34 Importantly, pharmacologic inhibition of GLS reduces oncogenic transformation.Citation35 Some studies have also shown that the downregulation of GLUL, a key enzyme in glutamine synthesis, correlates with a worse prognosis in some tumors.Citation36,Citation37 Accordingly, our data indicate that when cocultured with fibroblasts under conditions that better mimic the tumor microenvironment, cancer cells display the downregulation of GLUL, suggesting a decreased need for glutamine neogenesis, and increase GLS and GLUD1, two key enzymes in glutamine catabolism. It will be important to evaluate the prognostic value of GLS and GLUL in the different tumor compartments (stromal vs. epithelial), especially as this relates to tumor progression.
The current study also sheds new lights on the mechanism(s) by which glutamine promotes tumor growth. Our data indicate that glutamine modulates Cav-1 expression in stromal cells. High glutamine induces autophagy and loss of Cav-1 in the stroma. We have previously shown that a loss of stromal Cav-1 is sufficient to increase the generation of ROS, increases autophagy and decreases mitochondrial mass in fibroblasts.Citation12 In addition, Cav-1 (-/-) null stromal tissue secretes high levels of glutamine.Citation19 Hence, both glutamine and a loss of Cav-1 increase autophagy in fibroblasts. Our findings are also consistent with previous studies showing that in astrocytes, the glutamine byproduct ammonia induces ROS and autophagy.Citation38,Citation39 The finding that glutamine induces Cav-1 downregulation in the fibroblasts cocultured with cancer cells is of critical importance to understand breast tumorigenesis. In fact, loss of Cav-1 expression in cancer-associated fibroblasts is a marker of a lethal tumor-promoting microenvironment, associated with poor prognosis in breast cancer patients.Citation40,Citation41
Functionally, we show that glutamine and fibroblasts cooperate to sustain high mitochondrial activity in epithelial cancer cells and to protect epithelial cancer cells from autophagy and apoptotic cell death. Importantly, we also show that glutamine and stromal cells act synergistically to confer tamoxifen-resistance. Mechanistically, glutamine leads to decreased autophagy in the epithelial cancer cells with the upregulation of TIGAR. Previous studies have shown that the protein TIGAR inhibits glycolysis, apoptosis and autophagy and protects cells from oxidative stress.Citation42,Citation43
Our findings are consistent with a previous report showing that glutamine-derived ammonia does not alter mitochondrial activity and membrane polarity in the epithelial tumor compartment.Citation6 It was also shown that the addition of glutamine or α-ketoglutarate protects epithelial cells from TNF-induced cell death.Citation6
It is well-known that the oncogene c-Myc regulates glutamine metabolism. c-Myc via miR23a/b leads to increased glutaminase activity,Citation4 and increased expression of glutamine transporters.Citation44 Also, by increasing glutamine metabolism, c-Myc protects transformed cells from apoptosis.Citation45 It has been difficult to reconcile under the conventional Warburg Effect framework that c-Myc promotes aerobic glycolysis, glucose uptakeCitation46 and causes decreased mitochondrial function, and that at the same time promotes glutamine metabolism, leading to increased mitochondrial function. It is possible that in a subset of tumors, epithelial cancer cells with upregulation of c-Myc have increased uptake of glutamine with the generation of α-ketoglutarate and ammonia. α-Ketoglutarate then promotes mitochondrial activity in epithelial cells, while ammonia diffuses out of the cancer cells and is taken up by the stroma where it promotes autophagy, inhibits mitochondrial activity and promotes aerobic glycolysis. Further experiments to evaluate if c-Myc plays a role in this type of metabolic-coupling will be required.
In conclusion, our study uncovers a novel role for glutamine metabolism in the regulation of autophagy in the tumor microenvironment, and clearly shows that glutamine differentially affects individual cell-types within the tumor microenvironment. In tumor epithelial cells, glutamine augments mitochondrial biogenesis, decreases autophagy and protects against apoptosis. By contrast, glutamine increases autophagy and decreases Cav-1 expression in the tumor stromal compartment.
Materials and Methods
Materials.
Antibodies used were as follows: Beclin-1 (Novus Biologicals, NBP1-00085), Cathepsin B (Santa Cruz Biotech, sc-13985), caveolin-1 (Santa Cruz Biotech, sc-894), Cytokeratin 8/18 (Fitzgerald Industries International, 20R-CP004), GLS (Proteintech, 12855-1-AP), GLUD1 (Abnova, H00002746-M01), GLUL (glutamate-ammonia ligase, Sigma, HPA007316), Lamp-1 (Santa Cruz Biotech, E5 sc-17768), mitochondrial intact surface antibody (Chemicon International, MAB1273), SLC6A14 (Sigma Prestige, HPA003193), SLC7A5 (Abnova, MAB1711), TIGAR (Abcam, ab37910). Drugs, media and reagents were as follows: Chloroquine (Sigma, C6628), 4,6-diamidino-2-phenylindole (DAPI) (D3571), Tamoxifen (Sigma), Prolong Gold Antifade mounting reagent (Invitrogen, P36930), GlutaMAX-1 (Gibco, 35050), D-(+)-Glucose solution (Sigma, G8769), RPMI-1640 without L-Glutamine and Glucose (MP Biomedicals, C1601), Annexin V-APC (BD, 550474), propidium iodide (Invitrogen, P21493).
Cell cultures.
The human breast adenocarcinoma cell line (MCF7 cells) was purchased from ATCC. Human skin fibroblasts immortalized with human telomerase reverse transcriptase (hTERT-BJ1) were originally purchased from Clontech, Inc. Both cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% Fetal Bovine Serum and Penicillin 100 units/mL-Streptomycin 100 µg/mL (Invitrogen) in a 37°C, 5% CO2 incubator.
Co-culture system.
Fibroblasts and MCF7 cells were plated onto glass coverslips in 12-well plates in 1 mL of growth media. Cells were seeded at a 3:1 fibroblast to MCF7 cell ratio and the total number of cells per well was 1 × 105. Cultures of fibroblasts and MCF7 cells alone were plated in parallel with the same number of cells of a given type as in coculture (7.5 × 104 fibroblasts and 2.5 × 104 MCF7 cells). After 24 h, the media was changed to RPMI-1640 without glucose and glutamine containing 10% NuSerum (a low protein alternative to FBS, BD Bioscience) and 1% Pen-Strep. The media was supplemented either with 25 mM glucose and 0 mM glutamine (High Glucose, or Glutamine −) or with 10 mM glutamine and 5 mM glucose (High Glutamine, or Glutamine +). Cells were kept in these media for 3–5 d. Chloroquine (25 µM) or vehicle alone (control conditions) was added on the fourth day and mitochondrial mass and apoptosis were assessed after 24 h. For the tamoxifen experiments, tamoxifen (12 µM) or vehicle alone (control conditions) was added on the fourth day and apoptosis was assessed after 24 h.
Immunofluorescence.
Cells were washed in PBS with 10 mM CaCl2 and 1 mM MgCl2 three times and fixed with 2% PFA. After fixation, cells were permeabilized with 0.1% Triton X-100 and 0.2% BSA in PBS for 10 min. For some antibodies, cells were permeabilized with cold methanol for 10 min. Then, cells were quenched in PBS with 25 mmol/L NH4Cl for 10 min. After quenching, cells were rinsed with PBS and then incubated with the primary antibodies for 1 h. After washing, cells were incubated with anti-mouse or anti-rabbit secondary fluorochrome conjugated antibodies for 30 min. After rinsing with PBS, cells were stained with DAPI to stain the nucleus and mounted using Prolong anti-fade reagent.
Confocal microscopy.
Images were taken with a Zeiss LSM510 meta confocal system. Images were acquired with a 40× objective with a 405 nM Diode excitation laser with a band pass filter of 420–480 nm, a 488 nm Argon excitation laser with a band pass filter of 505–550, and a 543 nm HeNe excitation laser with a 561–604 nm filter.
Western blot analysis.
Cells were washed with cold PBS and harvested into cold lysis buffer (10 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 60 mM octyl-glucoside), with protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Roche Applied Science). After rotating for 40 min at 4°C, samples were centrifuged at 13,000 xg for 15 min to remove cellular debris. Protein concentrations were determined by BCA reagent (Pierce). Thirty micrograms of protein were loaded and separated by 10% acrylamide SDS-PAGE and transferred to a 0.2 µm nitrocellulose membrane (Fisher Scientific). After blocking in 5% milk buffer for 30 min at room temperature, membranes were incubated with the primary antibodies for 1 h at room temperature. Then, the membranes were washed and incubated with the horseradish peroxidase-conjugated secondary antibodies for 30 min at room temperature. After washing, the bound antibodies were visualized with an enhanced-chemiluminescence (ECL) substrate.
Annexin-V and propidium iodide cell death detection.
Cell apoptosis was quantified by flow cytometry using PI and Annexin V-APC, as previously described with minor modifications.Citation24 Briefly, GFP-positive MCF7 cells were plated alone or together with fibroblasts in 10% FBS DMEM. Next day, the media was changed to either High Glucose or High Glutamine media. After 3–5 d, cells were collected by centrifugation and re-suspended in 400 µl of Annexin-V binding buffer with Annexin V-APC conjugate and propidium iodide. Cells were then incubated in the dark for 5 min and analyzed by flow cytometry using a PE Texas Red signal detector (for propidium iodide), an APC signal detector (for Annexin V), and a GFP signal detector (to select out the GFP-positive MCF7 cell population).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| CAFs | = | cancer-associated fibroblasts |
| Cav-1 | = | caveolin-1 |
| GLUD1 | = | glutamate dehydrogenase 1 |
| GLUL | = | glutamate-ammonia ligase |
| GLS | = | glutaminase |
| K8/18 | = | cytokeratin 8/18 |
| PI | = | propidium iodide |
| ROS | = | reactive oxygen species |
| TCA | = | tricarboxylic acid |
Figures and Tables
Figure 1 Glutamine increases the mitochondrial mass of MCF7 cells cocultured with fibroblasts. (A) Fibroblast-MCF7 cell cocultures were maintained in media containing high glutamine (right) or no glutamine (left) for 5 d. Cells were then fixed and immuno-stained with antibodies directed against the intact mitochondrial membrane (red) and K8/18 (green). Nuclei were counterstained with DAPI (blue). The upper panels show the mitochondrial staining (red channel). The bottom panels show the K8/18 staining (green channel) to identify the MCF7 cell population. Note that high glutamine increases the mitochondrial mass specifically in MCF7 cells as compared with cocultures maintained without glutamine. However, glutamine does not promote mitochondrial biogenesis in the fibroblasts. Original magnification, 40x. (B) Fibroblasts and MCF7 cells were maintained as single cell-type cultures in high glutamine media for 5 d. Cells were fixed and immunostained with antibodies directed against the intact mitochondrial membrane (red) and K8/18 (green). Nuclei were counter-stained with DAPI (blue). The upper panels show the mitochondrial staining (red). The bottom panels show K8/18 staining (green) labeling MCF7 cells. Note that when cultured as single cell-types in high glutamine conditions, the mitochondrial mass is slightly higher in fibroblasts than in MCF7 cells. Original magnification, 40×.
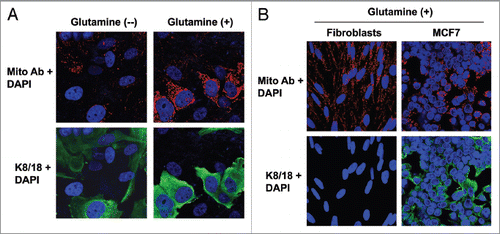
Figure 2 Glutamine induces the downregulation of Cav-1 in the fibroblast compartment. MCF7 cells and fibroblasts were cocultured for 5 d in media containing either high glutamine (right) or no glutamine (left). Cells were fixed and immunostained with antibodies against Cav-1 (red) and K8/18 (green). Nuclei were counter-stained with DAPI (blue). K8/18 staining is specific for MCF7 cells and Cav-1 is specific for fibroblasts since MCF7 cells lack Cav-1 expression. Note that Cav-1 expression is decreased in the fibroblast compartments of samples cultured with high glutamine, compared with no glutamine media. Original magnification, 40×.
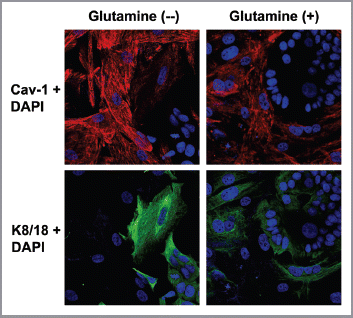
Figure 3 MCF7 cells in coculture show increased glutamine catabolism, but decreased glutamine synthesis. To evaluate glutamine metabolism, MCF7 cells were cultured alone or with fibroblasts for 5 d in high glutamine media. Then, cells were fixed and immunostained with antibodies against K8/18 (green) and GLUL (A, red) or GLS (B, red) or GLUD1 (C, red). Nuclei were counter-stained with DAPI (blue). K8/18 was used to distinguish MCF7 cells from fibroblasts. (A) Glutamine neosynthesis is decreased in MCF7 cells in coculture. GLUL catalyzes the synthesis of glutamine from glutamate and ammonia. Note that GLUL is highly expressed in single cultures of MCF7 cells. However, GLUL expression is decreased in MCF7 cells cultured with fibroblasts. (B and C) Glutamine catabolism is increased in MCF7 cells in coculture. GLS (B) and GLUD1 (C) catalyze the first and second steps respectively of the catabolism of glutamine. Note that both GLS and GLUD1 are upregulated in MCF7 cells in coculture, compared with MCF7 cells alone. Original magnification, 40× for all.
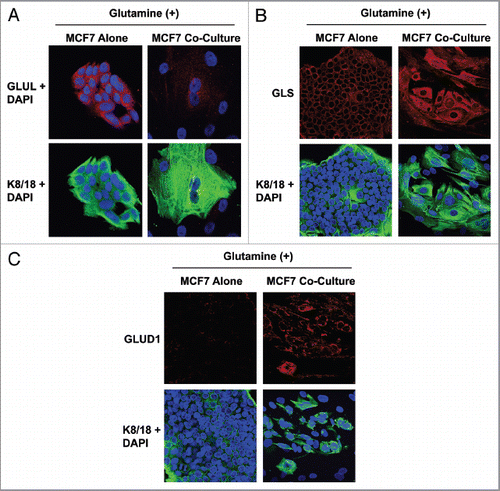
Figure 4 MCF7 cells in coculture show increased expression of the transporter for glutamine uptake, but decreased expression of the transporter for glutamine extrusion. MCF7 cells were cultured alone or with fibroblasts for 5 d in high glutamine media. Then, cells were fixed and immunostained with antibodies against K8/18 (green) and SLC6A14 (A, red) or SLC7A5 (B, red). Nuclei were counter-stained with DAPI (blue). K8/18 was used to distinguish MCF7 cells from fibroblasts. Note that SLC6A14, the transporter that facilitates glutamine uptake, is highly upregulated in MCF7 cells in coculture, compared with MCF7 cells alone. Conversely, SLC7A5, the transporter that facilitates glutamine extrusion, is greatly decreased in MCF7 cells in coculture, compared with MCF7 cells alone. Original magnification, 40×.
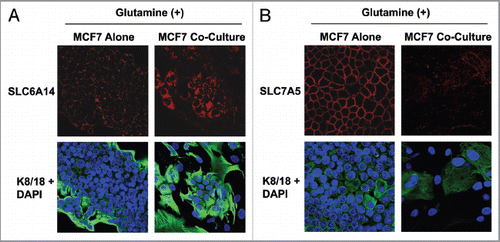
Figure 5 Chloroquine abolishes the glutamine-induced increase in mitochondrial mass in cocultured MCF7 cells, and augments apoptosis. (A) Fibroblasts were cocultured with MCF7 cells for 4 d in high glutamine media. Then, cells were treated either with 25 µM chloroquine (right) or vehicle alone (left) for 24 h. Cells were fixed and immunostained with antibodies against K8/18 (green) and the intact mitochondrial membrane (red). Nuclei were counter-stained with DAPI (blue). K8/18 specifically stains MCF7 cells (lower panels). The upper panels show the red channel only. Note that chloroquine treatment abolishes the glutamine-induced increase of mitochondrial mass of cocultured MCF7 cells. Original magnification, 40×. (B) MCF7 cells were cultured alone or in coculture with fibroblasts in the presence of chloroquine or vehicle alone (CTRL) under high glutamine conditions. Apoptosis was measured with PI staining. The percentage of apoptotic MCF7 cells (PI positive) is shown. Note that chloroquine does not induce apoptosis in MCF7 cells cultured alone, but does induce a 1.8-fold increase in apoptosis in MCF7 cells cocultured with fibroblasts.
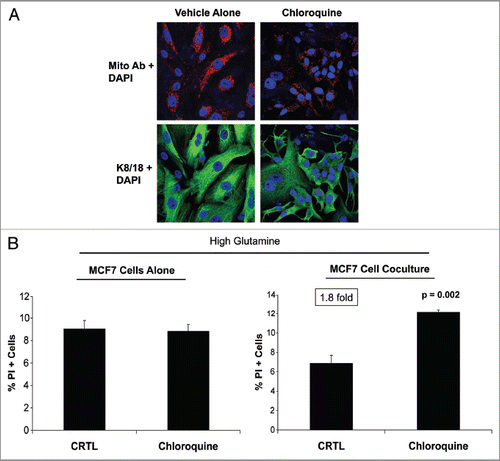
Figure 6 Glutamine decreases autophagy in MCF7 cells, but increases autophagy in fibroblasts. Single cultures of fibroblasts (A) and MCF7 cells (B) were incubated with high glutamine or no glutamine media for 3 d. Cells were then lysed and subjected to immuno-blot analysis with a panel of autophagy markers. Equal loading was assessed by β-actin immunoblotting. Note that high glutamine strongly induces the expression of autophagy markers (Lamp-1 and Cathepsin B) in the fibroblasts (A). However, high glutamine decreases the expression of autophagy markers (Beclin-1 and Lamp-1) in MCF7 cells (B). These results suggest that the glutamine-mediated effects on autophagy are compartment-specific.
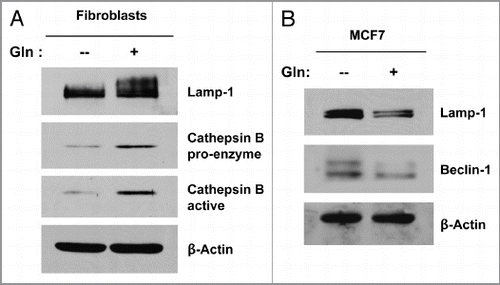
Figure 7 Glutamine protects MCF7 cells against apoptosis, with increased TIGAR expression. MCF7 cells were cultured alone in media containing high glutamine or high glucose (without glutamine) for 3 d. (A) Apoptosis was measured with annexin V and PI staining. The left graph shows the percentage of Annexin V-positive cells (early and late apoptosis). The right graph shows the percentage of total cell death (Annexin V-positive and/or PI-positive). Note that MCF7 cells cultured alone with high glutamine show an up to 2-fold decrease in apoptosis, as compared with MCF7 cells maintained in high glucose media. P values are as shown. (B) Cells were lysed and subjected to immuno-blot analysis with antibodies directed against the anti-apoptotic protein TIGAR. Equal loading was assessed by β-actin immunoblotting. Note that high glutamine increases the expression of TIGAR in MCF7 cells, compared with high glucose media.
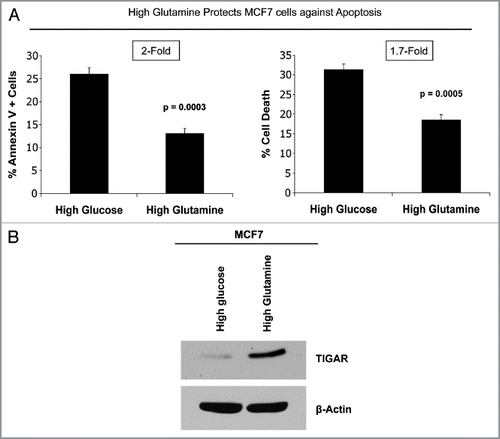
Figure 8 Glutamine protects MCF7 cells from tamoxifen-induced apoptosis. MCF7 cells either in homotypic culture or cocultured with fibroblasts were maintained either in high glutamine (Gln) or no glutamine conditions for 4 d. Then, cells were treated with tamoxifen (12 µM) or vehicle alone (CTR) for 24 h. Apoptosis was measured with annexin V and PI staining. The percentage of apoptotic or dead MCF7 cells (Annexin V positive and/or PI positive) is shown. (A and B) For simplicity of explanation, the same graph is shown twice in (A and B). (A) MCF7 cells alone. MCF7 cells alone cultured with high glutamine show a 3.3-fold decrease in apoptosis compared with cells cultured with no glutamine (first and second bars). Interestingly, high glutamine protects MCF7 cells from tamoxifen-induced apoptosis. Note that MCF7 cell tamoxifen-induced apoptosis is decreased by 1.5-fold in high glutamine conditions, as compared with no glutamine (third and fourth bars). Cocultured MCF7 cells. Note that under untreated conditions, cocultured MCF7 cells with high glutamine show a 1.9-fold decrease in apoptosis compared with cells cultured with no glutamine (fifth and sixth bars). However, high glutamine greatly protects cocultured MCF7 cells against tamoxifen-induced apoptosis. Note that upon tamoxifen treatment, cocultured MCF7 cells in high glutamine show a 3.4-fold decrease in apoptosis, compared with cells cultured with no glutamine (seventh and eighth bars). These results indicate that glutamine protects MCF7 cells fromtamoxifen-induced apoptosis, and that glutamine and fibroblasts cooperate to confer tamoxifen-resistance. P values are as shown. (B) Fibroblasts cooperate with glutamine to confer tamoxifen-resistance. In untreated control conditions (CTR), cocultured MCF7 cells maintained with glutamine show a 1.5-fold reduction in apoptosis compared with MCF7 cells alone (second and sixth bars). However, upon tamoxifen treatment in the presence of glutamine, fibroblasts induce a 4.3-fold decrease in MCF7 cell apoptosis (fourth and eighth bars). P values are as shown.
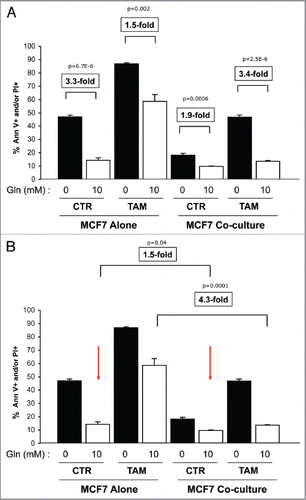
Figure 9 Glutamine addiction and stromal autophagy: a vicious cycle. Our current data suggest that metabolic-coupling occurs between epithelial cancer cells and stromal cells. Cancer-associated fibroblasts undergo an autophagic program, leading to the generation and secretion of high glutamine levels into the tumor microenvironment. Cancer cells accumulate glutamine and convert it to glutamate and ammonia. Glutamate is further catabolized to α-ketoglutarate, which enters the TCA cycle and increases the mitochondrial activity of epithelial cancer cells. Epithelial cancer cells are protected from apoptosis by the upregulation of TIGAR, an endogenous inhibitor of glycolysis, apoptosis and autophagy. The other glutamine byproduct, ammonia, freely diffuses into the microenvironment, and induces autophagy and glutamine production in CAFs. Finally, the use of autophagy inhibitors such as chloroquine can uncouple the epithelial and stromal compartments, resulting in decreased mitochondrial activity in epithelial cancer cells ( and ).
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Related Research Data
References
- DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313 - 324; PMID: 19881548; http://dx.doi.org/10.1038/onc.2009.358
- Bode BP, Fuchs BC, Hurley BP, Conroy JL, Suetterlin JE, Tanabe KK, et al. Molecular and functional analysis of glutamine uptake in human hepatoma and liver-derived cells. Am J Physiol Gastrointest Liver Physiol 2002; 283:G1062 - G1073; PMID: 12381519
- Eagle H. Nutrition needs of mammalian cells in tissue culture. Science 1955; 122:501 - 514; PMID: 13255879; http://dx.doi.org/10.1126/science.122.3168.501
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762 - 765; PMID: 19219026; http://dx.doi.org/10.1038/nature07823
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104:19345 - 19350; PMID: 18032601; http://dx.doi.org/10.1073/pnas.0709747104
- Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010; 3:ra31; PMID: 20424262; http://dx.doi.org/10.1126/scisignal.2000911
- Mariño G, Kroemer G. Ammonia: a diffusible factor released by proliferating cells that induces autophagy. Sci Signal 2010; 3:pe19; PMID: 20516476; http://dx.doi.org/10.1126/scisignal.3124pe19
- Holroyde CP, Skutches CL, Boden G, Reichard GA. Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 1984; 44:5910 - 5913; PMID: 6388829
- Tayek JA. A review of cancer cachexia and abnormal glucose metabolism in humans with cancer. J Am Coll Nutr 1992; 11:445 - 456; PMID: 1506607
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008; 118:3930 - 3942; PMID: 19033663
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276; PMID: 20814239; http://dx.doi.org/10.4161/cc.9.16.12553
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533; PMID: 20855962; http://dx.doi.org/10.4161/cc.9.17.12928
- Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res 2006; 66:632 - 637; PMID: 16423989; http://dx.doi.org/10.1158/0008-5472.CAN-05-3260
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001; PMID: 19923890; http://dx.doi.org/10.4161/cc.8.23.10238
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 2034; PMID: 19411448; http://dx.doi.org/10.2353/ajpath.2009.080873
- Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci 2011; 102:1590 - 1596; PMID: 21585620; http://dx.doi.org/10.1111/j.1349-7006.2011.01985.x
- Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol 2011; 32:787 - 799; PMID: 21584795; http://dx.doi.org/10.1007/s13277-011-0181-6
- Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8:2420 - 2424; PMID: 19556867; http://dx.doi.org/10.4161/cc.8.15.9116
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505; PMID: 20861672; http://dx.doi.org/10.4161/cc.9.17.12721
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433; PMID: 20562526; http://dx.doi.org/10.4161/cc.9.12.12048
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 1783; PMID: 21558814; http://dx.doi.org/10.4161/cc.10.11.15659
- Ogbureke KU, Fisher LW. SIBLING expression patterns in duct epithelia reflect the degree of metabolic activity. J Histochem Cytochem 2007; 55:403 - 409; PMID: 17210923; http://dx.doi.org/10.1369/jhc.6A7075.2007
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther 2010; 10:537 - 542; PMID: 20861671; http://dx.doi.org/10.4161/cbt.10.6.13370
- Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: Therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011; 10:2521 - 2528; PMID: 21768775; http://dx.doi.org/10.4161/cc.10.15.16584
- Bröer S, Brookes N. Transfer of glutamine between astrocytes and neurons. J Neurochem 2001; 77:705 - 719; PMID: 11331400; http://dx.doi.org/10.1046/j.1471-4159.2001.00322.x
- Waagepetersen HS, Sonnewald U, Schousboe A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: functional implications. Neuroscientist 2003; 9:398 - 403; PMID: 14580123; http://dx.doi.org/10.1177/1073858403254006
- Deitmer JW, Broer A, Broer S. Glutamine efflux from astrocytes is mediated by multiple pathways. J Neurochem 2003; 87:127 - 135; PMID: 12969260; http://dx.doi.org/10.1046/j.1471-4159.2003.01981.x
- Deitmer JW. Strategies for metabolic exchange between glial cells and neurons. Respir Physiol 2001; 129:71 - 81; PMID: 11738647; http://dx.doi.org/10.1016/S0034-5687(01)00283-3
- Bacci A, Sancini G, Verderio C, Armano S, Pravettoni E, Fesce R, et al. Block of glutamate-glutamine cycle between astrocytes and neurons inhibits epileptiform activity in hippocampus. J Neurophysiol 2002; 88:2302 - 2310; PMID: 12424271; http://dx.doi.org/10.1152/jn.00665.2001
- Parry-Billings M, Leighton B, Dimitriadis GD, Curi R, Bond J, Bevan S, et al. The effect of tumour bearing on skeletal muscle glutamine metabolism. Int J Biochem 1991; 23:933 - 937; PMID: 1773899; http://dx.doi.org/10.1016/0020-711X(91)90082-X
- Chen MK, Espat NJ, Bland KI, Copeland EM 3rd, Souba WW. Influence of progressive tumor growth on glutamine metabolism in skeletal muscle and kidney. Ann Surg 1993; 217:655 - 666; discussion 66–7 PMID: 8099476; http://dx.doi.org/10.1097/00000658-199306000-00007
- Szeliga M, Obara-Michlewska M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009; 55:71 - 75; PMID: 19428809; http://dx.doi.org/10.1016/j.neuint.2009.01.008
- Knox WE, Horowitz ML, Friedell GH. The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer Res 1969; 29:669 - 680; PMID: 5251291
- Linder-Horowitz M, Knox WE, Morris HP. Glutaminase activities and growth rates of rat hepatomas. Cancer Res 1969; 29:1195 - 1199; PMID: 4307782
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18:207 - 219; PMID: 20832749; http://dx.doi.org/10.1016/j.ccr.2010.08.009
- Matsuno T, Goto I. Glutaminase and glutamine synthetase activities in human cirrhotic liver and hepatocellular carcinoma. Cancer Res 1992; 52:1192 - 1194; PMID: 1346587
- Dal Bello B, Rosa L, Campanini N, Tinelli C, Torello Viera F, D'Ambrosio G, et al. Glutamine synthetase immunostaining correlates with pathologic features of hepatocellular carcinoma and better survival after radio-frequency thermal ablation. Clin Cancer Res 2010; 16:2157 - 2166; PMID: 20233882; http://dx.doi.org/10.1158/1078-0432.CCR-09-1978
- Bai G, Rama Rao KV, Murthy CR, Panickar KS, Jayakumar AR, Norenberg MD. Ammonia induces the mitochondrial permeability transition in primary cultures of rat astrocytes. J Neurosci Res 2001; 66:981 - 991; PMID: 11746427; http://dx.doi.org/10.1002/jnr.10056
- Murthy CR, Rama Rao KV, Bai G, Norenberg MD. Ammonia-induced production of free radicals in primary cultures of rat astrocytes. J Neurosci Res 2001; 66:282 - 288; PMID: 11592125; http://dx.doi.org/10.1002/jnr.1222
- Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res 2011; 13:213; PMID: 21867571; http://dx.doi.org/10.1186/bcr2892
- Pavlides S, Vera I, Gandara R, Sneddon S, Pestell R, Mercier I, et al. Warburg meets autophagy: cancer associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy and aerobic glycolysis. Antioxid Redox Signal 2011; In press PMID: 21883043; http://dx.doi.org/10.1089/ars.2011.4243
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107 - 120; PMID: 16839880; http://dx.doi.org/10.1016/j.cell.2006.05.036
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009; 28:3015 - 3026; PMID: 19713938; http://dx.doi.org/10.1038/emboj.2009.242
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 2008; 105:18782 - 18787; PMID: 19033189; http://dx.doi.org/10.1073/pnas.0810199105
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007; 178:93 - 105; PMID: 17606868; http://dx.doi.org/10.1083/jcb.200703099
- Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987; 235:1492 - 1495; PMID: 3103217; http://dx.doi.org/10.1126/science.3103217