Abstract
The epidermal growth factor receptor (EGFR) is a ubiquitously expressed receptor tyrosine kinase (RTK) and is recognized as a key mediator of tumorigenesis in many human tumors. Currently there are five EGFR inhibitors used in oncology, two monoclonal antibodies (panitumumab, and cetuximab) and three tyrosine kinase inhibitors (erlotinib, gefitinib, and lapatinib). Both strategies of EGFR inhibition have demonstrated clinical successes, however many tumors remain non-responsive or acquire resistance during therapy. To explore potential molecular mechanisms of acquired resistance to cetuximab we previously established a series of cetuximab-resistant clones by chronically exposing the NCI-H226 NSCLC cell line to escalating doses of cetuximab. Cetuximab-resistant clones exhibited a dramatic increase in steady-state expression of EGFR, HER2, and HER3 receptors as well as increased signaling through the MAPK and AKT pathways. RNAi studies demonstrated dependence of cetuximab-resistant clones on the EGFR signaling network. These findings prompted investigation on whether or not cells with acquired resistance to cetuximab would be sensitive to the EGFR targeted TKI erlotinib. In vitro, erlotinib was able to decrease signaling through the EGFR axis, decrease cellular proliferation, and induce apoptosis. To determine if erlotinib could have therapeutic benefit in vivo, we established cetuximab-resistant NCI-H226 mouse xenografts, and subsequently treated them with erlotinib. Mice harboring cetuximab-resistant tumors treated with erlotinib exhibited either a tumor regression or growth delay as compared to vehicle controls. Analysis of the erlotinib treated tumors demonstrated a decrease in cell proliferation and increase rates of apoptosis. The work presented herein suggests that 1) cells with acquired resistance to cetuximab maintain their dependence on EGFR and 2) tumors developing resistance to cetuximab can benefit from subsequent treatment with erlotinib, providing rationale for its use in the setting of cetuximab resistance.
Introduction
The epidermal growth factor receptor (EGFR) is a member of the HER family of receptor tyrosine kinases (RTKs) and consists of four members: EGFR (ErbB1/HER1), HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4). The EGFR is a RTK that serves to control various cellular activities including migration, proliferation and survival. When activated at the cell surface by various cognate ligands, EGFR homo- or hetero-dimerizes with other HER family members, leading to the activation of its intrinsic kinase and subsequent phosphorylation of tyrosine residues on its C-terminal tail.Citation1 These phosphorylated residues serve as docking sites for numerous adaptor proteins that act as initiators of several signal transduction pathways.Citation2,Citation3 Notably, the SH2 domain containing protein Grb2 binds to phospho-tyrosine residues on EGFR and recruits the guanine nucleotide exchange factor SOS to the cell surface. SOS promotes the exchange of GDP for GTP on the small GTPase protein Ras, which is responsible for the activation of the MAPK pathway, ultimately resulting in progression through the cell cycle. Additionally, PI3K can bind to phospho-tyrosine residues on the EGFR and phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 recruits the serine/threonine kinase AKT to the cell surface, where it can become activated and regulate various cellular processes impacting both cell proliferation and survival.Citation4 The cellular activities regulated by EGFR are now widely accepted to play a major role in tumorigenesis.Citation5
Over the past 20 years, it has become evident that human cancers develop aberrant signaling through both the MAPK and PI3K/AKT pathways due to the overexpression and/or mutation of the EGFR.Citation6–Citation8 EGFR disregulated activity has been strongly associated with the development and progression of head and neck squamous cell carcinoma (HNSCC),Citation9–Citation11 non-small cell lung cancer (NSCLC),Citation12,Citation13 colorectal cancer (CRC),Citation14,Citation15 breast cancerCitation16–Citation18 and brain cancer.Citation19–Citation21 These findings have lead researchers to develop drugs that target the EGFR and prevent its signaling from the cell surface. One approach involves the use of small molecule tyrosine kinase inhibitors (TKIs) that bind to the ATP-binding site in the tyrosine kinase domain (TKD) of the EGFR. To date three anti-EGFR TKIs, erlotinib (OSI-774, Tarceva), gefitinib (ZD1839, Iressa) and lapatinib (GW572016, Tykerb), have been approved by the FDA for use in oncology. A second approach to EGFR inhibition includes the use of monoclonal antibodies (mAbs) that bind to EGFR's extracellular ligand-binding domain preventing both ligand binding and dimerization.Citation22–Citation24 Currently, two mAbs against the EGFR have been approved by the FDA for use in oncology including the human:murine chimeric mAb cetuximab (IMC-225, Erbitux) and the fully humanized mAb panitumumab (Vectibix). Cetuximab has exhibited clinical success in the setting of metastatic CRC (mCRC) and HNSCC, as both a monotherapy or in combination with chemotherapy/radiation treatments.Citation25–Citation27 Cetuximab has also been shown to have antitumor effects in the setting of NSCLC,Citation28,Citation29 however it has not yet been FDA approved for treatment of this cancer.
Both approaches to EGFR inhibition show considerable clinical promise. However, increasing evidence suggests that patients who initially respond to EGFR inhibitors may subsequently become refractory.Citation30 Therefore, an improved understanding of molecular mechanisms of acquired resistance to EGFR inhibitors may provide valuable leads to enhance the efficacy of this class of agents. The identification of catalytic domain EGFR mutations that predict response to EGFR-TKIs in selected lung cancer patients represents a landmark development in the EGFR field.Citation31–Citation33 Although EGFR TKD mutations appear to correlate with response to the TKIs erlotinib and gefitinib, no such correlation exists for cetuximab response.Citation34
In this report we investigated whether cetuximab-resistant tumors would be sensitive to the EGFR TKI erlotinib. To test this hypothesis we established a series of cetuximab-resistant cell lines in vitro by chronically treating the NCI-H226 NSCLC cell line with increasing amounts of cetuximab for a prolonged period of time.Citation35,Citation36 Individual clones with acquired resistance to cetuximab were treated with erlotinib resulting in decreased activation of EGFR, HER2 and HER3, along with a decrease in the activation of the downstream signaling molecules AKT and MAPK. This lead to decreased proliferation and increased apoptosis in all cetuximab-resistant clones tested. To build upon these in vitro findings, we developed acquired resistance to cetuximab in vivo by treating the cetuximab-sensitive NSCLC cell line NCI-H226 with cetuximab until resistant tumors manifested. Cessation of cetuximab treatment and subsequent erlotinib treatment resulted in tumor growth delay and tumor regression. Immunohistochemical analysis of xenograft tumors demonstrated a decrease in cell proliferation and an increase in apoptosis. The results presented herein suggest that tumors with acquired resistance to cetuximab may still rely on EGFR for growth and survival and can be successfully targeted via alternative EGFR TKIs. Collectively these findings suggest that treating tumors that acquire resistance to cetuximab with erlotinib may prove to be efficacious.
Results
NSCLC cell lines with acquired resistance to cetuximab remain addicted to the EGFR signaling network.
We have previously described the development and characterization of the NCI-H226 NSCLC line (H226) with acquired resistance to cetuximab.Citation36 Briefly, H226 was continuously exposed to increasing concentrations of cetuximab over 6 mo. Following the development of heterogeneous populations of cetuximab-resistant clones, we isolated individual sub-clones of cetuximab-resistant cells. This process resulted in six stable cetuximab-resistant clones designated HC1, HC4, HC5, HC6, HC7 and HC8. We chose to further study cetuximab-resistant clones HC1, HC4 and HC8 because these three clones were representative of a series of cetuximab-resistant clones. The cetuximab-sensitive parental line was designated HP. To confirm that established clones remained resistant to cetuximab we performed growth proliferation assays using the HC1, HC4 and HC8 resistant clones. Clones were treated with 100 nM cetuximab for 72 h (). All three clones tested displayed a robust cetuximab-resistant phenotype when challenged with 100 nM cetuximab as compared with the HP parental control.
Analysis of EGFR expression indicated that steady-state expression and activity of the EGFR was increased in all three cetuximab-resistant cell lines (HC1, HC4 and HC8) relative to the parental control (HP) (). Cetuximab-resistant cell lines also displayed increased EGFR expression levels compared with HP cells as demonstrated via confocal immunofluorescence microscopy (). In addition to increased expression of the EGFR, the known auto-phosphorylation site tyrosine 1173 (Y1173) was robustly phosphorylated in cetuximab-resistant cell lines as compared to the parental control (). Further, cetuximab-resistant clones displayed increased activated forms of the HER family members HER2 (Y1121/1122) and HER3 (Y1289), along with MAPK (Y202/Y204), AKT (S473) and S6K (S240/244) as detected by immunoblot analysis (). These data suggest that cetuximab-resistant clones exhibit robust activation of the EGFR signaling cascade.
To determine if cells with acquired resistance to cetuximab depend on EGFR signaling we performed proliferation assays using small interfering RNAs (siRNA) targeting EGFR ( and E). All three cetuximab-resistant lines displayed growth inhibitory effects at both 10 and 25 nM siEGFR. Collectively these data indicate that cells with acquired resistance to cetuximab remain dependent on EGFR activated signaling pathways that result in increased proliferative potential.
NSCLC cell lines with acquired resistance to cetuximab are sensitive to erlotinib in vitro.
We hypothesized that cetuximab-resistant clones may be susceptible to other forms of therapy that target the EGFR since these cells remained dependent on the EGFR signaling pathway for sustained growth and survival. To test this hypothesis we looked at the expression of several signaling proteins in cetuximab-resistant cell lines treated with 100 nM cetuximab or erlotinib (250 or 750 nM) for 24 h (). EGFR, HER2 and HER3 phosphorylation levels were decreased at both doses of erlotinib examined in all cetuximab-resistant lines, whereas cetuximab had little impact on the levels of phosphorylated EGFR, HER2 and HER3. Additionally, erlotinib inhibited signaling through both the AKT and MAPK pathways as demonstrated via a decrease in their phosphorylation levels. To determine if erlotinib could impact the proliferative potential of cetuximab-resistant clones we performed growth proliferation studies using four different doses of erlotinib ranging from 250 nM to 1 uM (). All cetuximab-resistant clones demonstrated statistically significant, dose dependent growth inhibitory effects. Treatment with 750 nM erlotinib reduced cetuximab-resistant cell proliferation rates to 40–60% compared with vehicle control treatment.
We further measured cell proliferation by detecting BrdU incorporation after treatment with 250 or 750 nM erlotinib for 24 h in HC1, HC4 and HC8 clones with acquired resistance to cetuximab (). Upon erlotinib treatment (at both 250 or 750 nM) a higher percentage of cetuximab-resistant clones exhibited G1 arrest as compared with cetuximab- or vehicle-treated cells, resulting in a decrease in cellular proliferation rate. Cetuximab-sensitive HP parental cells also demonstrated a higher percentage of cells halted in the G1 phase when treated with either cetuximab or erlotinib.
To determine if erlotinib induces apoptosis in cetuximabresistant clones we performed Annexin-V analysis after treatment with erlotinib for 48 h (). Both 500 nM and 1 uM erlotinib treatments resulted in a statistically significant increase in apoptosis of cetuximab-resistant clones. Thus, erlotinib seems to induce apoptosis in cells with acquired resistance to cetuximab. Taken collectively, these data demonstrate that erlotinib can inhibit EGFR signaling through the AKT and MAPK pathways, ultimately impacting cell cycle progression and survival of cetuximab-resistant clones.
NSCLC cell lines with acquired resistance to cetuximab are sensitive to erlotinib in vivo.
To confirm our in vitro results we performed a series of mouse xenograft studies of acquired resistance to cetuximab. To develop acquired resistance to cetuximab in vivo, we inoculated 60 mice with the NSCLC line NCI-H226 unilaterally with 2 × 106 tumor cells in the dorsal flank. Tumors were allowed to grow to 50–100 mm3, at which time 50 mice were treated with cetuximab (0.5 mg/mouse) twice weekly and 10 mice were treated with IgG control (0.5 mg/mouse) twice weekly by intraperitoneal injection. IgG treated tumors grew uninhibited, while cetuximab treated tumors demonstrated tumor control and delayed growth. We continued cetuximab treatment until tumors demonstrated resistance, which was defined as marked tumor growth in the presence of continued cetuximab therapy. At this time we established mouse pairs whose tumors manifested resistance in the same temporal relationship. The development of cetuximab resistance was noted when tumors were larger than 500 mm3, which occurred for most mice after 50–100 d of cetuximab treatment. Resistance was observed in 36 of 50 tumor xenografts (72%) treated with cetuximab. A total of 15 cetuximab-resistant xenograft pairs were selected for further study (30 mice total). Several mice treated with cetuximab remained sensitive (demonstrated by controlled tumor size) after 200 d post cetuximab start date, and were not used for further analysis (data not shown).
Erlotinib treatment studies were performed with mice bearing cetuximab-resistant xenografts. All cetuximab-resistant tumor mice were taken off cetuximab (, indicated by the black arrow), and subsequently treated with either erlotinib (70 mg/kg) or vehicle (carboxymethylcellulose: CMC) for 5 d per week by oral gavage. Eleven out of 15 (73%) cetuximab-resistant tumors treated with erlotinib demonstrated a tumor response, while four (27%) tumors failed to respond. In , we illustrate 8 out of the 15 pairs of xenografts in this study. Intriguingly, three xenografts treated with erlotinib demonstrated tumor regression (–C), while two xenografts exhibited a growth delay ( and E). A small subset of erlotinib treated xenografts showed only a minor growth delay with subsequent continued growth (–H). Collectively, these series of xenograft studies suggest that erlotinib may be effective in treating tumors that have become resistant to cetuximab.
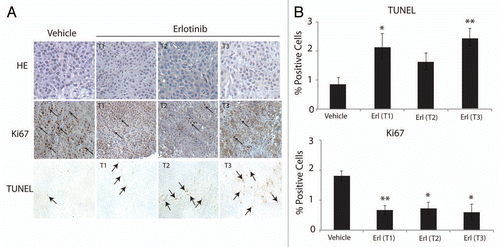
To further investigate the growth delay effect of cetuximab-resistant tumors by erlotinib we examined the rate of proliferation and apoptosis in excised tumor samples. We first measured cell proliferation (Ki67) in tumors treated with erlotinib (T1, T2, T3) or vehicle control via immunohistochemistry (IHC) ( and Ki67 parts). Erlotinib and vehicle treated tumors were collected 3–6 h after the last treatment and fixed in formalin. Each tumor treated with erlotinib demonstrated significantly reduced Ki67 positive staining as compared to the vehicle control, indicative of reduced cellular proliferation within the tumor. Next, we used this series of tumors to measure the levels of apoptosis in each tumor sample via terminal deoxynucleotidyl transferase-mediated dUTP nick and labeling (TUNEL) ( and TUNEL parts). These data show that erlotinib treated tumors exhibited increased amounts of apoptosis as compared to the vehicle controls, demonstrating the advantageous effect of erlotinib on cetuximab-resistant tumors.
Quantitation of IHC-positive staining for Ki67 and TUNEL is shown in . Ki67 or TUNEL positive cells in either vehicle- or erlotinib-treated cetuximab-resistant xenografts were enumerated in five random fields at 200×. Ki67 staining revealed that erlotinib treated tumors displayed statistically significant decreases in proliferation as compared with vehicle control (33–40% decrease in proliferation, respectively). Additionally, two of the three erlotinib treated tumors demonstrated statistically significant increases in apoptosis as compared with vehicle control: T1 demonstrated a 1.4-fold increase in apoptosis, and T3 demonstrated a 1.8-fold increase in apoptosis. Collectively these series of experiments demonstrate that erlotinib may lead to decreased tumor growth via inhibition of proliferation and induction of apoptosis.
Discussion
Cetuximab has exhibited promising antitumor activity in clinical trials as monotherapy and when used in combination with chemotherapy and/or radiation, particularly in the settings of mCRCCitation37–Citation41 and HNSCC.Citation42–Citation44 However, the development of acquired resistance to cetuximab is now well recognized and represents a major obstacle to successful treatment using this promising molecular targeting agent. With the phenomenon of acquired resistance to cetuximab becoming more prevalent it is important to understand both mechanisms of acquired resistance to cetuximab therapy and strategies to either (1) prevent or (2) circumvent/overcome this resistance. Our findings prompted us to investigate if other types of EGFR inhibitors, specifically erlotinib, a small molecule TKI directed towards the EGFR, could be advantageous in the setting of acquired resistance to cetuximab.
In this study we utilized both in vitro and in vivo models of acquired resistance to cetuximab to investigate potential strategies for circumventing this resistance. To accomplish this we established a series of cetuximab-resistant clones by exposing NCI-H226 cells to increasing concentrations of cetuximab over 6 mo. Cetuximab-resistant clones demonstrated increased total and active forms of EGFR and HER3, along with the downstream signaling proteins AKT and MAPK. Challenging cetuximab-resistant clones with cetuximab did not modulate the active forms of the proteins listed above except for MAPK, which was downregulated moderately. This suggests that pMAPK may not be a prominent effector molecule for this model of cetuximab resistance. Additionally, all cetuximab-resistant clones remained addicted to the EGFR signaling pathway, as demonstrated via siEGFR studies. These data support a previous report from our laboratory indicating that cetuximab-resistant clones overexpress EGFR and partner with other HER family members in order to escape cetuximab inhibition.Citation36 Although we found an increase in expression of the EGFR and its family members, Lu et al. reported opposite findings. In this study, they found that cetuximab-resistant DiFi5 colorectal cells expressed markedly lower protein levels of EGFR, attributed to an increased association of EGFR with Cbl, resulting in ubiquitination of EGFR and to its destruction.Citation45 Thus, there are likely to be multiple mechanisms of cetuximab-resistance in different cancer cell types.
The finding that cetuximab-resistant clones remained dependent on the EGFR signaling network prompted us to investigate whether these cell lines would be sensitive to an alternative EGFR inhibitor, the small molecule TKI erlotinib. We show that erlotinib can effectively inhibit EGFR signaling through the AKT and MAPK pathways in all three cetuximab-resistant clones. This resulted in both a growth inhibitory effect as demonstrated via cell proliferation assays and BrdU incorporation, and a pro-apoptotic effect by Annexin-V analysis. Inhibition of cell cycle progression may play a larger role than apoptosis in erlotinib's growth inhibitory effect since the pro-apoptotic effect was rather modest. Additionally, we show that erlotinib can have both anti-proliferative and pro-apoptotic effects when given sequentially to tumors that have acquired resistance to cetuximab in vivo. Xenograft studies presented here show that erlotinib can effectively delay growth of tumors that have acquired resistance to cetuximab; a novel finding that is highly translational. However, the longevity of tumor growth delay and/or tumor regression cannot be concluded from this study and thus requires further investigation. Regardless, the current study demonstrates that erlotinib treatment yields a clear growth delay and/or tumor regression in cetuximab-resistant tumors. Overall, the data presented herein suggests that the use of erlotinib after the development of acquired resistance to cetuximab may be beneficial.
The current data are corroborated by previous studies that report positive anticancer effects of TKI co-treatment with cetuximab. Matar et al. showed that the combination of cetuximab and gefitinib, an EGFR targeted TKI, has a greater growth inhibitory and pro-apoptotic effect in vulvar, colon, breast and prostate cancer cell lines.Citation46 In their vulvar squamous cell carcinoma xenograft model, tumors that expressed medium to high levels of EGFR responded the most to dual agent therapy. Further, Huang et al. validated these findings using both TKIs gefitinib and erlotinib in conjunction with cetuximab in HNSCC and NSCLC cells and xenograft models.Citation47 These findings were further confirmed in an in vitro model of hepatocellular cancer, where they showed a synergistic anti-proliferative effect of cetuximab and erlotinib co-treatment on both p53 wild type and mutant hepatocellular carcinoma cells.Citation48 Additionally, a clinical study demonstrated that erlotinib given to patients before and after treatment with cetuximab proved to be beneficial in pancreatic cancer.Citation49 The data presented in this current study is the first to demonstrate the antitumorigenic effects of the small molecule TKI erlotinib in the setting of acquired resistance to cetuximab in both an in vitro and in vivo model, providing further evidence for the use of erlotinib in the treatment of cetuximab-resistant tumors.
There are several possible explanations for why erlotinib may be an effective treatment for cetuximab-resistant tumors. The data presented here, and previously reported in references Citation36, Citation50 and Citation51, suggests that cetuximab-resistant clones cannot only bypass the inhibitory effects of cetuximab, but also in turn become more tumorigenic by overexpressing EGFR. Erlotinib's effectiveness in this setting may lie in the functional differences between small molecule TKIs and mAbs. The ability for erlotinib to cross the plasma membrane and directly bind to and block EGFRs tyrosine kinase seems to be an advantageous therapy for tumors with acquired resistance to cetuximab. Additionally erlotinib prevents the activation of other HER family members by shutting down EGFR's intrinsic kinase activity. There is now growing evidence for the intracellular signaling ability of EGFR from within the endosome,Citation52,Citation53 mitochondriaCitation54,Citation55 and nucleus.Citation56,Citation57 While mAbs are unable to cross the plasma membrane and target EGFRs intracellular signaling apparatus, TKIs have this potential. Thus, the potent anti-EGFR effects of erlotinib in the model presented in this study may be due the enhanced ability of TKIs to block the intracellular signaling of EGFR through the MAPK and AKT pathways. This hypothesis requires further investigation and is an active area of current research.
In conclusion the findings presented herein have potential clinical implications when considering optimal cancer therapeutic strategies in patients who manifest resistance to cetuximab therapy. It has become clear that EGFR remains active in cetuximab-resistant clones, and maintains its ability to signal through its tyrosine kinase. Thus, the use of alternative EGFR targeting agents shows promise in this arena. Currently, there are multiple on-going clinical trials of dual erlotinib and cetuximab therapy for various cancers (clinicaltrials.gov), and a recent phase I clinical trial of combinatory cetuximab and erlotinib treatment of solid malignancies was shown to be feasible.Citation58 Overall, the data presented in this current study supports the use of erlotinib subsequently in patients who become clinically resistant to cetuximab therapy.
Materials and Methods
Cell lines.
The human NSCLC line NCI-H226 was obtained from ATCC (Manassas, VA) and maintained in 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in RPMI 1640 (Mediatech Inc., Manassas, VA). Cell culture supplements were obtained from Life Technologies Inc. (Gaithersburg, MD).
Establishment of acquired resistance to EGFR targeted monoclonal antibody cetuximab.
The development of cells with acquired resistance has been previously described in reference Citation36. Briefly, the NCI-H226 cell line was continuously exposed to increasing amounts of cetuximab over a 6 mo time period. The exposure of cetuximab was progressively doubled every 10–14 d until 7–8 dose doublings were successfully achieved. We isolated individual sub-clones that continued to grow in increasing concentrations of cetuximab, resulting in six stable cetuximabresistant clones, three of which were used for analysis in this study: HC1, HC4 and HC8. In parallel we treated parental NCI-H226 cells (HP) with IgG in phosphate-buffered saline (PBS) vehicle for the same amount of time.
Compounds.
Erlotinib (OSI-774, Tarceva) was provided by OSI Pharmaceuticals Inc. (Melville, NY). Cetuximab (IMC-225, Erbitux) was purchased from the University of Wisconsin Pharmacy.
Antibodies.
All antibodies were purchased from commercial sources as indicated below: EGFR, HER3, HER2, AKT, pEGFR (Y1173), HRP-conjugated goat-anti-rabbit IgG and goat-anti-mouse IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). pHER2(Y1221/1222), pHER3(Y1289), pAKT(S473), p-MAPK (Y202/204), MAPK, p70S6K (S240/244) and S6K were obtained from Cell Signaling Technology (Beverly, MA). Alpha-tubulin was obtained from Calbiochem (San Diego, CA).
Immunoblotting analysis.
Following treatment, cells were lysed with RIPA buffer (1 M Tris-HCL, pH 7.4, 150 mM NaCl, 500 mM EDTA, 5% DOC, 0.1% Triton-X100, 1 mM DTT, 1 mM PMSF, 0.04 mM sodium orthovanadate, 2M β-glycerophosphate and 10 ug/mL of leupeptin and aprotinin). Lysates were sonicated and protein was quantitated using a standard Bradford absorbance assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were fractionated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA), which were subsequently incubated with the appropriate primary antibody overnight at 4µC. Proteins were detected via incubation with HRP-conjugated secondary antibodies and the ECL chemiluminescence detection system (GE Healthcare, Piscataway, NJ).
Immunofluorescent staining of EGFR.
5,000 cells per well were seeded on a 4-well glass Millicell EZ slide (Millipore). Cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min, washed three times in PBS, before permeabilization with PBS containing 0.1% Triton-X100 and 1% BSA for 20 min. Subsequently, the slides were washed three times with PBS and blocked in 3% BSA in PBS for 30 min. Cells were incubated with anti-EGFR antibody (1:100) overnight at 4µC. Samples were then washed in PBS and incubated with FITC-conjugated appropriate secondary antibody (Sigma, St. Louis, MO) for 1 h. Following washes with PBS, slides were incubated with Alexa Fluor 647 phalloidin (Invitrogen) diluted at 1:50 in PBS for 20 min protected from light. The slides were mounted using ProLong gold with DAPI antifade mounting solution (Invitrogen). Fluorescence microscopy and photography were performed using a FITC filter array on a Nikon 80i upright confocal microscope.
RNA interference studies.
EGFR siRNA was purchased from Dharmacon (Lafayette, CO). Transfection of Dharmacon siRNA into cell lines was achieved by using Dharmafect 1 (Dharmacon) or RNAiMax (Invitrogen) reagents and following the manufactures protocol.
Cell proliferation assay.
Cells were seeded at 1,200 cells per well in 200 ul of media on a 96 well plate, grown for 24 h, and then treated with drug for 72 h prior to analysis using the Cell Counting Kit 8 (Dojindo Molecular Technologies, Rockville, MD). Ten uL of CCK-8 solution was added to each well, and incubated for 2 h prior to absorbance analysis (A450 nm with plate reader). The percentage of cell growth was calculated by comparison of the A450 reading from treated vs. control wells. All treatments were preformed in quadruplicate.
BrdU cell cycle distribution analysis.
Cells were plated at a density of 600,000 per 100 mm plate and allowed to adhere overnight. The cells were treated with vehicle, 100 nM cetuximab, 250 or 750 nM erlotinib for 24 h. The following day the cells were pulsed with 10 uM BrdU for one hour. The cells were harvested by trypsinization, washed with cold PBS and fixed with 70% ethanol for 20 min. The cells were then labeled with a FITC-conjugated mouse anti-BrdU antibody and processed according to manufacturer recommendation (BD PharMingen, San Jose, CA). The cells were analyzed by flow cytometry (BD FACScan). ModFit Software (Verity Software House, Topsham, ME) was used to analyze the data.
Annexin-V apoptosis assay.
600,000 cells were plated in 100 mm plates, and after 24 h of incubation treated with either vehicle, 100 nM cetuximab, 500 nM or 1 uM erlotinib for 48 h and harvested after trypsinization. Next, cells were washed with PBS, resuspended in binding buffer (BD Biosciences PharMingen) and stained with FITC Annexin-V (FITC Annexin-V apoptosis detection kit, BD Biosciences PharMingen). The cells were analyzed by flow cytometry (BD FACScan). All experimental arms were done in triplicate and displayed as averages with standard error bars.
Mouse xenograft model.
Athymic nude mice (4- to 6-week-old males) were purchased from the Harlan laboratories (Indianapolis, IN). All animal procedures and maintenance were conducted in accordance with the institutional guidelines of the University of Wisconsin. NCI-H226 cells were injected in the dorsal flank of the mouse at respective day 0 (2 × 106 cells). Once tumors reached 50–100 mm3, mice were randomized and started on their respective treatments (cetuximab or IgG control). Cetuximab dose for all experiments was 0.5 mg (20 mg/kg) intraperitoneally injected twice weekly. Tumor diameters were measured with digital calipers and tumor volumes were calculated by the formula: (π/6 × (large diameter) × (small diameter)Citation2. Tumors were monitored for cetuximab resistance that was defined as marked tumor growth in the presence of continued cetuximab therapy. Once cetuximab-resistant tumors reached a volume >500 mm3, mice were paired according to similar time points of resistance. At this point cetuximab therapy was ceased and one mouse of the pair was treated with vehicle (carboxymethylcellulose: CMC) and the second mouse of the pair treated with erlotinib (70 mg/kg), 5 d a week by oral gavage for the indicated times. Mice that remained sensitive (demonstrated by controlled tumor size) to cetuximab after 200 d post cetuximab start date were not used for further analysis.
Mouse tumor collection and Immunohistochemistry (Ki67 and TUNEL assays).
Tumors were harvested from mice at 3 and 6 h after the final erlotinib treatment, fixed in 10% neutral buffered formalin and then embedded in paraffin. Five-micron sections were used for hematoxylin-eosin (H&E) staining and immunohistochemistry (Ki67 and TUNEL assays). Samples were deparaffinized and antigen retrieval was performed in citrate buffer (pH = 6.0). Samples were then incubated with Ki67 primary antibody (AbCam, Cambridge, MA). Samples were washed in PBS and incubated in secondary antibody for 1 h followed by a 30 min incubation in ABC buffer provided by Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). DAB staining was done using Ultravision Plus Detection System (Lab Vision Corporation, Fremont, CA). For TUNEL, we used the ApopTag Plus Peroxidase in situ apoptosis detection kit from Millipore. Samples were prepared according to manufacturer's recommended protocol, with the use of proteinase K (20 ug/mL) for antigen retrieval. Images were captured using Nikon Fluorescence microscope. ImageJ was used to obtain total number of cells (via thresholding that was maintained across all samples). Color deconvolution was used to identify the positive staining and was thresholded across all image samples. Five images per tumor sample were taken, and all images were averaged and standard error mean was calculated.
Statistical analysis.
Student's t-test was used to evaluate the significance of change in percent of cell growth, BrdU cell cycle distribution and Annexin-V apoptosis assay. Statistical analysis to determine the significance of positive immunohistochemistry Ki67 and TUNEL staining of erlotinib or vehicle treated cetuximab-resistant tumors sections were also done with t-test.
Abbreviations
| DMSO | = | dimethyl sulfoxide |
| EGFR | = | epidermal growth factor receptor |
| HER2 | = | epidermal growth factor receptor 2 |
| HER3 | = | epidermal growth factor receptor 3 |
| H&E | = | hematoxylin and eosin stain |
| HNSCC | = | head and neck squamous cell carcinoma |
| IHC | = | immunohistochemistry |
| mCRC | = | metastatic colorectal cancer |
| mAbs | = | monoclonal antibodies |
| NSCLC | = | non-small cell lung cancer |
| PIP2 | = | phosphatidylinositol-(4,5)-bisphosphate |
| PIP3 | = | phosphatidylinositol-(3,4,5)-trisphosphate |
| RTK | = | receptor tyrosine kinase |
| SDS-PAGE | = | SDS-polyacrylamide gel electrophoresis |
| SCC | = | squamous cell carcinoma |
| SiRNA | = | small interfering RNA |
| TUNEL | = | terminal deoxynucleotidyl transferase-mediated dUTP nick and labeling |
| TKD | = | tyrosine kinase domain |
| TKI | = | tyrosine kinase inhibitor |
Figures and Tables
Figure 1 Cetuximab-resistant clones remain dependent on the EGFR signaling pathway. (A) Cetuximab growth response using cetuximab-resistant clones established from the non-small cell lung cancer (NSCLC) line NCI-H226. Parental NCI-H226 cell line, HP, and cetuximab-resistant cell lines (HC1, HC4, HC8) were treated with the indicated concentration of cetuximab. Growth was measured 72 h post treatment using the growth proliferation assay described in materials and methods. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05. (B) Cetuximab-resistant cell lines overexpress HER family members and have increased AKT, MAPK and S6 activity. Parental cells (HP) and cetuximab-resistant clones (HC1, HC4, HC8) were lysed and fractionated on SDS-PAGE followed by immunoblotting for the indicated proteins. α-Tubulin was used as a loading control. (C) Expression of EGFR in cetuximab-resistant clones by confocal immunofluorescence microscopy. EGFR expression was visualized via immunofluorescent staining in the parental HP cell line and cetuximab-resistant cell lines (HC1, HC4, HC8). The left column is the merged image of EGFR; Red, phalloidin staining of the cytoskelton; Green, EGFR staining; Blue, DNA staining; Magnification, ×400. (D) EGFR is effectively knocked down in cetuximab-resistant cell lines. Cetuximab-resistant cell lines were plated and treated with either 25 nM EGFR siRNA or scrambled siRNA. Seventy-two hours later protein was collected and fractioned by SDS-PAGE and immunoblotted for EGFR. α-Tubulin was used as a loading control. (E) Cetuximab-resistant cell lines are dependent on EGFR. Cetuximab-resistant cell lines were plated and treated with either 10 or 25 nM of EGFR siRNA, or 25 nM scrambled siRNA. Proliferation was measured at 72 h after treatment using the proliferation assay described in materials and methods. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05, **p ≤ 0.001.
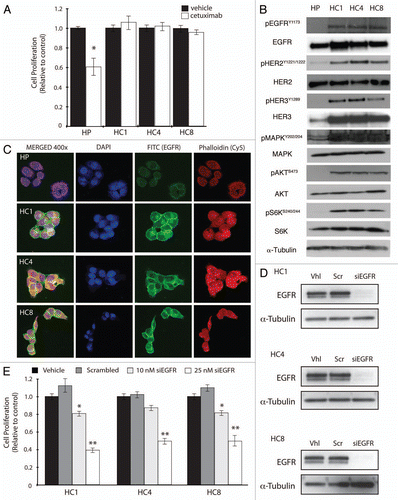
Figure 2 Cetuximab-resistant cell lines are sensitive to the EGFR targeted TKI erlotinib. (A) HER family members and downstream signaling molecules AKT and MAPK are effectively inhibited with erlotinib treatment in cetuximab-resistant cell lines. Cetuximab-resistant clones (HC1, HC4, HC8) were treated with vehicle (DMSO), cetuximab (100 nM) or erlotinib (250 or 750 nM) for 24 h. Whole cell protein lysates were fractionated on SDS-PAGE followed by immunoblotting for the indicated proteins. α-Tubulin was used as a loading control. (B) Erlotinib inhibits the proliferation of cetuximab-resistant cell lines. Parental cells (HP) and cetuximab-resistant cell lines (HC1, HC4, HC8) were plated and allowed to adhere for 24 h prior to vehicle (DMSO), cetuximab (100 nM) or erlotinib treatment: 0.25 µM, 0.75 µM or 1 uM. Growth was measured at 72 h after drug treatment using the growth proliferation assay described in experimental methods and plotted as a percentage of growth relative to untreated control cells. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05, **p ≤ 0.001. (C) Erlotinib treatment induces G1 cell cycle phase arrest in cetuximab-resistant cell lines. Cetuximab-resistant cell lines (HC1, HC4, HC8) were plated and grown for 24 h prior to treatment with vehicle (DMSO), cetuximab (100 nM) or erlotinib (250 or 750 nM) for 24 h. Cell cycle phase distribution was analyzed as described in the materials and methods. Data points are represented as mean ± SEM (n = 3). *p ≤ 0.05, **p ≤ 0.001.
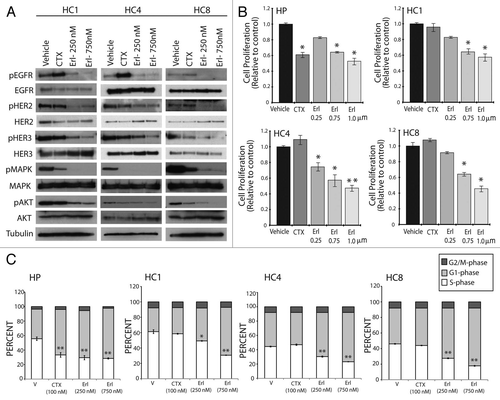
Figure 3 Erlotinib induces apoptosis in cells with acquired resistance to cetuximab. Parental cell line (HP) or cetuximab-resistant cell lines (HC1, HC4, HC8) were plated and allowed to adhere for 24 h prior to treatment with vehicle (DMSO), cetuximab (100 nM) or erlotinib (500 nM or 1 uM) for 48 h prior to Annexin-V analysis via flow cytometry. Annexin-V analysis was described in the materials and methods. Data points are represented as mean ± SEM (n = 3). *p ≤ 0.05, **p ≤ 0.001.
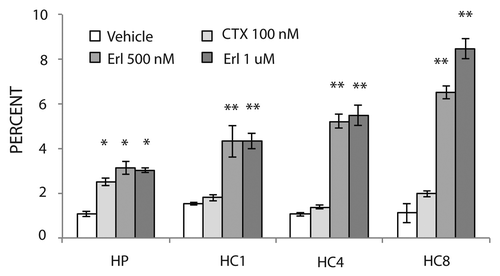
Figure 4 Erlotinib can delay growth of cetuximab-resistant tumors in vivo. Mice were injected with NCI-H226 and grown to 50–100 mm3. All mice were randomized to treatment or control groups and treated with either 0.5 mg/mouse (20 mg/kg) of cetuximab or IgG intraperitoneally twice per week. Tumors were monitored for cetuximab resistance that was defined as marked tumor growth in the presence of continued cetuximab therapy. Once cetuximab-resistant tumors reached a volume >500 mm3, mice were paired according to similar time points of resistance. At this point each mouse was taken off cetuximab and treated with either vehicle (■) or 70 mg/kg of erlotinib (◊), 5 d a week by oral gavage. The black arrow designates the starting time point of erlotinib or vehicle treatment. The average tumor volume of mice treated with IgG (▴) is included in all parts for comparison purposes.
Figure 5 Erlotinib decreases proliferation and enhances apoptosis of cetuximab-resistant tumors. (A) Cetuximab-resistant tumor samples after treatment with either erlotinib (Ctx+Erl) or vehicle (Ctx+Veh) were prepared and analyzed for proliferation (Ki67) and apoptosis (TUNEL). Erlotinib treated cetuximab-resistant tumors are designated by T1, T2 and T3. The T1, T2 and vehicle treated cetuximab-resistant tumors were collected 3 h after final erlotinib treatment, and the T3 erlotinib treated cetuximab-resistant tumor was collected 6 h post final erlotinib treatment. Black arrows denote positive staining DNA on TUNEL part, and positive staining nuclei in Ki67 part. H&E stained sections (magnification, ×200). (B) Quantitation of IHC-positive staining for Ki67 and TUNEL stained tumor samples. Graph of the TUNEL (upper) and Ki67 (lower) expression as percentage of positive cells (five random fields, one section for each sample, *p ≤ 0.05, **p ≤ 0.001). ImageJ was used to quantify the positive staining.
Acknowledgments
This project was supported, in part, by grant P30CA014520 from the National Cancer Institute, grant 1UL1RR025011 from the Clinical and Translational Science Award program of the National Center for Research Resources and the National Institutes of Health (D.L.W.) and by grant RSG-10-193-01-TBG from the American Cancer Society (D.L.W.). Fluorescence microscopy and photography were supported in part by a core grant to the Waisman Center from the National Institute of Child Health and Human Development (P30 HD03352).
References
- Citri A, Yarden Y. EGF-ERBB signaling: towards the systems level. Nat Rev Mol Cell Biol 2006; 7:505 - 516; PMID: 16829981; http://dx.doi.org/10.1038/nrm1962
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 2004; 4:937 - 947; PMID: 15573115; http://dx.doi.org/10.1038/nrc1503
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550 - 562; PMID: 19629070; http://dx.doi.org/10.1038/nrc2664
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550 - 562; PMID: 19629070; http://dx.doi.org/10.1038/nrc2664
- Sergina NV, Moasser MM. The HER family and cancer: emerging molecular mechanisms and therapeutic targets. Trends Mol Med 2007; 13:527 - 534; PMID: 17981505; http://dx.doi.org/10.1016/j.molmed.2007.10.002
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127 - 137; PMID: 11252954; http://dx.doi.org/10.1038/35052073
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene 2000; 19:6550 - 6565; PMID: 11426640; http://dx.doi.org/10.1038/sj.onc.1204082
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37:9 - 15; PMID: 11597399; http://dx.doi.org/10.1016/S0959-8049(01)00231-3
- Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 2002; 62:7350 - 7356; PMID: 12499279
- Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol 2006; 24:2666 - 2672; PMID: 16763281; http://dx.doi.org/10.1200/JCO.2005.04.8306
- Grandis JR, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst 1998; 90:824 - 832; PMID: 9625170; http://dx.doi.org/10.1093/jnci/90.11.824
- Veale D, Ashcroft T, Marsh C, Gibson GJ, Harris AL. Epidermal growth factor receptors in non-small cell lung cancer. Br J Cancer 1987; 55:513 - 516; PMID: 3038157; http://dx.doi.org/10.1038/bjc.1987.104
- Piyathilake CJ, Frost AR, Manne U, Weiss H, Bell WC, Heimburger DC, et al. Differential expression of growth factors in squamous cell carcinoma and precancerous lesions of the lung. Clin Cancer Res 2002; 8:734 - 744; PMID: 11895903
- Saif MW. Colorectal cancer in review: the role of the EGFR pathway. Expert Opin Investig Drugs 2010; 19:357 - 369; PMID: 20095919; http://dx.doi.org/10.1517/13543781003593962
- Yasui W, Sumiyoshi H, Hata J, Kameda T, Ochiai A, Ito H, et al. Expression of Epidermal Growth-Factor Receptor in Human Gastric and Colonic Carcinomas. Cancer Res 1988; 48:137 - 141; PMID: 2446740
- Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker?. Cancer J 2010; 16:23 - 32; PMID: 20164687; http://dx.doi.org/10.1097/PPO.0b013e3181d24fc1
- Bhargava R, Gerald WL, Li AR, Pan Q, Lal P, Ladanyi M, et al. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol 2005; 18:1027 - 1033; PMID: 15920544; http://dx.doi.org/10.1038/modpathol.3800438
- Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett 1998; 425:145 - 150; PMID: 9541025; http://dx.doi.org/10.1016/S0014-5793(98)00224-5
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985; 313:144 - 147; PMID: 2981413; http://dx.doi.org/10.1038/313144a0
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl 1985; 3:161 - 172; PMID: 3011820
- Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, et al. Analysis of the phosphatidylinositol-3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res 2003; 63:2742 - 2746; PMID: 12782577
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301 - 311; PMID: 15837620; http://dx.doi.org/10.1016/j.ccr.2005.03.003
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010; 28:1254 - 1261; PMID: 20100961; http://dx.doi.org/10.1200/°CO.2009.24.6116
- Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res 1995; 1:1311 - 1318; PMID: 9815926
- Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu DS, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040 - 2048; PMID: 18003960; http://dx.doi.org/10.1056/NEJMoa071834
- Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chien CRC, Makhson A, et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N Engl J Med 2009; 360:1408 - 1417; PMID: 19339720; http://dx.doi.org/10.1056/NEJMoa0805019
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 1127; PMID: 18784101; http://dx.doi.org/10.1056/NEJMoa0802656
- Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009; 373:1525 - 1531; PMID: 19410716; http://dx.doi.org/10.1016/S0140-6736(09)60569-9
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008; 359:1367 - 1380; PMID: 18815398; http://dx.doi.org/10.1056/NEJMra0802714
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:73; PMID: 15737014; http://dx.doi.org/10.1371/journal.pmed.0020073
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101:13306 - 11311; PMID: 15329413; http://dx.doi.org/10.1073/pnas.0405220101
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 2139; PMID: 15118073; http://dx.doi.org/10.1056/NEJMoa040938
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 1500; PMID: 15118125; http://dx.doi.org/10.1126/science.1099314
- Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst 2005; 97:1185 - 1194; PMID: 16106023; http://dx.doi.org/10.1093/jnci/dji238
- Benavente S, Huang S, Armstrong EA, Chi A, Hsu KT, Wheeler DL, et al. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin Cancer Res 2009; 15:1585 - 1592; PMID: 19190133; http://dx.doi.org/10.1158/1078-0432.CCR-08-2068
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008; 27:3944 - 3956; PMID: 18297114; http://dx.doi.org/10.1038/onc.2008.19
- Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:2311 - 2319; PMID: 18390971; http://dx.doi.org/10.1200/°CO.2007.13.1193
- Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360:1408 - 1417; PMID: 19339720; http://dx.doi.org/10.1056/NEJMoa0805019
- Saltz LB, Lenz HJ, Kindler HL, Hochster HS, Wadler S, Hoff PM, et al. Randomized phase II trial of cetuximab, bevacizumab and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol 2007; 25:4557 - 4561; PMID: 17876013; http://dx.doi.org/10.1200/°CO.2007.12.0949
- Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040 - 2048; PMID: 18003960; http://dx.doi.org/10.1056/NEJMoa071834
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351:337 - 345; PMID: 15269313; http://dx.doi.org/10.1056/NEJMoa033025
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006; 354:567 - 578; PMID: 16467544; http://dx.doi.org/10.1056/NEJMoa053422
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 1127; PMID: 18784101; http://dx.doi.org/10.1056/NEJMoa0802656
- Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an eastern Cooperative Oncology Group study. J Clin Oncol 2005; 23:8646 - 8654; PMID: 16314626; http://dx.doi.org/10.1200/°CO.2005.02.4646
- Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res 2007; 67:8240 - 8247; PMID: 17804738; http://dx.doi.org/10.1158/0008-5472.CAN-07-0589
- Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tabernero J, et al. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res 2004; 10:6487 - 6501; PMID: 15475436; http://dx.doi.org/10.1158/1078-0432.CCR-04-0870
- Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004; 64:5355 - 6362; PMID: 15289342; http://dx.doi.org/10.1158/0008-5472.CAN-04-0562
- Huether A, Hopfner M, Baradari V, Schuppan D, Scherubl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol 2005; 70:1568 - 1578; PMID: 16226226; http://dx.doi.org/10.1016/j.bcp.2005.09.007
- Epstein RJ, Leung TW. Tumor resensitization to erlotinib following brief substitution of cetuximab. Cancer Chemother Pharmacol 2008; 62:1111 - 1112; PMID: 18283460; http://dx.doi.org/10.1007/s00280-008-0698-6
- Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801 - 3813; PMID: 19684613; http://dx.doi.org/10.1038/onc.2009.234
- Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther 2009; 8:696 - 703; PMID: 19276677; http://dx.doi.org/10.4161/cbt.8.8.7903
- Wang Y, Pennock S, Chen X, Wang Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol 2002; 22:7279 - 7290; PMID: 12242303; http://dx.doi.org/10.1128/MCB.22.20.7279-90.2002
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol 2004; 16:400 - 406; PMID: 15261672; http://dx.doi.org/10.1016/j.ceb.2004.06.005
- Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol 2004; 24:7059 - 7071; PMID: 15282306; http://dx.doi.org/10.1128/MCB.24.16.7059-71.2004
- Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem 2009; 284:36592 - 36604; PMID: 19840943; http://dx.doi.org/10.1074/jbc.M109.000760
- Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3:802 - 808; PMID: 11533659; http://dx.doi.org/10.1038/ncb0901-802
- Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer 2006; 94:184 - 188; PMID: 16434982; http://dx.doi.org/10.1038/sj.bjc.6602941
- Guarino MJ, Schneider CJ, Hosford MA, Brahmer JR, Rudin CM, Finckenstein FG, et al. Dual inhibition of the epidermal growth factor receptor (EGFR) pathway with the combination of cetuximab and erlotinib: A phase I study in patients with advanced solid malignancies. Oncologist 2009; 14:119 - 124; PMID: 19182243; http://dx.doi.org/10.1634/theoncologist.2008-0124