 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Pancreatic cancer is one of the most lethal of human malignancies, and potent therapeutic options are lacking. Inhibition of cell cycle progression through pharmacological blockade of cyclin-dependent kinases (CDK) has been suggested as a potential treatment option for human cancers with deregulated cell cycle control. Dinaciclib (SCH727965) is a novel small molecule multi-CDK inhibitor with low nanomolar potency against CDK1, CDK2, CDK5 and CDK9 that has shown favorable toxicity and efficacy in preliminary mouse experiments, and has been well tolerated in Phase I clinical trials. In the current study, the therapeutic efficacy of SCH727965 on human pancreatic cancer cells was tested using in vitro and in vivo model systems. Treatment with SCH727965 significantly reduced in vitro cell growth, motility and colony formation in soft agar of MIAPaCa-2 and Pa20C cells. These phenotypic changes were accompanied by marked reduction of phosphorylation of Retinoblastoma (Rb) and reduced activation of RalA. Single agent therapy with SCH727965 (40 mg/kg i.p. twice weekly) for 4 weeks significantly reduced subcutaneous tumor growth in 10/10 (100%) of tested low-passage human pancreatic cancer xenografts. Treatment of low passage pancreatic cancer xenografts with a combination of SCH727965 and gemcitabine was significantly more effective than either agent alone. Gene Set Enrichment Analysis identified overrepresentation of the Notch and Transforming Growth Factor-beta (TGF-beta) signaling pathways in the xenografts least responsive to SCH727965 treatment. Treatment with the cyclin-dependent kinase inhibitor SCH727965 alone or in combination is a highly promising novel experimental therapeutic strategy against pancreatic cancer.
Introduction
Ductal adenocarcinoma of the pancreas (used synonymously with the term ‘pancreatic cancer’ throughout the following text) is the fourth most common cause of cancer-related mortality in the western world and one of the most devastating of human malignancies known to date. It is almost uniformly lethal even in early, organ-confined stages and accounts for approximately 33,000 deaths in the United States every year.Citation1 Its overall median 5-y survival rate of less than 5% is among the worst of all human cancers.Citation1,Citation2 Although recent decades have seen concerted research efforts aimed at better understanding of the underlying etiologic and pathophysiological mechanisms and at development of novel experimental therapeutic strategies, the overall prognosis of pancreatic cancer has not improved significantly and the extremely poor patient survival rates have remained largely unchanged. Therefore, development of novel experimental therapeutic strategies directed against pancreatic cancer, assessment of their efficacy in relevant preclinical in vivo models and rapid translation into clinical application is an urgent necessity.
Aberrant activation of cyclin-dependent kinases (CDKs) and dysregulation of cell cycle progression is a hallmark of many human cancers. Thus, manipulation of cell cycle progression by means of small molecule inhibitors has long been suggested as a potential treatment option for rapidly dividing cancer cells.Citation3 However, unfavorable toxicity profiles and severe adverse effects have often prevented successful clinical application of candidate cell cycle inhibitors identified by means of in vitro assays.Citation4 SCH727965 (Dinaciclib) is a potent novel small molecule inhibitor designed to inhibit various cyclin-dependent kinases, including those involved in regulation of cell cycle progression. SCH727965 has been shown to inhibit CDK1, CDK2, CDK5 and CDK9 with low nanomolar potency. It has shown a favorable safety profile and pharmacokinetics in mice, and has been shown to inhibit tumor growth in several preclinical xenograft models.Citation5 In early clinical trials, SCH727965 has been well tolerated, and has shown PET evidence of activity.Citation6,Citation7 SCH727965 is currently in Phase II clinical evaluation for melanoma and multiple myeloma.
In the present study, we show that monotherapy with SCH727965 is highly active in diminishing pancreatic cancer growth in murine subcutaneous xenograft model systems, and that the combination with gemcitabine results in significant accentuation of tumor growth inhibition in the orthotopic setting. Moreover, gene set enrichment analysis (GSEA) of basal global mRNA expression profiles suggests that resistance to this compound may be associated with activation of other oncogenic pathways in pancreatic cancer, including the Notch and Transforming Growth Factor-β (TGFβ) signaling pathways, providing the seedbed for potential combinatorial therapies to overcome treatment resistance.
Results
SCH727965 inhibits growth of pancreatic cancer cells in vitro.
The effects of SCH727965 treatment on viability and growth of pancreatic cancer cells in vitro were determined. In vitro cell growth of pancreatic cancer cells was inhibited by SCH727965 in a dose-dependent manner. Upon incubation with SCH727965 for 72 h, the GI50s were approximately 10 and 20 nM for MIAPaCa-2 and Pa20C cells, respectively (). These results are consistent with studies of SCH727965 in other cancer cell lines.Citation5 In soft agar assays, 5 to 10 nM of SCH727965 significantly reduced colony formation and anchorage independent growth of MIAPaCa-2 cells (). Moreover, in vitro cell migration of Pa20C and MIAPaCa-2 cells was significantly reduced by SCH727965-concentrations starting from 2–5 nM, as demonstrated using BD FluoroChrom, modified Boyden Chamber and wound healing assays (, D and ).
SCH727965 antagonizes activation of RalA.
RalA is an effector of the Ras signal transduction pathway,Citation8 and has been shown to be critically important for tumorigenicity of human pancreatic cancer cells.Citation9,Citation10 We have previously shown that activation of RalA can be blocked by functional inactivation of Cyclin-Dependent Kinase 5 (CDK5) in pancreatic cancer cells, possibly through interference with a proposed linear signaling axis via p35/CDK5-RalA/B downstream of oncogenic KRAS. We have shown that this decrease in Ral activation is at least partially responsible for the decreased tumorigenicity we have found to result from inhibition of CDK5 in these cells.Citation11 SCH727965 is a potent inhibor of CDK5 (IC50 1 nM).Citation5 Therefore, we hypothesized that SCH727965 treatment would lead to reduced RalA activation. We found that incubation of MIAPaCa-2 cells with 2–10 nM of SCH727965 led to substantial reduction of active RalA-GTP levels. This effect was partially rescued by enforced overexpression of the constitutively active Ral guanine nucleotide exchange factor Rgl2-CAAX in this in vitro model system (). Since the Ral signal transduction pathway is a centrally important effector of dysregulated KRAS in pancreatic cancer,Citation9,Citation10 this suggests that SCH727965 may inhibit RAS-Ral mediated tumorigenicity in pancreatic cancer.
Blockade of Rb-phosphorylation in vitro by SCH727965.
The Retinoblastoma (Rb) protein is a well-known gate keeper of cell cycle progression. Not unexpectedly, treatment of MIAPaCa-2 cells with SCH727965 reduced phosphorylation of Rb at Ser807 and Ser811 in a dose-dependent manner, while GAPDH and total Rb levels remained unchanged, as observed using protein gel blot analysis ().
SCH727965 inhibits the in vivo growth of a panel of low-passage pancreatic cancer xenografts.
These encouraging results prompted us to further study the potential therapeutic efficacy of SCH727965 in pancreatic cancer models in vivo. Our group has previously described the suitability of patient-derived low-passage subcutaneous pancreatic cancer xenografts as an effective platform model system for translational research and drug testing.Citation12,Citation13 Therefore we employed this valuable resource to further study the extent of growth inhibition achievable by SCH727965 treatment, and molecular markers associated with sensitivity and resistance. Treatment with SCH727965 given as twice weekly i.p. doses of 40 mg/kg for 4 weeks caused significant tumor growth inhibition (TGI) in 10/10 (100%) of low-passage subcutaneous xenografts tested. However, the extent of TGI varied considerably among different xenografts studied () A > 40% growth inhibition upon treatment with SCH727965 was observed in 8/10 xenograft models of pancreatic cancer (). Relative growth inhibition at the end of treatment for each xenograft is given in . Of interest, xenografts derived from less differentiated cancers had more pronounced growth inhibition upon SCH727965 treatment than xenografts established from well-differentiated cases. However, growth retardation due to treatment with SCH727965 was not consistently accompanied by any discernible morphological change in the tumor microarchitecture (data not shown).
Molecular surrogate markers of therapeutic response to SCH727965.
SCH727965 is a multi-CDK inhibitor with the potential to inhibit cell proliferation and cell cycle progression.Citation4 In line with this mechanism, we found significant reduction of nuclear immunolabeling of the surrogate marker of proliferation Ki67 in SCH727965-treated Panc286 and Panc219 xenografts as compared with mock-treated control tumors (). These two xenografts had shown the most pronounced therapeutic response in terms of growth retardation upon treatment with SCH727965 in our panel representing ten cases of pancreatic cancer ( and ). As opposed to this, little to no reduction of Ki67 nuclear immunolabeling upon treatment with SCH727965 was found in the two most resistant pancreatic cancer xenografts, Panc291 and JH033 (). Immunolabeling of phospho-Rb(Ser807/811) was significantly reduced both in sensitive Panc286 as well as—albeit to a lesser degree—in resistant JH033 xenografts ().
Combination treatment of orthotopic pancreatic cancer xenografts with SCH727965 and gemcitabine.
Gemcitabine is currently the standard first line therapy for pancreatic cancer, but it results in overall median survival of only ∼5–6 mo.Citation14,Citation15 We examined the combination efficacy of SCH727965 and gemcitabine in an orthotopic xenograft model of pancreatic cancer (13), derived from Panc265, a low-passage xenograft that demonstrates modest sensitivity to gemcitabine. Freshly harvested 1–2 mm3 pieces of Panc265 were implanted orthotopically into the pancreata of nude mice under anesthesia, and xenograft establishment (“primary tumor”) was assessed by ultrasound. Mice were randomized into 4 treatment groups, for treatment with vehicle-only control, SCH727965, gemcitabine, or a combination of SCH727965 and gemcitabine. shows that although the Panc265 xenograft is resistant to SCH727965 monotherapy, the combination was significantly more effective than either SCH727965 or gemcitabine alone. There was little or no tumor growth in the group treated with both SCH727965 and gemcitabine. This result suggests that the combination of SCH727965 and gemcitabine may be promising for treatment of pancreatic cancer.
Basal global gene expression analysis and gene set enrichment analysis (GSEA) identifies signaling pathways overrepresented in SCH727965-sensitive vs. resistant pancreatic cancer xenografts.
Basal global gene expression analysis was performed on duplicate samples derived from each of the low-passage pancreatic cancer xenografts used in this study by means of Affymetrix cDNA microarrays.Citation16 Normalized global mRNA expression data of the 2 most SCH727965-sensitive xenografts (Panc286 and Panc219) were then compared with the teo most resistant lines (Panc291 and JH033) by means of the publicly available Gene Set Enrichment Analysis tool (http://www. broadinstitute.org/gsea/). Of note, using this analysis, regulation of cell cycle progression (HSA04110) was one of the top ten signaling pathways included in this analysis and identified as being overrepresented in terms of basal steady-state mRNA expression levels in sensitive xenografts (), supporting the validity of this analysis tool in this given setting. Likewise, the list of ten pathways receiving highest scores as likely being overrepresented in xenografts most resistant to treatment with SCH727965 in vivo included the Notch (HSA04330) and Transforming Growth Factor (TGF)β (HSA04350) signaling pathways (), which have previously been shown to be involved in pancreatic carcinogenesis and tumor progression, and have been demonstrated to be promising novel therapeutic targets for pancreatic cancer.Citation17–Citation20 Enrichment plots demonstrating overrepresentation of components of the Notch and TGFβ signaling pathways, respectively, in SCH727965-resistant as compared with SCH727965-sensitive low-passage pancreatic cancer xenografts are depicted in . Average SMAD4 mRNA was significantly higher in SCH727965 resistant versus sensitive xenografts. Moreover, in sensitive xenograft lines, higher SMAD4 mRNA expression was found after treatment with SCH727965, compared to mock treated controls; this suggests induction or selection of SMAD4 expression by SCH727965 treatment (). It will therefore be a fascinating aim of future experimental studies to investigate whether therapeutic resistance to SCH727965 might be overcome by concomitant therapeutic blockade of either of these pathways.
Discussion
Pancreatic cancer remains one of the worst killers among human cancers, and new therapeutic options are urgently needed. In the current study we show that the novel multi-CDK inhibitor SCH727965 has considerable inhibitory effects on human pancreatic cancer cells in both in vitro and in vivo model systems. In vitro, SCH727965 inhibited growth, migration and colony formation of human pancreatic cancer cells through inhibition of cell cycle progression and decrease in Rb phosphorylation. These mechanisms were confirmed in vivo using immunohistochemistry on drug-treated low-passage subcutaneous pancreatic cancer xenograft tissue specimens. Important for pancreatic cancer, SCH727965 appears to significantly antagonize a critical effector pathway downstream of aberrant Ras signaling. Mutations within the KRAS2 gene are found in more than 90% of pancreatic cancers,Citation21 and they are among the earliest genetic aberrations observed in low-grade PanIN lesions during the multistep progression model culminating in the development of a fully invasive pancreatic cancer phenotype.Citation22 Taken together, these observations mark oncogenic Ras signaling as a prime therapeutic target in pancreatic cancer. In preclinical studies, disruption of KRAS2 function via RNA interference, antisense DNA or expression of dominant negative KRASN17 attenuates the tumorigenicity of pancreatic cancer cell lines.Citation23–Citation25 Unfortunately, strategies to inhibit the Ras pathway directly in patients have been largely unsuccessful, as exemplified by the lack of clinical activity of farnesyltransferase inhibitors (FTIs), which interfere with critical post-translational modification of RAS proteins.Citation26 Ras signaling through the Ral pathway has emerged as a critical mediator of the malignant phenotype in pancreatic cancer cells,Citation9,Citation10 but there have been no effective strategies to inhibit Ral signaling. Our data presented here indicate that SCH727965 can block Ral activation in pancreatic cancer cells, potentially through inhibition of CDK5, which we have shown is an important determinant of Ras-mediated Ral activation in pancreatic cancer cells.Citation11
In vivo, SCH727965 treatment dramatically reduced growth of a panel of 10 low-passage pancreatic cancer xenografts. There was a considerable amount of variation in the growth inhibitory effect of SCH727965 between the individual low-passage xenografts included in this study. It is tempting to speculate whether these differences in therapeutic response reflect variations in the underlying genetic alterations that might cause differences in the basal proliferation rates. Our own group and others have previously demonstrated that therapeutic response to drug treatment can be predicted by analysis of global gene expression patterns in many cases.Citation16,Citation27–Citation30 Here, we found that the publicly available Gene Set Enrichment Analysis tool (www.broadinstitute.org/gsea/) was able to readily identify control of cell cycle progression as one of the most highly enriched signaling pathways in SCH727965-sensitive vs. -resistant subcutaneous pancreatic cancer xenografts. In the xenografts showing the least sensitivity toward SCH727965 in terms of growth inhibition, the Notch- and TGFβ signaling pathways were among the top ten candidates thus identified. Both of these pathways are involved in pancreatic carcinogenesis and progression, and have long been suggested as promising therapeutic targets in pancreatic as well as in many other cancers.Citation17,Citation19,Citation31–Citation34 It is therefore tempting to speculate that concomitant pharmacological inhibition of either of these pathways, for which experim ental small molecule inhibitors are readily available to date, might prove to be a suitable strategy to overcome resistance to SCH727965 and enhance therapeutic potency in future studies.
In conclusion, these current data suggest that treatment with the novel, well-tolerated cyclin-dependent kinase inhibitor SCH727965 appears to be a promising new treatment option for pancreatic cancer with novel inhibitory effects on Ras-mediated Ral activation. Rapid evaluation of this strategy in a clinical setting therefore appears to be well justified.
Materials and Methods
Cell lines.
The pancreatic cancer cell line MIAPaCa-2 was obtained from the American Type Culture Collection (www.atcc.org), and the low-passage cell lines Pa14C, Pa16C and Pa20C were generated at our institution.Citation21 Cells were maintained in a humidified atmosphere supplemented with 5% CO2 in DMEM culture media containing 10% FBS (both Invitrogen, Carlsbad, CA), 1x MEM vitamins solution (Sigma-Aldrich, St. Louis, MO), 1x non-essential amino acid solution (Biofluids, Camarillo, CA), 1% sodium pyruvate solution, 1x Pen/Strep (both Biofluids) and 5 µg/ml Plasmocin (Invivogen, San Diego, CA).
In vitro cell growth assays.
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assays (Promega, Madison, WI) were done as previously described in reference Citation35 and Citation36.
Migration assays.
Wound healing and modified Boyden chamber assays were done as previously described in reference Citation11, Citation35 and Citation37. FluoroBlok migration assays (BD Biosciences, Bedford, MA) were performed following the standard procedure as recommended by the manufacturer.
Colony formation assays.
Colony formation in soft agar was assessed as previously described in reference Citation17 and Citation35. In brief, soft agar assays were set up in 6-well plates, each well containing a bottom layer of 1% agarose (Invitrogen), a middle layer of 0.6% agarose including 10,000 cells, and a top layer of medium only. Mixtures in each well were supplemented with SCH727965 at the indicated concentrations or solvent only, and the plates were incubated for 3 weeks. Next, 1.5 ml of 0.5% Wright's staining solution was added to each well. After incubation at 4°C overnight, colonies were visualized by trans-UV illumination and counted using analysis software QuantityOne (BioRad, Hercules, CA).
RalA activation assays.
RalA activation assays were done as previously described in reference Citation11. In brief, subconfluent cells were serum starved at 0.5% FBS over night and treated with SCH727965 for 30 min. Protein extraction and RalA activation assays were then performed using the RalA activation assay kit following the standard procedure recommended by the manufacturer (Upstate, Temecula, CA).
Protein gel blot analysis.
Protein gel blot analyses were performed as described previously in reference Citation11. Antibodies used for protein gel blotting were Phospho-Rb(Ser807/811) (#9308), Rb (#9313) and GAPDH (#2118; all Cell Signaling Technology, Danvers, MA).
Generation of subcutaneous and orthotopic xenografts and drug treatment.
All animal experiments conformed to the guidelines of the Animal Care and Use Committee of Johns Hopkins University and animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care. Subcutaneous murine xenografts of low-passage patient-derived human pancreatic cancers were generated as previously described elsewhere in reference Citation11 and Citation13. Xenografts from ten different cases were randomly picked and tumor tissues were serially transplanted into both flanks of ten male CD1 nu/nu athymic mice each (n = 20 xenograft tumors per case). Ten days after subcutaneous implantation, mice for each case were randomized into two groups of five mice with similar average xenograft tumor volumes and assigned to receive treatment with SCH727965 or solvent only (n = 10 xenografts per arm). In the treatment arm, 40 mg/ kg SCH727965 in 20% (w/v) hydroxypropyl-β-cyclodextrin (HPBCD) were given by intraperitoneal injection twice weekly. Mice in the control arms received mock treatment with solvent only. Total body weights were determined weekly, and xenograft tumor volumes were measured weekly using digital callipers as previously described in reference Citation38. After 4 weeks of treatment, all mice were euthanized and tumor tissues harvested. Relative growth inhibition (GI) of xenograft tumors was calculated as:
with TuVEOT: = average tumor volume at the end of treatment.
For studies examining the effect of SCH727965 and gemcitabine in pancreatic cancer, low passage xenograft tissue from a modestly gemcitabine sensitive pancreatic cancer, Panc265, was minced and implanted orthotopically in the pancreas of athymic nude mice, as described in reference Citation35. Mice were randomized by tumor size, estimated by ultrasound immediately preceding initiation of therapy (day 10 post-implantation), into 4 groups (n = 8 per group): control, gemcitabine, SCH727965 and SCH727965 + gemcitabine. Control mice received solvent only, SCH727965 in HPBCD was administered at 40 mg/kg twice weekly (Monday and Thursday), gemcitabine was given at 20 mg/kg twice weekly (Tuesday and Friday), and the combination group received both gemcitabine and SCH727965. All dosing was done intraperitoneally for 3 weeks; tumors were harvested and weighed at the end of treatment.
Tissue samples were preserved in formalin solution for histology and immunohistochemistry, for RNA-extraction and subsequent quantitative real-time RT-PCR analysis tissue specimens were snap-frozen in liquid nitrogen and stored at −80°C until used.
Immunohistochemistry.
For Ki67 (MIB1) staining of formalin-fixed paraffin-embedded tissue sections, anti-Ki67 primary antibody (clone K2, Ventana Medical Systems, Tucson, AZ) was used in combination with a Ventana Benchmark Autostainer as described in reference Citation39. Phospho-Rb(Ser807/811) was stained using a rabbit anti-human polyclonal antibody (#9308, CellSignaling Technology, Danvers, MA) at a dilution of 1:300 following the standard recommendations provided by the manufacturer.
Expression microarrays and gene set enrichment analysis.
Basal whole genome mRNA expression patterns were assessed using Affymetrix U133A Plus 2.0 expression microarrays and analyzed using the publicly available Gene Set Enrichment Analysis (GSEA) toolCitation29 as previously described in reference Citation11 and Citation16.
Quantitative real-time RT-PCR analysis.
RNA extraction, reverse transcription and quantitative real-time PCR were performed as previously described elsewhere in reference Citation35, Citation36 and Citation40. Primer sequences are available upon request. Relative quantification of mRNA transcript levels was done using the 2deltaCt method.Citation41
Statistical analysis.
Kruskal-Wallis analysis was performed using SPSS version 15.0.1 for Microsoft Windows, 2-tailed t-test was performed using GraphPad Prism for Windows version 5. p < 0.05 was regarded as statistically significant. Results in bar diagrams are plotted as means and standard deviations if not otherwise indicated.
Disclosure of Potential Conflicts of Interest
R.B. is an employee of Merck Research Laboratories, Kenilworth, NJ.
Figures and Tables
Figure 1 Cyclin dependent kinase inhibitor SCH727965 reduces growth, colony formation and motility of pancreatic cancer cells in vitro. (A) Treatment with SCH727965 causes dose-dependent growth retardation of pancreatic cancer cells in vitro as observed in MTT assays. (B) SCH727965 significantly reduced colony formation of MIAPaCa-2 cells in soft agar at concentrations of 5 and 10 nM, respectively. Incubation with SCH727965 inhibited migration of MIAPaCa-2 and Pa20C cells in BD FluoroBlok migration assays (C), and Pa20C cells in wound healing assays (D).
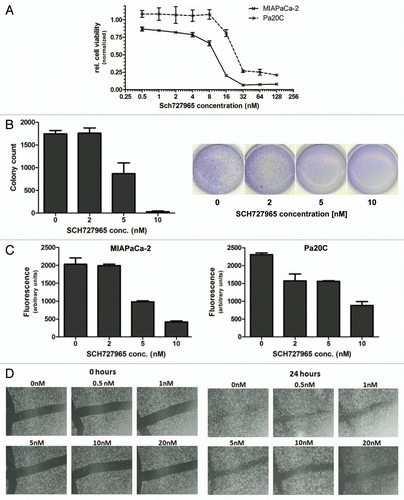
Figure 2 Modified Boyden chamber assays show decreased in vitro cell motility of Pa20C cells after treatment with SCH727965 for 72 h.
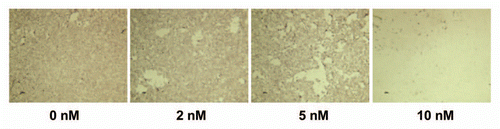
Figure 3 In vitro inhibition of RalA activation and Rb-phosphorylation by SCH727965. SCH727965-treatment for 30 min blocks activation of RalA in MIAPaCa-2 cells; RalA activation is partially rescued by enforced expression of Rgl2-CAAX (A). SCH727965 incubation for 16 h reduces phosphorylation of Rb in a dose-dependent manner as shown using protein gel blot analysis (B).
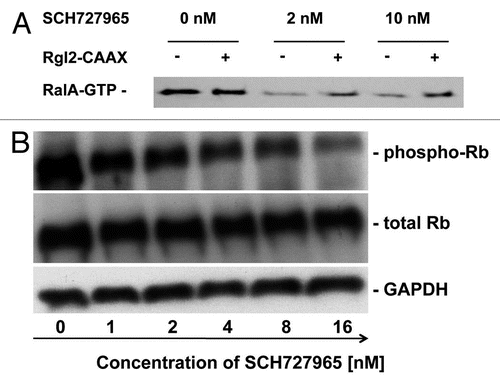
Figure 4 Spectrum of in vivo growth inhibition by SCH727965 in a panel of ten subcutaneous low-passage pancreatic cancer xenografts. Ten low-passage subcutaneous pancreatic cancer xenografts were treated with SCH727965 (n = 10 per line) or solvent only (n = 10 in each line) for 4 weeks. Growth curves show varying degrees of xenograft growth inhibition upon SCH727965-treatment. The figure shows an example with little (A) and another case with high (B) sensitivity toward SCH727965. Representative examples of xenograft tumors harvested at the end of treatment are shown for the respective lines (left tumors: controls; right tumors: SCH727965-treated). Representative histologies of control (left) and SCH727965-treated tumors (right) are shown as H&E stained tissue sections. (C) Mean inhibition of xenograft growth upon treatment with SCH727965 as compared with mock-treated controls in 10 individual xenograft lines. The diagram represents means and standard deviations of growth inhibition for each respective xenograft line.
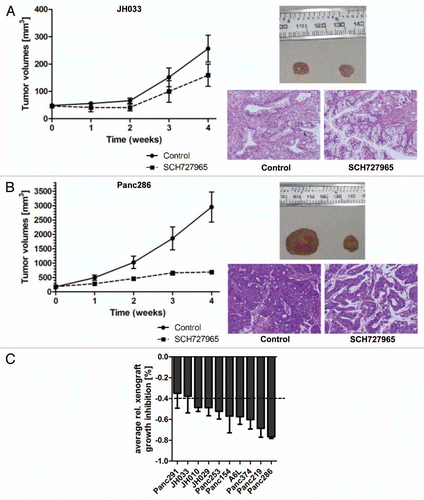
Figure 5 Surrogate biomarker of growth retardation in pancreatic cancer xenografts treated with SCH727965. (A) Tissue specimens from xenografts that showed most vs. least pronounced growth retardation upon treatment with SCH727965 in vivo were stained for Ki67 expression by immunohistochemistry. The figure shows representative sections of mock-treated controls and SCH727965-treated xenografts for each case. (B) Phospho-Rb(Ser807/811) staining of xenograft tissue specimens by IHC at cessation of treatment.
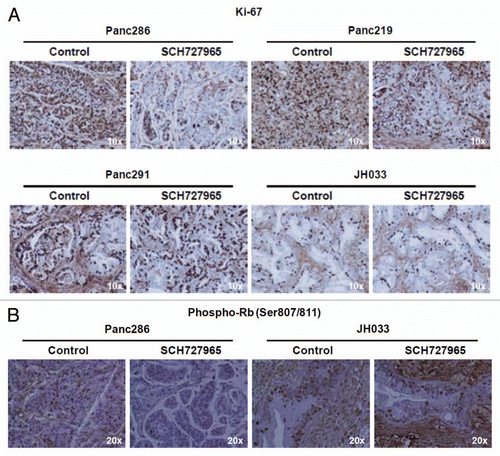
Figure 6 Combination treatment of orthotopic pancreatic cancer xenografts with SCH727965 and gemcitabine. The low passage pancreatic cancer xenograft Panc265 was implanted orthotopically into nude mice. Ten days later, tumor volume was measured by ultrasound, and mice (eight per group) were randomized into four treatment groups (vehicle-only control, SCH727965 only, gemcitabine only, and combination SCH727965 and gemcitabine), and treatment was initiated. SCH727965 (40 mg/kg) and gemcitabine (20 mg/kg) were each given twice weekly, as described in Materials and Methods. After 3 weeks of treatment, tumors were harvested and weighed. (A) relative tumor growth, calculated as (final tumor volume minus initial tumor volume/initial tumor volume); (B) average tumor weight (in grams) upon culmination of therapy. *p < 0.05; **p < 0.01; ***p < 0.001. (C) Representative macroscopic picture of mock treated intrapancreatic xenograft tumor (white arrowheads) at the end of treatment as compared with (D) combination treatment with SCH727965 plus gemcitabine. Note the different scales (indicated in mm at the right side of each image) in (C and D).
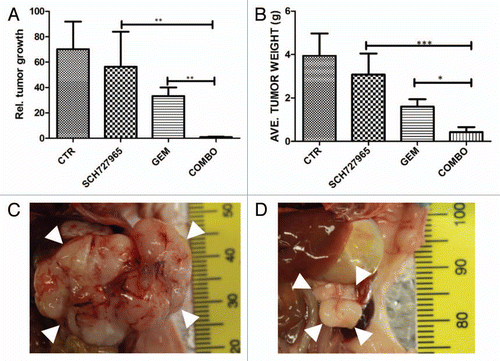
Figure 7 Gen Set Enrichment Analysis (GSEA) shows enrichment of the Notch and TGFβ pathways in SCH727965-resistant xenografts. Top, the primary result of the gene set enrichment analysis is the enrichment score (ES). GSEA calculates the ES by walking down the ranked-ordered list of genes, increasing a running-sum statistic when a gene is in the gene set and decreasing it when it is not. Middle, rank ordered list of genes (horizontal line in red and blue color). The top of this list (red color) contains genes upregulated in resistant cases. The bottom of the list (blue color) contains downregulated genes in sensitive cases. Anytime a gene from the gene set is found along the list, a vertical black bar is plotted (hit). If most of the hits are at the top of the list, then this gene set is enriched in resistant cases, if they are found at the bottom of the list then they are enriched in sensitive cases; if, however, they are distributed homogenously across the rank ordered list of genes, then that gene set is not enriched in any of the gene expression profiles (resistant vs. sensitive). These figures show enrichment of the TGFβ and Notch pathways in resistant cases. Bottom, value of the ranking metric along the list of the ranked genes.
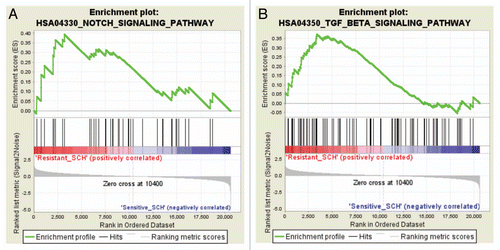
Figure 8 SMAD4 mRNA expression in xenograft tissue specimens at the end of treatment. (A) Steady-state SMAD4 mRNA levels were significantly higher in mock-treated xenografts that were resistant to SCH727965 (Panc291 and JH033) as compared with xenografts that were SCH727965 sensitive (Panc286 and Panc219). Ten tissue samples from each xenograft line were included in the analysis and mRNA expression levels were determined by means of quantitative real-time RT-PCR. (B) SMAD4 mRNA expression levels in sensitive and (C) resistant xenografts after treatment with SCH727965 or solvent, respectively.
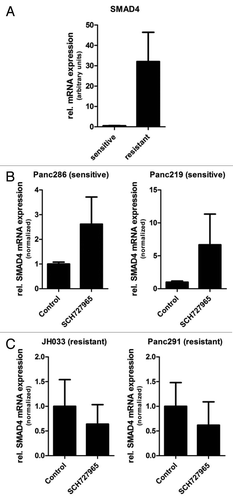
Table 1 Relative growth inhibition at the end of treatment caused by SCH727965 treatment in low-passage xenografts
Table 2 Top ten signaling pathways overrepresented in pancreatic cancer xenograft lines sensitive to SCH727965
Table 3 Top ten signaling pathways overrepresented in pancreatic cancer xenograft lines resistant to SCH727965
Acknowledgments
The authors thank Schering-Plough Biopharma (Palo Alto, CA) for synthesizing the cyclin-dependent kinase inhibitor Dinaciclib for this study.
This work was supported by NIH R01CA134767 to B.D.N. and A.M. B.D.N. was supported by NIH R01CA085567, and grants from the Flight Attendant Medical Research Foundation (FAMRI) and the National Pancreas Foundation. A.M. was supported by the Sol Goldman Pancreatic Cancer Research Center, the Michael Rolfe Foundation for Pancreatic Cancer Research, and NIH R01CA113669. G.F. was supported by BONFOR grant number O-111.0001.2, by the German Cancer Foundation (Deutsche Krebshilfe) grant number 109215 and by the European Community's Seventh Framework Program (FP7-2007–2013) under grant agreement HEALTH-F2-2011-256986. H.A. received generous support from the D'Amato Family.
References
- ACS. Cancer Facts & Figures 2007
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin 2006; 56:106 - 130; PMID: 16514137; http://dx.doi.org/10.3322/canjclin.56.2.106
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153 - 166; PMID: 19238148; http://dx.doi.org/10.1038/nrc2602
- Dickson MA, Schwartz GK. Development of cell cycle inhibitors for cancer therapy. Curr Oncol 2009; 16:36 - 43; PMID: 19370178
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 2010; 9:2344 - 2353; PMID: 20663931; http://dx.doi.org/10.1158/1535-7163.MCT-10-0324
- Shapiro GI, Bannerji R, Small K, Black S, Statkevich P, Abutarif M, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the novel cyclin-dependent kinase inhibitor SCH 727965 administered every 3 weeks in subjects with advanced malignancies. J Clin Oncol 2008; 26 suppl abstr 3532
- Nemunaitis M, Saltzman M, Rosenberg MA, Khaira D, Small K, Kirschmeier P, et al. A phase I doseescalation study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of SCH 727965, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Clin Oncol 2009; 27 suppl abstr 3535
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev 2002; 16:2045 - 2057; PMID: 12183360; http://dx.doi.org/10.1101/gad.993902
- Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 2005; 7:533 - 545; PMID: 15950903; http://dx.doi.org/10.1016/j.ccr.2005.04.030
- Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol 2006; 16:2385 - 2394; PMID: 17174914; http://dx.doi.org/10.1016/j.cub.2006.10.023
- Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, et al. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res 2010; 70:4460 - 4469; PMID: 20484029; http://dx.doi.org/10.1158/0008-5472.CAN-09-1107
- Jimeno A, Feldmann G, Suarez-Gauthier A, Rasheed Z, Solomon A, Zou GM, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther 2009; 8:310 - 314; PMID: 19174553; http://dx.doi.org/10.1158/1535-7163.MCT-08-0924
- Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006; 12:4652 - 4661; PMID: 16899615; http://dx.doi.org/10.1158/1078-0432.CCR-06-0113
- Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15:2403 - 2413; PMID: 9196156
- Storniolo AM, Enas NH, Brown CA, Voi M, Rothenberg ML, Schilsky R. An investigational new drug treatment program for patients with gemcitabine: results for over 3000 patients with pancreatic carcinoma. Cancer 1999; 85:1261 - 1268; PMID: 10189130; http://dx.doi.org/10.1002/(SICI)1097-0142(19990315)85:6<1261::AIDCNCR7>3.3.CO;2-K
- Jimeno A, Tan AC, Coffa J, Rajeshkumar NV, Kulesza P, Rubio-Viqueira B, et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res 2008; 68:2841 - 2849; PMID: 18413752; http://dx.doi.org/10.1158/0008-5472.CAN-07-5200
- Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res 2009; 15:2291 - 2301; PMID: 19258443; http://dx.doi.org/10.1158/1078-0432.CCR-08-2004
- Truty MJ, Urrutia R. Basics of TGFbeta and pancreatic cancer. Pancreatology 2007; 7:423 - 435; PMID: 17898532; http://dx.doi.org/10.1159/000108959
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008; 10:111 - 122; PMID: 18258927; http://dx.doi.org/10.2353/jmoldx.2008.070115
- De La O JP, Murtaugh LC. Notch signaling: where pancreatic cancer and differentiation meet?. Gastroenterology 2009; 136:1499 - 1502; PMID: 19327730; http://dx.doi.org/10.1053/j.gastro.2009.03.022
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801 - 1806; PMID: 18772397; http://dx.doi.org/10.1126/science.1164368
- Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3:157 - 188; PMID: 18039136; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154305
- Aoki K, Yoshida T, Sugimura T, Terada M. Liposome-mediated in vivo gene transfer of antisense K-ras construct inhibits pancreatic tumor dissemination in the murine peritoneal cavity. Cancer Res 1995; 55:3810 - 3816; PMID: 7641198
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002; 2:243 - 247; PMID: 12242156; http://dx.doi.org/10.1016/S1535-6108(02)00122-8
- Hirano T, Shino Y, Saito T, Komoda F, Okutomi Y, Takeda A, et al. Dominant negative MEKK1 inhibits survival of pancreatic cancer cells. Oncogene 2002; 21:5923 - 5928; PMID: 12185592; http://dx.doi.org/10.1038/sj.onc.1205643
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 2004; 22:1430 - 1438; PMID: 15084616; http://dx.doi.org/10.1200/JCO.2004.10.112
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347:1999 - 2009; PMID: 12490681; http://dx.doi.org/10.1056/NEJMoa021967
- Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, et al. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics 2006; 7:278; PMID: 17074082; http://dx.doi.org/10.1186/1471-2164-7-278
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545 - 15550; PMID: 16199517; http://dx.doi.org/10.1073/pnas.0506580102
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34:267 - 273; PMID: 12808457; http://dx.doi.org/10.1038/ng1180
- De La O JP, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci USA 2008; 105:18907 - 18912; PMID: 19028876; http://dx.doi.org/10.1073/pnas.0810111105
- Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2009; 136:1741 - 1796; PMID: 10.1053/j.gastro.2009.01.008
- Mythreye K, Blobe GC. The type III TGFbeta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci USA 2009; 106:8221 - 8226; PMID: 19416857; http://dx.doi.org/10.1073/pnas.0812879106
- Kabashima A, Higuchi H, Takaishi H, Matsuzaki Y, Suzuki S, Izumiya M, et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int J Cancer 2009; 124:2771 - 2779; PMID: 19296540; http://dx.doi.org/10.1002/ijc.24349
- Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67:2187 - 2196; PMID: 17332349; http://dx.doi.org/10.1158/0008-5472.CAN-06-3281
- Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008; 57:1420 - 1430; PMID: 18515410; http://dx.doi.org/10.1136/gut.2007.148189
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425:846 - 851; PMID: 14520411; http://dx.doi.org/10.1038/nature01972
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130:1120 - 1133; PMID: 17889654; http://dx.doi.org/10.1016/j.cell.2007.07.019
- Bisht S, Feldmann G, Koorstra JB, Mullendore M, Alvarez H, Karikari C, et al. In vivo characterization of a polymeric nanoparticle platform with potential oral drug delivery capabilities. Mol Cancer Ther 2008; 7:3878 - 3888; PMID: 19074860; http://dx.doi.org/10.1158/1535-7163.MCT-08-0476
- Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther 2008; 7:2725 - 2735; PMID: 18790753; http://dx.doi.org/10.1158/1535-7163.MCT-08-0573
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method. Methods 2001; 25:402 - 408; PMID: 11846609; http://dx.doi.org/10.1006/meth.2001.1262