Abstract
Spectral imaging systems are capable of detecting and quantifying subtle differences in light quality. In this study we coupled spectral imaging with fluorescence and white light microscopy to develop new methods for quantifying autophagy and apoptosis. For autophagy, we employed multispectral imaging to examine spectral changes in the fluorescence of LC3-GFP, a chimeric protein commonly used to track autophagosome formation. We found that punctate autophagosome-associated LC3-GFP exhibited a spectral profile that was distinctly different from diffuse cytosolic LC3-GFP. We then exploited this shift in spectral quality to quantify the amount of autophagosome-associated signal in single cells. Hydroxychloroquine (CQ), an anti-malarial agent that increases autophagosomal number, significantly increased the punctate LC3-GFP spectral signature, providing proof-of-principle for this approach. For studying apoptosis, we employed the Prism and Reflector Imaging Spectroscopy System (PARISS) hyperspectral imaging system to identify a spectral signature for active caspase-8 immunostaining in ex vivo tumor samples. This system was then used to rapidly quantify apoptosis induced by lexatumumab, an agonistic TRAIL-R2/DR5 antibody, in histological sections from a preclinical mouse model. We further found that the PARISS could accurately distinguish apoptotic tumor regions in hematoxylin and eosin-stained sections, which allowed us to quantify death receptor-mediated apoptosis in the absence of an apoptotic marker. These spectral imaging systems provide unbiased, quantitative and fast means for studying autophagy and apoptosis and complement the existing methods in their respective fields.
Introduction
Spectral imaging exposes nuances in light quality that cannot otherwise be detected by the human eye. Spectral imaging was developed by NASA and the remote Earth sensing community for monitoring climate and temperature change, and has since been adapted for use in a variety of academic and commercial applications. In the field of biomedical research, spectral analysis has been used for discerning malignant from normal tissue,Citation1 separating vascular from non-vascular regions in skin and tumors,Citation2,Citation3 and identifying cultured cells in different phases of the cell cycle.Citation4 Spectral imaging systems are highly versatile with endless applicability. Their quantitative nature and high resolving power make them ideal for use in cellular assays that measure autophagy and apoptosis, two forms of programmed cell death (PCD) that are critical to normal biological development,Citation5,Citation6 but also contribute to pathological conditions including neurodegenerative disease,Citation7 heart diseaseCitation8 and especially cancer.Citation9,Citation10
Autophagy (Latin for “self-eating”; PCD type II) is a process by which cells engulf portions of their cytoplasm and deliver them to lysosomes for degradation. While this is a destructive process that is commonly observed in dying cells, autophagy also functions as a survival mechanism, allowing cells to survive under stressful conditions.Citation11 For example, autophagy was shown to protect cancer cells from hypoxia, nutrient deprivation, and DNA damage-induced cell death.Citation12–Citation15 Recently autophagy indices were also shown to predict the invasive potential and aggressiveness of human melanomas in vitro as well as in animal models and patients.Citation16 Autophagy is studied using a compliment of assays, one of which involves fluorescence imaging of the chimeric protein, myosin light chain 3-green fluorescent protein (LC3-GFP), which tracks autophagosomes, the functional units of autophagy. At the onset of autophagy, LC3-I (a cytosolic protein) is conjugated to phosphatidylethanolamine (PE), and this lipified form of LC3, LC3-II, integrates into the outer membrane of autophagosomes.Citation17 Cytoslic LC3-GFP emits diffuse fluorescence, whereas autophagosome-associated LC3-GFP appears punctate. This is a useful system for identifying and quantifying those cells within a field that exhibit punctate LC3-GFP expression and are therefore actively undergoing autophagy. However, this method has limitations. For one, investigator bias is applied in developing criteria that define LC3-GFP puncta. Second, it is difficult to accurately determine the number of LC3-GFP puncta (i.e. autophagosomes) per cell, as opposed to the number of cells per field with detectable puncta.Citation18 As autophagy is homeostatic process, 100 % of cells could theoretically exhibit some basal level of autophagy. Hence, imaging systems that distinguish and enumerate punctate LC3-GFP structures at the subcellular level, without subjectivity, would potentially strengthen this assay.
Apoptosis (Greek for “a falling off”; PCD type I), is a controlled and deliberate form of cell death performed by the caspase family of proteases.Citation19 While cells may die through apoptosis-independent pathways,Citation20 many tumoricidal agents are effective due to their ability to initiate apoptosis. On the other hand, resistance to apoptosis is a hallmark of cancer that may arise due to the induction/expression of anti-apoptotic genes and/or aberrant signaling of pro-survival cellular pathways.Citation21 Apoptosis is associated with cell morphological features that distinguish it from others forms of cell death, such as necrosis.Citation22 Apoptosis induces chromatin condensation and nuclear fragmentation in its early and late stages, respectively. Apoptosis also reduces cytosolic volume and induces membrane blebbing and the formation of apoptotic bodies, or vesicles that form around apoptotic cells, which are thought to protect surrounding cells from toxic cellular contents. As these distinctive morphological effects make apoptosis easily visible to a trained eye, it is also conceivable that apoptosis is associated with quantifiable spectral changes.
Results
Quantitative multi-spectral imaging of the autophagosome marker, LC3.
In order to mimic the phenotypic changes of autophagy in LC3-GFP-expressing melanoma cell lines, we used 4-aminoquinoline, hydroxychloroquine (CQ). CQ inhibits late-stage autophagosome-lysosome fusion, leading to the accumulation of autophagosomes, an effect that resembles the induction of autophagy. CQ treatment increased the total levels of both LC3 isoforms, I and II, in C8161 and A375P melanoma cell lines ( and data not shown), and induced aggregation of LC3-GFP but not the GFP vector control (). CQ was therefore a useful tool compound for inducing autophagy-associated changes, like LC3 aggregation. We then set out to develop a multi-spectral imaging-based approach to rapidly and quantitatively monitor LC3-GFP localization in cultured cells.
We first imaged C8161 cells, which we noted to have relatively high autophagosome numbers under standard cell culture conditions ( and C). For our imaging system, we employed the Nuance Multispectral Imaging (MSI) System (CRi) fitted to a Nikon AZ100 multizoom stereomicroscope, with an excitation wavelength of 475nm and long-pass emission filter (500–750nm). We detected emitted wavelengths of light with three distinct spectral profiles corresponding to background fluorescence (black pseudocolor), diffuse LC3-GFP fluorescence (green pseudocolor), and punctate LC3-GFP fluorescence (red pseudocolor; ). By comparison, only one spectra (in addition to background) was detected for the emission profile of GFP in GFP vector control cells (Fig. S2). Although the punctate LC3-GFP fluorescence exhibited the same peak emission wavelength (∼525nm) as diffuse LC3-GFP, it emitted a more narrow range of spectra. This difference in emission profile then allowed us to distinguish cytosolic from autophagosome-associated LC3 and measure autophagosome numbers based on spectral criteria. To test this approach we treated cells with CQ to increase autophagosome numbers and induce high levels of LC3-GFP puncta. Eight hours of CQ treatment resulted in a visible decrease in diffuse GFP fluorescence and an increase in GFP puncta. By MSI, control cultures were composed primarily of the green/diffuse LC3-GFP spectra, whereas CQ-treated cells exhibited a marked decrease in the green spectra and an increase in the red spectra that corresponded to punctate LC3-GFP structures (). CQ had no effect on the spectral profile of GFP in GFP vector control cells (data not shown). Using MSI quantitative software, we quantified the amount of red spectra per cell, which indicated the amount of LC3-GFP puncta, or autophagosomes, per cell. CQ treatment significantly increased the red spectra in C8161 (p < 0.001) and A375P (p = 0.02) cells (). These data demonstrate the utility of MSI in conjunction with the LC3-GFP assay for measuring autophagosomes, making MSI a useful tool in the study of autophagy.
Hyper-spectral imaging of death receptor-mediated apoptosis in histological tumor samples.
We next tested whether spectral imaging could detect and quantify apoptosis in paraffin-embedded tumor sections. We used the Prism and Reflector Imaging Spectroscopy System (PARISS) hyperspectral imaging system fitted to a Zeiss Axioskop upright microscope with white light source, and examined sections from HCT15 colon cancer xenografts that were harvested from control mice or mice treated with lexatumumab, a TRAIL receptor agonistic antibody that specifically activates TRAIL-R2/DR5 and induces apoptosis in cancer cells.Citation23 Tumor sections were immuno-stained for detection of cleaved caspase-8 (CC8), an apical caspase in the extrinsic cell death pathway that is directly activated by TRAIL death receptors and therefore serves as a marker of their activity. After 24 h of treatment, lexatumumab induced significant levels of caspase-8 activation, which was qualitatively evident by gross examination of CC8-stained slides (). CC8-positive regions were specific to tumor regions undergoing apoptosis, as positively stained cells exhibited morphological hallmarks of apoptosis (i.e. fragmented/pycnotic nuclei and cytosolic retraction; and S3). We then scanned sections using the PARISS imager to discern CC8-stained from non-stained regions. We used a 0.99 correlation coefficient, which determines the level of spectral resolution: a correlation coefficient of 1.00 has the highest spectral resolution and will therefore identify the maximum number of distinguishable spectra. Using this correlation coefficient, the PARISS detected three distinct spectra in lexatumumab-treated tumors (). Spectra corresponded to patches of dense cellularity (green pseudocolor), less dense stromal regions (red pseudocolor), and regions positive for CC8 chromagenic staining (blue pseudocolor; ). We used low magnification (2.5X) and collected spectral data from five random, 1mm-wide strips of tumor using 10 micron increments (i.e. 100 acquisitions per scan). Using these parameters, we were able to rapidly sample a major portion (5mm) of the tissue. The amount of blue specta, corresponding to CC8-positive regions, was significantly higher in tumors from lexatumumabtreated mice. On average, CC8-associated spectra from lexatumumab-treated tumors accounted for 24.5 ± 3.8% of the total spectra per 1mm field, compared with 1.5 ± 0.7% for tumors from control-treated mice (p = 0.0003; ). These findings demonstrate that spectral imaging, and the PARISS system in particular, is a rapid method for quantifying chromagenic staining of death receptor-induced caspase-8 activation in paraffinembedded histological sections.
Detection of an apoptosis-specific spectral signature in H&E-stained tumor samples.
Tumor regions that were rich in CC8 staining exhibited H&E staining patterns that were distinct from CC8-negative regions ( and S3). This would be expected due to the distinctive morphological changes that occur in apoptotic cells. Qualitatively, we noted that regions corresponding to intense CC8 staining in serial sections were composed of cells with smaller, more intensely stained nuclei. These regions also contained cells with reduced cytoplasm, which was evident from the absence of eosin staining (). We hypothesized that the PARISS could quantify apoptosis in H&E-stained tumor sections by detecting spectral changes associated with differential staining patterns in apoptotic vs. healthy tumor tissue. We tested this possibility using a magnification of 20X and a correlation coefficient of 0.98, and found that PARISS recognized four distinct spectra in H&E-stained HCT15 tumors from mice treated with lexatumumab (Fig. S4). shows images from serial sections that were stained for CC8 to identify regions of lexatumumab-induced apoptosis, and H&E in the absence of any immunostaining. A spectral pseudocolor map was then constructed from PARISS scanning of the H&E image. Notably, CC8-positive regions were enriched with a unique spectra (blue pseudocolor). In addition, CC8-positive regions contained high levels of spectra (red pseudocolor) that corresponded to small, intensely stained nuclei of apoptotic cells with condensed DNA. The two other spectra, denoted by green and gray pseudocolors, corresponded to tumor cell cytoplasm and surrounding tumor stromal tissue, respectively. At higher magnification (; 40X), all spectra and the structures they corresponded to were more apparent. Using higher magnification, we quantified the percent composition of spectra in four random fields and compared spectral data between control and lexatumumab treatment groups (). The blue spectra, which co-localized with CC8-positive regions and was therefore titled the caspase spectra, accounted for 5.2 ± 1.3% of spectra in tumors from lexatumumab-treated mice and was virtually undetectable in tumors from control mice (0.13 ± 0.03%; p < 0.001). The DNA spectra (red pseudocolor) was also significantly elevated in the lexatumumab-treated group compared with controls (24.4 ± 2.3% vs. 9.6 ± 1.9%; p = 0.003). In healthy tumor cells, the DNA spectra appeared patchy and disjointed, which was likely indicative of interphase cells with uncondensed chromatin. By contrast, the DNA spectra were solid and more intense in the nuclei of regions that co-localized with CC8 staining, which might be predicted in apoptotic cells where chromatin has condensed and fragmented. The cytosolic spectra (green pseudocolor) significantly decreased with lexatumumab treatment from 56.2 ± 3.2% to 30.9 ± 4.2% (p = 0.003), suggesting a loss of cytosolic volume, another morphological hallmark of apoptosis. The stromal spectra (gray pseudocolor) were no different between the control and lexatumumab treatment groups at approximately 40% of the total specta per field.
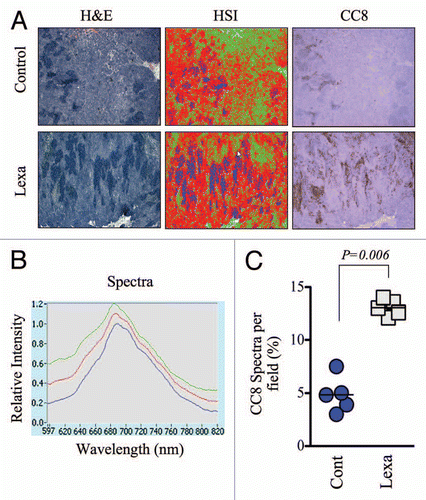
Therapeutic responses are known to occur with intratumor heterogeneity, and tissue analyses using highly magnified specimens may fail to account for this variability. In order to obtain a global view of apoptosis from a wide area of tumor tissue, we used lower magnification hyperspectral imaging in H&E-stained specimens. We acquired PARISS spectral data from 2.5X-magnified fields, to which we fit the apoptosis-specific spectra that were described above. At this lower magnification, cellular and subcellular resolution was lost, and the spectral library that was generated for 20 and 40X fields was not useful for detecting apoptotic regions. However, it is worth noting that areas of high cellularity (i.e. rich in the green, cytosolic spectra) could still be distinguished from stroma (gray spectra; Fig. S5) at the lower magnification. We developed a new spectral library specifically for 2.5X-magnified H&E-stained sections, using a less stringent correlation coefficient of 0.97. This new library contained 3 primary spectra that matched areas of dense non-cellullar stromal tissue (green pseudocolor), cell-dense tumor tissue (red pseudocolor) and those that tightly colocalized with CC8 staining in serially sections (blue pseudocolor; and B). We quantified the amount of blue spectra in a 5 mm-wide portion of the tumor by scanning five 1 mm sections. The average amount of blue spectra corresponding to apoptotic regions was significantly higher in the lexatumumab group (p = 0.006, ), thereby validating this technique for the assessment of apoptosis in broad tumor areas.
Discussion
In this report we developed spectral imaging methods for quantifying autophagosome numbers in vitro using the LC3-GFP autophagy assay, and for analyzing apoptosis in ex vivo specimens from preclinical colon cancer models. To our knowledge this is the first time spectral imaging has been used for these specific purposes. Spectral imaging does not replace the current methods used for the study of autophagy and apoptosis. However, because it is fast, unbiased and quantitative it may be used to improve and complement those existing methodologies.
For applying spectral imaging to the study of autophagy, we used multispectral imaging in combination with LC3-GFP fluorescence imaging, a widely used autophagy assay. This assay is based on the localization of LC3, which is expressed in the cytosol during conditions of low autophagy and then incorporates into autophagosomal membranes at the onset of autophagy. LC3-GFP shifts from a diffuse to punctate appearance when it migrates from the cytosol into autophagosomes. While the human eye can spatially discriminate punctate from diffuse GFP, it is unable to tease apart minor spectral changes that occur when chimeric GFP migrates from the cytosol to autophagosomal membranes. Spectral imaging systems, on the other hand, acquire data at individual wavelengths, rather than simultaneously across a whole spectrum of wavelengths, and are therefore capable of detecting subtle changes in light spectra. We found that cytosolic LC3-GFP had a distinctly broader range of emission spectra than autophagosome-localized LC3-GFP, although they emit at the same peak wavelength. This shift in spectral profile may be understood by considering that punctate LC3-GFP becomes more concentrated when bound to autophagosomes. The conformation of GFP fluorophores and their associated emission spectra may be altered upon migration into autophagosomes, where the chemical environment is distinctly different from the cytosol. Although the exact chemical/physical basis for the changed LC3-GFP spectra upon autophagy is not clear, the more narrow emission spectra of punctate LC3-GFP allowed us to spectrally classify and quantify autophagosomal density in cultured melanoma cells.
The introduction of spectral imaging to the LC3-GFP assay improves this method of autophagy measurement for the following reasons. First, spectral analysis reduces investigator bias, as the data are collected and analyzed by imaging software. Second, manual methods for quantifying cells with punctate LC3-GFP cannot accurately resolve the number of individual puncta per cell. As autophagy is a homeostatic process, every cell in a field of view could potentially have some detectable level of autophagy, in which case the number of cells per field with LC3 puncta may be 100%. It is the number of autophagosomes per cell that is critical to measuring levels of autophagy. The spectral-based method we developed quantifies the density of LC3-GFP puncta at the subcellular level, and therefore overcomes this limitation. It may also be useful for quantifying low or basal levels of autophagy, which may be below the detection threshold of current methods.
The LC3-GFP assay is critical to the study of autophagy. However, it measures autophagosmes and not autophagy, per se, which is a dynamic process. Therefore, the LC3-GFP assay must be accompanied by other autophagy assays such as electron microscopy, the LC3 turnover assay, and measurement of longlived protein degradation. Autophagosome numbers increase in response to compounds that stimulate autophagy but also those that inhibit the late stages of autophagy. Hydroxychloroquine (CQ) is an example of an autophagy inhibitor that blocks autolysomsome formation leading to an accumulation of autophagosomes. In this regard, CQ induced an autophagy-like phenotype, and served as a tool compound that allowed us to develop this spectral-based quantitative system for tracking autophagosomes using the LC3-GFP assay.
The study of apoptosis is critical to evaluating the effects of therapeutics like TRAIL receptor-targeted agents that exact their effects through activation of the extrinsic apoptotic pathway. There are a plethora of widely accepted assays that measure apoptosis in vitro, however, the options for detecting and quantifying apoptosis in ex vivo tumor samples are more limited. As caspases execute the apoptotic cell death program, activated caspases serve as markers for apoptosis in immunohistochemical staining procedures. Caspase-8, for example, is an initiator caspase that is cleaved/activated by pro-apoptotic TRAIL receptors, making it a reliable indicator of death receptor-mediated apoptotic signaling. In this study, we demonstrated the utility of PARISS hyperspectral imaging for detecting cleaved caspase-8 positive regions in colon cancer histological sections from mice treated with lexatumumab, a TRAIL-R2/DR5 agonistic antibody. As this system quantified image pixels with spectral characteristics that were specific to chromagen that precipitated in regions of CC8 positivity, this system could also be applied to staining for other markers that are detected by chromagenic immunostaing methods. One of the advantages to using PARISS for this purpose is that it minimizes user subjectivity, which is introduced by manual methods that require investigators to count marker positive cells or approximate the percentage of tissue that stained positively. A second advantage was the ability to scan low magnification images, which allowed us to sample an entire tumor section. This is important because apoptotic responses to therapy often exhibit intratumor heterogeneity, and higher magnification images may not accurately represent the overall tumor response. Finally, the data acquisition with PARISS was fast: a 1 mm-wide section of tumor could be analyzed in 1–2 min, whereas manual methods that require acquisition and analysis of multiple high magnification images are more time-consuming. We used only one agent in our study, but preclinical studies that test multiple agents and combinations of agents, at multiple doses and dosing schedules, are considerably more complex. These types of experiments that use immunohistochemical analysis of biomarker expression as an endpoint would benefit from the rapid and quantitative data acquisition capability of PARISS.
Apoptosis is associated with a unique set of morphological features. We reasoned that changes in cell morphology, such as those associated with the induction of apoptosis, would alter the absorption and retention of H&E cellular stains leading to quantifiable spectral changes. In support of this hypothesis, the PARRIS hyperspectral imager recognized unique spectral elements that co-localized with CC8 staining in serial sections. This apoptotic spectral profile was then used to quantify apoptosis in tumor sections that were stained with only H&E in the absence of immunostaining for activated caspases or other apoptotic markers. We exploited the spectral differences between apoptotic and non-apoptotic cells to quantify apoptosis in response to the TRAIL-R2/DR5 activating antibody lexatumumab in a preclinical model of colon cancer. We assessed apoptosis in high magnification images with subcellular resolution in addition to lower magnification images. Lower magnification images were useful in obtaining a view of apoptosis in the whole tumor section, which is important for capturing and accounting for response heterogeneity that is known to exist in human tumors.
In summary, we have presented two unbiased spectral imaging methods for detection of autophagy and apoptosis, two types of programmed cell death widely present in normal development and physiology as well as in pathological conditions such as cancer. As autophagy and apoptosis are conventionally detected and measured with time-consuming and/or labor-intensive procedures, the development of faster and more convenient imaging methods may enhance the efficiency and effectiveness of their study in biomedical research.
Materials and Methods
Cell lines and reagents.
C8161 and A375P melanoma cell lines were a gift from Dr. Mary Hendrix of Northwestern University, and were cultured in RPMI medium supplemented with 10% FBS and gentamycin. HCT15 cells were purchased from ATCC and cultured in DMEM supplemented with 10% FBS and gentamycin. All cells were grown at 37°C and 5% CO2. C8161 and A375P stably expressing LC3-GFP or GFP control vectors were generated by pooling single cell clones after retrovirus transfection, as described previously.Citation16 Hydroxychloroquine was purchased from Sigma-Aldrich. Antibodies to LC3 and cleaved caspase-8 were purchased from Novus Biologics and Cell Signaling, respectively. Lexatumumab was generously provided by Dr. Robin Humphreys (Human Genome Sciences Inc.).
In vivo studies.
Mice were housed and maintained in accordance with the Penn State College of Medicine Institutional Animal Care and Use Committee and state and federal guidelines for the humane treatment and care of laboratory animals. Five week old, female nude mice (NU/NU) were injected subcutaneously with 2×106 HCT15 cells suspended in PBS/Matrigel (v:v) at a final volume of 200µl. After 21 d, animals with size-matched tumors were administered one bolus dose of lexatumumab (10mg/kg; i.v.) or vehicle (PBS) in a volume of 100µl. After 24 h, animals were sacrificed and tumors were excised and processed for histological or immunohistochemical analysis.
Multispectral imaging of LC3-GFP localization.
Melanoma cell cultures were imaged using the Nuance Multispectral Imaging System (CRi) and related software. Fluorescence images were captured using a Nikon AZ100 multizoom stereomicroscope with 40X magnification and an excitation wavelength of 475nm. Emission spectra were collected at 10nm increments using a long pass filter covering wavelengths between 500 and 750nm. Punctate and diffuse LC3-GFP fluorescence were assigned individual pseudocolors as their spectral profiles were distinct. Separate spectra were uncoupled using the system's unmixing feature. For quantification, the average signal per exposure was documented for the first 15 regions of interest (cells) that were identified using the “measure” function of the software.
PARISS hyperspectral imaging of cleaved caspase-8 (CC8) and H&E staining.
Tumor sections from control mice or mice treated with the agonistic TRAIL-R2/DR5 monoclonal antibody, lexatumumab, were immunstained with a CC8 specific antibody and counterstained with hematoxylin. Sections were imaged using a white light source and a Zeiss Axioskop upright microscope fitted with the Prism and Reflector Imaging Spectroscopy System (PARISS). The specifications of the PARISS system were described previously.Citation4 A correlation coefficient of 0.99 was used to generate a library of distinct spectra for 2.5X-magnified images. Data were collected in 10 micron slices. One hundred acquisitions (slices) were analyzed per scan and thus 1 mm of tumor width was analyzed per scan. Spectral data from five random fields (a total of 5 mm of tumor tissue) were collected. For hyperspectral analysis of H&E-stained tumor tissues, a correlation coefficient of 0.98 and magnification of 20X were used to generate a spectral library. This spectral library was then used for data acquisition of 20 and 40X magnified images. An additional spectral library was constructed for analyses of lower magnification images (2.5X, correlation coefficient of 0.97). H&E stained sections and hyperspectral pseudomaps were compared with serial sections that were stained for CC8, which provided the ability to match spectral data to tumor regions with known levels of death receptor-mediated apoptosis.
Figures and Tables
Figure 1 Punctate, autophagosome-associated LC3-GFP exhibits a unique spectral profile. (A) C8161 cells were treated with hydroxychloroquine (CQ; 25µM) for 16 h. Protein gel blots are shown. Ran is included as a loading control. (B) C8161-LC3-GFP and C8161-GFP-vector control cell lines were treated with CQ as in (A). Fluorescence images are shown. (C) Untreated C8161 cells were imaged using conventional fluorescence microscopy (top panel, far left) or multispectral imaging (MSI; top panel, second from left). Distinct spectra, represented by red and green pseudocolors, which are displayed in the spectral library (bottom panel), were uncoupled and shown as separate images (top, right). (D) C8161-LC3-GFP cells were treated with CQ (25µM) for 16 h. Control and CQ-treated cultures were analyzed by MSI. Fluorescence (top) and MSI unmixed images (bottom) are shown. Single cells (insets) were magnified to show co-localization of LC3-GFP puncta and red spectra. (E) The average signal, normalized to exposure time, for red spectra per cell was quantified in 15 single cells using MSI quantitative software. Histograms show the distribution of single cell, red spectra averages for control and CQ-treated C8161-LC3-GFP and A375P-LC3-GFP cells. Statistical significance was determined using the Student's t-test.
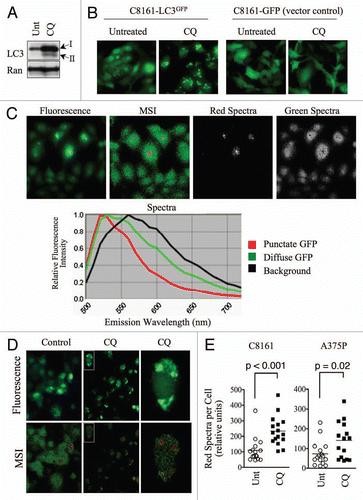
Figure 2 Hyperspectral quantification of cleaved caspase-8 immunostaining to assess preclinical response to lexatumumab. (A) Mice bearing HCT15 human colon cancer xenografted tumors were treated with lexatumumab (10mg/kg, i.v.) for 24 h. Tumors were then processed and stained for cleaved caspase-8 (CC8). Representative 10X images are shown. (B) A high magnification (80X) image from a lexatumumab-treated tumor is shown to emphasize the co-localization of CC8 staining and morphological markers of apoptosis, such as nuclear condensation and fragmentation. The arrow denotes a CC8-positive cell with pycnotic nucleus and the arrowhead points to a non-apoptotic, CC8-negative cell with a healthy, intact nucleus. (C) CC8-stained tumor sections from lexatumumab-treated mice were analyzed using the PARISS hyperspectral imager. The spectral library derived from a 2.5X-magnified field is shown. (D) HCT15 tumors from control and lexatumumab-treated mice were pseudocolored using the spectral library in (C). White light (bright field) and corresponding hyperspectral images are shown. (E) The percentage of blue spectra (CC8-positive regions) was quantified using the PARISS in five random, 2.5X fields. The histogram shows the distribution of the percentages derived from each scan for control and lexatumumab treatment groups. Statistical significance was calculated using a Student's t-test.
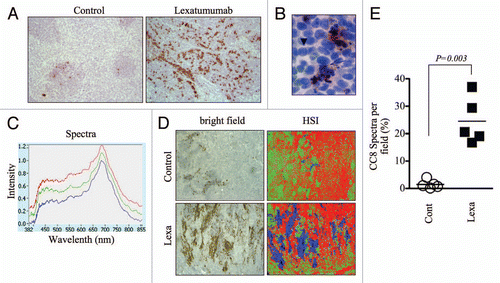
Figure 3 Characterization of an apoptosis-specific spectral signature in H&E-stained tumor sections. (A) Serial HCT15 tumor sections from mice treated with lexatumumab (10mg/kg; 24 h) were immunostained for CC8 or H&E, in the absence of chromagenic staining for an apoptotic marker. Spectral analysis using the PARISS was then performed on the H&E-stained section. Images are shown are 20X-magnified. (B) Serial sections from HCT15 tumors were stained and analyzed as in (A), with the exception that 40X magnification was employed. Images are shown for CC8 immunostaining, H&E staining and HS I pseudo-maps for control and lexatumumab treatments. (C) Spectral data from (B) are represented in histograms comparing individual spectra for control and lexatumumab treatment groups. Statistical significance was determined by Student's t-test.
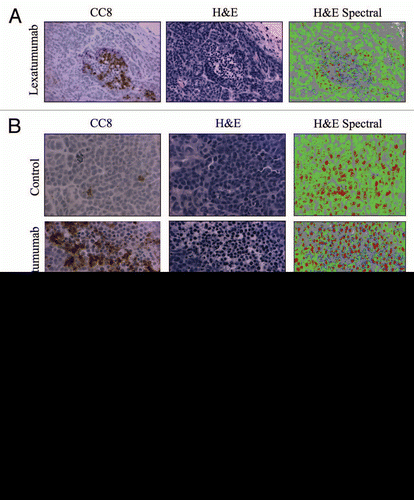
Figure 4 Global analysis of tumor apoptosis using the apoptosis spectral signature in conjunction with low magnification images of ex vivo tumor specimens. (A) Low magnification (2.5X) images from H&E-stained HCT15 tumors were analyzed by HS I using the PARISS . The HSI pseudo-map was derived from the spectral library shown in . Images from CC8-stained serial sections are included to demonstrate the overlap of lexatumumab-mediated apoptosis and expression of the apoptotic spectral signature. (B) The spectral library generated using the PARISS for 2.5X magnified tumor sections from lexatumumabtreated mice is shown. (C) Blue spectral elements corresponding to apoptotic regions were quantified by the PARISS in five random 1mm-wide scans. The percentage of apoptotic spectra, which co-localized with CC8 staining for each scan are shown. The difference between control and lexatumumab treatments was significant (p = 0.006), as determined by a Student's t-test.
Acknowledgments
We thank the G.I. morphology core of University of Pennsylvania School of Medicine [P30 DK50306] for their assistance in the preparation and analysis of histological samples. This work is partially supported by the Network of Translational Research in Optical Imaging (NTROI) (U54 CA105008, PI: W.S. El-Deiry), the University of Research Foundation (L.Z.L) and the Grant #IRG-78-002-29 from the American Cancer Society at the University of Pennsylvania. We would also thank Drs. Julian Lum and Ravi Amaravadi for assistance in the GFP-LC3 cell transfection and selection.
References
- Dicker DT, Lerner J, Van Belle P, Barth SF, Guerry D 4th, Herlyn M, et al. Differentiation of normal skin and melanoma using high resolution hyperspectral imaging. Cancer Biol Ther 2006; 5:1033 - 108; PMID: 16931902; http://dx.doi.org/10.4161/cbt.5.8.3261
- Tumeh PC, Lerner JM, Dicker DT, El-Deiry WS. Differentiation of vascular and non-vascular skin spectral signatures using in vivo hyperspectral radiometric imaging: implications for monitoring angiogenesis. Cancer Biol Ther 2007; 6:447 - 453; PMID: 17387267; http://dx.doi.org/10.4161/cbt.6.3.4019
- Mayes P, Dicker D, Liu Y, El-Deiry WS. Non-invasive vascular imaging in fluorescent tumors using multispectral unmixing. Biotechniques 2008; 45:459 - 460 461 - 464; PMID: 18855773; http://dx.doi.org/10.144/000112946
- Dicker DT, Lerner JM, El-Deiry WS. Hyperspectral image analysis of live cells in various cell cycle stages. Cell Cycle 2007; 6:2563 - 2570; PMID: 17912031; http://dx.doi.org/10.4161/cc.6.20.4912
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998; 94:727 - 737; PMID: 9753320; http://dx.doi.org/10.1016/S0092-8674(00)81732-8
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 830; PMID: 20811354; http://dx.doi.org/10.1038/ncb0910-823
- Bredesen DE, Rao RV, Mehelen P. Cell death in the nervous system. Nature 2006; 443:796 - 802; PMID: 17051206; http://dx.doi.org/10.1038/nature05293
- Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res 2008; 103:343 - 351; PMID: 18703786; http://dx.doi.org/10.1161/CIRCRESAHA.108.175448
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autopagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 734; PMID: 16148885; http://dx.doi.org/10.1038/nrc1692
- Brown JM, Attardi LD. The role of apoptosis in cancer evelopment and treatment response. Nat Rev Cancer 2005; 5:231 - 237; PMID: 15738985; http://dx.doi.org/10.1038/nrc1560
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 1010; PMID: 18971948; http://dx.doi.org/10.1038/nrm2529
- Sato K, Tsuchiahara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677 - 9684; PMID: 17942897; http://dx.doi.org/10.1158/0008-5472.CAN-07-1462
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892 - 10903; PMID: 18281291; http://dx.doi.org/10.1074/jbc.M800102200
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 510; PMID: 16990848; http://dx.doi.org/10.1038/sj.cdd.4402039
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548 - 558; PMID: 16946731; http://dx.doi.org/10.1038/sj.cdd.4402030
- Ma XH, Piao S, Wang D, Mcafee Q, Nathanson KL, Lum JJ, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478 - 3489; PMID: 21325076; http://dx.doi.org/10.1158/1078-0432.CCR-10-2372
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 5728; PMID: 11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 326; PMID: 20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 2001; 276:7320 - 7326; PMID: 11058599; http://dx.doi.org/10.1074/jbc.M008363200
- Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene 2008; 27:6452 - 6461; PMID: 18955972; http://dx.doi.org/10.1038/onc.2008.311
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; PMID: 10647931; http://dx.doi.org/10.1016/S0092-8674(00)81683-9
- Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci 2004; 19:124 - 128; PMID: 15143207; http://dx.doi.org/10.1152/nips.01519.2004
- Georgakis GV, Li Y, Humphreys R, Andreeff M, O'Brien S, Younes M, et al. Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Br J Haematol 2005; 130:501 - 510; PMID: 16098063; http://dx.doi.org/10.1111/j.1365-2141.2005.05656.x