 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Mito-CP11, a mitochondria-targeted nitroxide formed by conjugating a triphenylphosphonium cation to a five-membered nitroxide, carboxy-proxyl (CP), has been used as a superoxide dismutase (SOD) mimetic. In this study, we investigated the antiproliferative and cytotoxic properties of submicromolar levels of Mito-CP11 alone and in combination with fluvastatin, a well known cholesterol lowering drug, in breast cancer cells. Mito-CP11, but not CP or CP plus the cationic ligand, methyl triphenylphosphonium (Me-TPP+), inhibited MCF-7 breast cancer cell proliferation. Mito-CP11 had only minimal effect on MCF-10A, non-tumorigenic mammary epithelial cells. Mito-CP11, however, significantly enhanced fluvastatin-mediated cytotoxicity in MCF-7 cells. Mito-CP11 alone and in combination with fluvastatin inhibited nuclear factor kappa-B activity mainly in MCF-7 cells. We conclude that mitochondria-targeted nitroxide antioxidant molecules (such as Mito-CP11) that are non-toxic to non-tumorigenic cells could enhance the cytostatic and cytotoxic effects of statins in breast cancer cells. This strategy of combining mitochondria-targeted non-toxic molecules with cytotoxic chemotherapeutic drugs may be successfully used to enhance the efficacy of antitumor therapies in breast cancer treatment.
Introduction
Previously, we and others have shown that hydrophobic statins (e.g., simvastatin and fluvastatin) inhibit the proliferation of breast cancer cells (MCF-7, MDA-MB-231), inducing cytotoxicity in these cells.Citation1–Citation3 Statin-mediated cytostatic effects were attributed to a G1 cell cycle arrest and to decreased levels of cyclins D and E, cell cycle proteins regulating the G1-S transition of the cell cycle.Citation4 Statins induced apoptosis in several proliferating tumor cells, such as leukemia, lymphoma and neuroblastoma cells.Citation5 The cytotoxic effects of statins were shown to be independent of their lipid-lowering properties.Citation6 Although the molecular mechanisms by which hydrophobic statins exert cytotoxicity in breast cancer cells are not fully known, published data suggest that several factors, including statins' ability to stimulate inducible nitric oxide synthase, nitric oxide and block isoprenylation of many proteins (e.g., small GTPases), may play a role.Citation1 Agents inhibiting the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase activity and the farnesylation of Ras protein were shown to decrease breast cancer cell proliferation.Citation7 Doxorubicinand arsenic trioxide-induced cytotoxicity in hepatocellular carcinoma cells was enhanced in the presence of lovastatin.Citation8 Other studies implicated a role for increased oxidative stress induced by statins in tumor cells.Citation9 Lipophilic antioxidants (tocopherols and tocotrienols) greatly potentiated statin-mediated cytostatic and cytotoxic effects in breast cancer cells.Citation10 These findings suggest an intriguing connection between antioxidants, reactive oxygen species (ROS) and statin-mediated breast cancer cell cytotoxicity.
There is increasing interest on the role of oxidants and antioxidants in tumor cell proliferation.Citation11 Reports indicate that low endogenous levels of ROS enhance cancer cell proliferation and tumorigenesis, whereas high levels of ROS cause cellular damage.Citation12 ROS regulate multiple signaling cascades in cancer cells, including those involving nuclear factor-kappaB (NFκB),Citation13,Citation14 a redox-sensitive transcription factor that promotes the survival and proliferation of cancer cells.Citation15–Citation17 Recent research shows that ROS (superoxide and hydrogen peroxide) are also responsible for invadopodia formation that regulates extracellular matrix degradation in tumor metastasis.Citation18 These functions of ROS in cancer cells provide a basis for the inhibition of tumor progression and metastasis by ROS scavengers.Citation19 Supplementation with antioxidants and antioxidant enzymes (superoxide dismutase and glutathione peroxidase) decreased cellular proliferation and enhanced the chemotherapeutic activity of antitumor agents.Citation20 ROS (e.g., superoxide and hydrogen peroxide) are generated from several sources, the most prominent being mitochondria and the NADPH oxidase multi-protein complex (NOX).Citation21 Overexpression of NOX1, one of the isoforms of the NADPH oxidases, elevates ROS levels, cellular transformation and tumor growth in vivo.Citation22 Alternatively, suppression of manganese super-oxide dismutase (MnSOD), a key mitochondrial superoxide dis-mutating enzyme, by siRNA stimulates tumor cell proliferation in vitro, and promotes ovarian tumor growth in vivo.Citation23 Reports also suggest that mitochondria control NOX1-mediated redox signaling and breast tumorigenesis.Citation24 These findings indicate that mitochondrial targeting of antioxidants could inhibit tumor cell proliferation.Citation25
Nitroxides are a group of antioxidant molecules that exhibit superoxide dismutase (SOD) mimetic and radical scavenging activity.Citation26 Mitochondrial targeting of a five-membered nitroxide such as the carboxy proxyl (CP) involves coupling untargeted, parent nitroxides (e.g., CP) to a triphenylphosphonium (TPP+) cation through a linker alkyl chain (). TPP+-conjugated nitroxides (e.g., Mito-CP11, ) were shown to accumulate in the mitochondria of endothelial cells and enhance superoxide dismutation in mitochondria, mimicking the MnSOD-like activity.Citation27 In addition to the antioxidant activity, nitroxides (e.g., tempol and tempo) induce divergent apoptotic signal transduction pathway in breast tumor cells.Citation28
In this study, we characterize the antiproliferative and cytotoxic effects of mitochondria-targeted nitroxides, alone and in combination with fluvastatin. Results indicate that mitochondria-targeted five-membered nitroxides such as Mito-CP11 are more effective in inducing antiproliferative effects in MCF-7 human breast cancer cells than in normal mammary epithelial cells, MCF-10A. Consistent with the antiproliferative effects of Mito-CP11 in MCF-7 cells, we found that Mito-CP11 significantly diminishes NFκB activity in MCF-7 cells, but not in MCF-10A cells. In addition, Mito-CP11 enhanced fluvastatin-induced cytostatic and cytotoxic effects in breast cancer cells. These findings demonstrate that mitochondria-targeted nitroxides are novel inhibitors of breast cancer cell survival and proliferation, and may be useful in potentiating the cytostatic and cytotoxic effects of other chemotherapeutic agents.
Results
Antiproliferative effects of Mito-CP11 on breast cancer cells and non-cancerous mammary epithelial cells.
We examined the effects of Mito-CP11, the “untargeted” parent nitroxide, CP, and the methyltriphenylphosphonium cation (Me-TPP+, ) ligand on MCF-7 and non-tumorigenic mammary epithelial cells, MCF-10A (). Cells were treated with CP and Mito-CP11 (0.5 and 1.0 µmol/L) for 48 h. The “untargeted” nitroxide CP did not affect the breast cancer cell proliferation, whereas Mito-CP11 caused a dose-dependent decrease in the viability of breast cancer cells (MCF-7) but much less so in MCF-10A cells (). Cells were also treated with CP, CP plus Me-TPP+ and Me-TPP+ for 48 h, and cell proliferation monitored by the ability to form colonies ( and S1). Based on the lack of effect of these agents (Fig. S1), we conclude that the MitoCP11-mediated antiproliferative effect is not due to the nitroxide and/or the TPP+ moiety alone, and that linking both moieties by the 11-carbon linker chain is essential for the antiproliferative effects of Mito-CP11 observed in breast cancer cells. Results also showed that Mito-CP4 formed by linking the Me-TPP+ to CP moiety through a 4-carbon linker chain was not effective (data not shown).
To further verify these results, we measured the [3H]-thymidine uptake as an indication of DNA synthesis. The rate of thymidine uptake is increased during the S-phase of DNA synthesis, and is often used as an indicator of cell cycle progression. Figure S2 shows the effect of Mito-CP11 on the radiolabeled thymidine uptake in two breast cancer cell lines, MCF-7 and MDA-MB-231, as well as in noncancerous MCF-10A cells. Mito-CP11 treatment inhibited the [3H]-thymidine uptake in both breast cancer cells in a dose-dependent manner (Fig. S2). In the case of MCF-10A cells, higher concentrations of Mito-CP11 were needed for inhibition the [3H]-thymidine uptake, suggesting selective differential effects of Mito-CP11 in the normal mammary epithelial cells. The decreased thymidine uptake in Mito-CP11-treated breast cancer cells is due to decreased proliferation rather than to altered activity of nucleoside transporters.
Mito-CP11 exacerbates fluvastatin-mediated antiproliferative effects in breast cancer cells: Inhibitory effect of mevalonate.
MCF-7 and MCF-10A cells were treated with Mito-CP11, fluvastatin and Mito-CP11 plus fluvastatin for 48 h, followed by removal of media and re-plating. Consistent with other results obtained with fluvastatin and Mito-CP11, Mito-CP11 treatment induced a significant decrease in the clonogenic growth of MCF-7 cells (). Mito-CP11 (0.5 µM) treatment alone inhibited the colony formation in MCF-7 cells by ∼25% and fluvastatin (1 µM) caused a similar inhibition in colony formation. However, the colony formation was inhibited by 80% in MCF-7 cells in the presence of both Mito-CP11 (0.5 µM) and fluvastatin (1 µM). Similar results were obtained with MDA-MB-231 cells (data not shown). These results again implicate a novel role for Mito-CP11 in enhancing fluvastatin-mediated cytostatic effects in breast cancer cells.
We then investigated the effects of mevalonate, an immediate metabolic product of the acetyl CoA/HMG-CoA reductase reaction, to determine whether mevalonate reverses the antiproliferative effects of fluvastatin and fluvastatin plus Mito-CP11. As shown in , mevalonate partially reversed the anti-proliferative effects of fluvastatin and Mito-CP11, suggesting a role of either cholesterol biosynthesis or protein isoprenylation in mediating the enhanced antiproliferative effects of fluvastatin and Mito-CP11. However, as squalene (precursor of cholesterol) did not significantly affect the results observed with fluvastatin and Mito-CP11 (not shown), we attribute the restorative effects of mevalonate to the protein isoprenylation pathway. The antiproliferative effect of Mito-CP11 alone in MCF-7 cells was not affected by mevalonate, suggesting that the protein isoprenylation pathway is not involved in Mito-CP11-induced cytostatic effects in MCF-7 cells. Mevalonate had no effect on colony formation and cell survival in MCF-10A cells treated with fluvastatin and Mito-CP11 (Fig. S3).
Effects of Mito-CP and fluvastatin on cell survival measurements in breast cancer and control cells.
We monitored the effects of Mito-CP11 and CP in the presence and absence of fluvastatin on the viability of breast cancer and control non-transformed epithelial cells. Cells were treated for 48 h and cell survival measured by the MTT assay. Mito-CP11 plus fluvastatin treatment caused a significant decrease in cell viability in both MCF-7 and MCF-10A cells, as measured by direct cell counting (Fig. S4A) and MTT (Fig. S4B) assays. However, when the cells were washed free of these agents and incubated without the drugs for another 48 h, the observed cell survival was much higher in control MCF-10A cells as compared with MCF-7 cells subjected to Mito-CP11 plus fluvastatin treatments (Fig. S4B). Of note, CP and Me-TPP+ alone or together did not affect the cell survival or the antiproliferative effects of fluvastatin (Fig. S4C). We also tested the effects of Mito-CP11 and fluvastatin alone and together in H9C2, non-tumorigenic cardiomyocytes. Results indicated that these agents did not affect the cell survival or the proliferation in H9C2 cells (not shown). Next we investigated whether the differences in cell culture media composition contributed to the differential cytotoxic responses to fluvastatin and Mito-CP11 in MCF-7 and MCF-10A cells. MCF-7 cells in MCF10A culture media and MCF-10A in MCF-7 culture media were treated with Mito-CP and fluvastatin under similar conditions. Results showed that switching culture media instantly did not alter the responses to fluvastatin and Mito-CP11 (not shown).
From the results obtained using multiple approaches, including colony formation, [3H]-thymidine uptake, cell counting and MTT assays, we conclude that Mito-CP11 exacerbates the antiproliferative effects of fluvastatin in breast cancer cells.
Cytotoxic effects of Mito-CP11 and fluvastatin in MCF-7 and MCF-10A cells.
We examined whether Mito-CP11 used in combination with hydrophobic statins would exacerbate the cytotoxicity of either drug. The cytotoxicity was assessed using SYTOX Green, a non-permeable dye that becomes highly fluorescent after binding to nucleic acids in permeable dead cells. As shown in , Mito-CP11 (0.5 µM) alone did not cause any increase in the percentage of dead cells in MCF-7 and MCF-10A cells under conditions where Mito-CP11 elicited antiproliferative effects. However, Mito-CP11 dramatically enhanced fluvastatin-mediated cytotoxicity in MCF-7, but not in MCF-10A, cells. This increase in cytotoxicity observed in the presence of Mito-CP11 and fluvastatin was significantly mitigated by mevalonate. shows the fluorescence microscopy images of cells in the presence and absence of digitonin. Under similar experimental conditions (compare ), CP and Me-TPP+, either alone and in combination, did not elicit significant cytotoxicity as measured by SYTOX Green staining in fluvastatin-treated MCF-7 and MCF-10A cells (Fig. S5).
Uptake of Mito-CP11 into cell mitochondria.
We assessed the distributions of CP and Mito-CP11 in MCF-7 cells using EPR. As shown in , CP uptake into the mitochondria was very minimal. However, Mito-CP11 distribution was quite different. As shown in endothelial cells,Citation27 Mito-CP11 was found to accumulate in the mitochondria of both MCF-7 and MCF-10A cells. No detectable signal was observed in the cytosolic fraction. These results suggest that Mito-CP11 but not CP preferentially accumulates in mitochondria. The steady-state levels of Mito-CP11 in MCF-7 and MCF-10A were nearly identical (). We also observed that fluvastatin did not affect Mito-CP11 distribution in MCF-7 and MCF-10A cell mitochondria (data not shown). These results suggest that despite increased mitochondrial membrane potential in cancer cells, the steady-state levels of Mito-CP11 in MCF-7 and MCF-10A cell mitochondria are similar, and therefore the differential toxicity of Mito-CP11 in MCF-7 and MCF-10A is not due to increased uptake into MCF-7 mitochondria.
Inhibition of NFκB activity in MCF-7 cells.
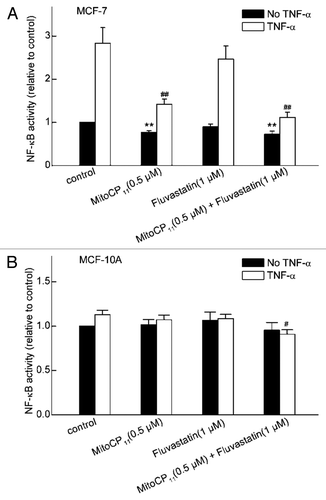
To identify mechanisms that contribute to the differential toxicity of Mito-CP11 in MCF-7 and MCF-10A cells, we examined Mito-CP11-dependent changes in NFκB activity. We focused on NFκB as a potential target of Mito-CP11 because NFκB promotes the survival and proliferation of breast cancer cells,Citation15–Citation17 and antioxidants can inhibit NFκB activity in several types of cancer.Citation13 We found that treatment of MCF-7 cells with Mito-CP11 inhibits moderately but significantly the basal NFκB activity and robustly inhibits TNFα-induced NFκB activity (). In contrast to the effects of Mito-CP11, fluvastatin does not significantly alter NFκB activity in MCF-7 cells (), indicating that fluvastatin and Mito-CP11 impact different signaling pathways to inhibit breast cancer cell proliferation. We found that Mito-CP11 does not significantly alter basal NFκB activity in MCF-10A cells (). MCF-10A cells did not detectably respond to TNFα (), which prohibited us from determining the effects of Mito-CP11 on TNFα-induced NFκB activity in these cells. Our finding that Mito-CP11 induces greater loss of basal NFκB activity in MCF-7 than in MCF10A cells, and the discovery that Mito-CP11 profoundly inhibits TNFα-induced NFκB activity in MCF-7 cells, support the conclusion that inhibition of the NFκB pathway contributes to the antiproliferative effects of Mito-CP11 in MCF-7 cells.
Chemopreventive effect of fluvastatin on DMBA-induced tumor incidence.
Previous research had revealed that female Dahl salt-sensitive (SS) rats exhibit a high incidence of mammary tumors in response to treatment with 7,12-dimethylbenz[a] anthracene (DMBA).Citation29 Administration with a single oral gavage of DMBA to SS females (48- to 55-d-old) yielded extensive mammary carcinoma (95%). Beginning 3 wks after DMBA administration, rats were palpated weekly for the detection of mammary tumors. The mean latency to appearance of the first palpable mammary cancer was 42 d (Fig. S6). Following 15 weeks after DMBA treatment, the average number of tumors detected per rat was 2.5 (Fig. S6).
We wished to evaluate whether administration of fluvastatin to rats would result in plasma level concentration sufficiently high to elicit chemopreventive effects. These studies were designed to test the effects of fluvastatin on inhibition of tumor formation, rather than its ability to cause tumor regression by inhibiting proliferation of already neoplastically transformed tumor cells (see Materials and Methods).
As seen in Figure S7, the latency of tumor development was significantly longer in rats treated with 4 mg/ml fluvastatin as compared with 1 mg/ml treated rats and control rats (p < 0.003). A considerable difference in tumor burden (i.e., the average number of palpable tumors per rat) between the control group and the group treated with 4 mg/ml fluvastatin at weeks 10–15 was noticeable. At week 15, rats were euthanized and autopsies were performed. Excised tumors were measured, enumerated and processed for standard histopathology. There was a significant decrease in tumor formation between the control group and the 4 mg/ml treated experimental group (an average of 5 vs. 2; p < 0.005) (data not shown). The majority of tumors detected were classifed histologically as determined by hematoxylineosin (H&E) staining as adenocarcinomas, and the remainder (10%) as adenomas (benign) (Fig. S7). These results suggest that daily administration of fluvastatin (4 mg/ml) to rats by oral gavage can lead to statin levels in vivo high enough to inhibit DMBA-induced tumor formation. Future studies will examine the chemotherapeutic effects of fluvastatin and Mito-CP11 combination in a suitable xenograft animal model of breast carcinogenesis.
Discussion
In this study, we tested the cytostatic and cytotoxic properties of Mito-CP11 and CP in MCF-7 human breast cancer cells and MCF-10A normal mammary epithelial cells. At sub-micromolar concentrations, Mito-CP11, a mitochondria-targeted five-membered ring nitroxide, exerted greater cytostatic effects in MCF-7 than MCF-10A cells. CP, a five-membered nitroxide, was not found to be cytostatic or cytotoxic over a wide range of concentrations in these cells.
Mito-CP11 and mitochondria-targeted compounds like Mito-Q are known to be preferentially taken up by the mitochondria.Citation27,Citation30 Lipophilic cations distribute their charge over a large surface area, allowing them to easily penetrate the lipid bilayers. The large negative membrane potential of 150–180 mV across the mitochondrial inner membrane is responsible for mitochondrial accumulation of the lipophilic triphenylphosphonium cations. Mito-Q was shown to accumulate 50- to 100-fold within mitochondria.Citation31 Nevertheless, the difference in ΔΨm detectable between normal and malignant cancerous cells is at least 60 mV. Thus, mitochondria-targeted compounds are sensitive to the higher ΔΨm in malignant cells, and selectively accumulate in their mitochondria. The increased toxicity of lipophilic cationic probes (e.g., rhodamine 123) was linked to enhanced accumulation of these probes into cancer cells. However, it was not clear whether the mitochondrial accumulation of rhodamine 123 was significantly higher in cancer cells as compared with noncancerous control cells. Several synthetic alkyl-lysophospholipid analogs (e.g., edelfosine) exhibited selective antitumor action that is attributed to accumulation of the phospholipid ether in tumor cells.Citation32 In this study, we measured the steady-state concentrations of Mito-CP11 in mitochondrial fractions of MCF-7 and MCF10A cells. As shown in , the intensity of the EPR signal in both mitochondrial fractions (i.e., from MCF-7 and MCF-10A cells) was nearly identical. Based on this result, we conclude that Mito-CP11-induced differential toxicity is not simply due to differential uptake of Mito-CP11 into MCF-7 and MCF-10A mitochondria, and that other factors may be involved.
Recently, we reported Mito-CP11 acting as an antioxidant in bovine aortic endothelial cells treated with H2O2.Citation27 Due to selective uptake of Mito-CP11 in the mitochondria, it inhibited the cytochrome c release and caspase-3 activation in cells treated with peroxides. In SOD1G93A astrocytes, Mito-CP11 and Mito-Q at nanomolar concentrations prevented mitochondrial dysfunction, diminished superoxide production and restored motor neuron survival.Citation33 Nitroxides are known to react with various types of ROS like peroxyl, hydroxyl and superoxide radicals. Nitroxides catalyze the superoxide dismutation in a pH-dependent manner.Citation26 During this reaction, two molecules of superoxide dismutate to form O2 and H2O2. Recent reports have shown that traditional antioxidants such as ascorbic acid, exert strong pro-oxidant properties in response to the tumor microenvironment, increasing the levels of ROS like H2O2 which lead to cancer cell death.Citation34 Paradoxically, in normal cells, antioxidants protect from ROS-mediated cell damage.Citation11,Citation12
The proposed use of statins in breast cancer therapy remains controversial.Citation35–Citation37 Results from a meta analysis showed that statins had no effect on breast cancer. Previously, we and others showed that hydrophilic statins such as pravastatin did not induce cytotoxicity in breast cancer cells.Citation1,Citation2 Results from a new study conducted on a large population of postmenopausal women with breast cancer lead to the conclusion that the use of lipophilic or hydrophobic statins exhibit beneficial effects in breast cancer therapy.Citation37 However, when both hydrophobic and hydrophilic statins (e.g., pravastatin) were considered together as a class, no statistically significant beneficial effect was found.Citation37 Also, when pravastatin data were excluded in the randomized clinical trials used in the meta analysis, the results of the meta analysis were not statistically significant. These results clearly indicate that hydrophobic or lipophilic statins but not hydrophilic statins (pravastatin) should be considered in breast cancer therapy. Another important consideration is that statins, at concentrations that can be achieved in plasma (sub-micromolar levels) may not effectively act as a chemotherapeutic. The present cell culture shows that Mito-CP11 could sensitize breast cancer cells to fluvastatin-induced cytotoxicity. Thus, mitochondria-targeted nitroxides such as Mito-CP11 could act as an effective antitumor adjuvant in augmenting the chemotherapeutic or chemopreventive potential of statins.
A recent report suggests that long-term administration of the statins, atorvastatin and lovastatin, failed to inhibit rodent mammary carcinogenesis in Sprague-Dawley rats.Citation38 In this model, mammary tumors were induced by intravenous injection of methylnitrososourea (MNU). Statins were given in the diet. The results are, however, not in agreement with the inhibitory effects of fluvastatin in DMBA-induced mammary carcinogenesis as reported in the present study. We do not yet have a clear-cut explanation for the observed discrepancies between the two studies; however, it must be pointed out that both studies significantly differed in several aspects of experimentation design, most notably in rat strain and the type of carcinogen used to induce breast tumors.
However, the present data show that fluvastatin, when administered chronically at 4 mg/ml to DMBA-treated rats, can inhibit tumor formation in a breast cancer prevention animal model. Future studies should test the scope of the combined administration of fluvastatin and Mito-CP as potential chemotherapeutics in a xenograft animal model of breast carcinogenesis.
NFκB is a very plausible participant in the signaling pathways that are disrupted by Mito-CP11 in breast cancer cells. Numerous studies indicate that NFκB activity promotes breast cancer development and progression, due to the NFκB-dependent expression of proteins that promote cancer cell survival, proliferation and metastasis.Citation15–Citation17 Many of these proteins are induced by TNFα, which is a well-known activator of NFκB in breast cancer and other types of cancer.Citation11,Citation18 The importance of NFκB in promoting the malignant phenotype is further indicated by reports that NFκB activity is higher in breast cancer cell lines, including MCF-7 cells, than in the mammary epithelial MCF-10A cells.Citation16,Citation17,Citation39,Citation40 Based on these pro-proliferative functions of NFκB in breast cancer, our finding that Mito-CP11 inhibits NFκB activity in MCF-7 cells provides a plausible mechanism to account for the antiproliferative effects of Mito-CP11 in MCF-7 cells. It is likely that the antioxidant properties of Mito-CP11 are responsible for this loss of NFκB activity, since antioxidants have been found to inhibit NFκB activity in multiple cell types.Citation13 Although Mito-CP11 accumulates mainly in the mitochondria, it is also present in the cytoplasm,Citation31 where redox-sensitive kinases that regulate NFκB are located.Citation13,Citation14 Thus, the antioxidant effects of Mito-CP in the cytoplasm, and potentially in other organelles, could disrupt NFκB signaling pathways. Intriguingly, we found that Mito-CP11 inhibits basal NFκB activity more in MCF-7 than MCF-10A cells, which might account in part for the ability of Mito-CP11 to inhibit proliferation more in MCF-7 than MCF-10A cells. We also found that Mito-CP11 potently inhibits TNFα-induced activation of NFκB in MCF-7 cells. This effect of Mito-CP11 might contribute to its cytotoxic actions, since agents that inhibit the TNFα-induced activation of NFκB decrease the survival of cancer cells.Citation13 Taken together, these findings provide evidence that inactivation of NFκB by Mito-CP11, mostly likely due to its antioxidant properties, contributes to antiproliferative effects of this drug.
In addition to its antioxidant property, Mito-CP11 might also have unique functions due to the presence of the alkyl chain linked to the triphenylphosphonium group. Alkylphosphonium salts have previously been shown to exert antitumor activity in a variety of human tumor cell lines.Citation41 Several alkylphosphonium cations also suppressed tumor growth in animal xenograft models.Citation41 Some phosphonium cations exhibited cytotoxicity in drug-resistant ovarian cancer cell lines and other multidrug-resistant experimental tumors.Citation42 The proposed mechanism involves selective accumulation of lipophilic, cationic phosphonium compounds in mitochondria of neoplastic cells, leading to inhibition of mitochondrial respiration. The molecular target involved in enhanced cytostatic and cytotoxic effects of phosphonium compounds in tumor cells still remains to be established. In our experiments, however, the methyltriphenylphosphonium cation, at the concentrations used, did not affect cell proliferation. Recently, it was reported that mitochondrial ROS regulate cell proliferation via the extracellular signal-regulated kinase (ERK1/2) pathway.Citation43 Mitochondria-targeted antioxidants (MTAs) (Mito-CP11) but not “untargeted” nitroxides induced an increase in phosphorylated ERK1/2 in tumor cells.Citation43 The upstream MEK kinase (MAPK kinase) induced cell proliferation or cell death, depending upon the levels.Citation44 Previous data also suggest that oncogene-induced mitochondrial ROS serve as signaling molecules to dampen the ERK1/2 MAPK pathway to levels that promote cellular proliferation.Citation30 More recently, it was shown that Mito-Q (Co-enzyme Q10 conjugated to a triphenylphosphonium moiety) potently induces antiproliferative mechanism in breast cancer cells but not in healthy mammary cells.Citation45 It was proposed that Mito-Q, being a weak electrophile, induced cell cycle arrest and autophagy via Nrf2-regulated enzyme NQO1.Citation45 Thus, the molecular target involved in MTA-mediated cytostatic and cytotoxic effects in cancer cells is dependent on the MTA structure.
An alternative mechanism by which Mito-CP enhanced fluvastatin-induced breast cancer cell cytotoxicity may be related to statin's ability to inhibit glycolytic activity in breast cancer cells.Citation46 Using NMR metabonomic analysis, the investigators showed that incubation of MDA-MB-468 cells with lovastatin for 48 h strongly inhibited the glycolytic activity and decreased the de novo formation of 13C-alanine and 13C-lactate.Citation46 Mitochondria-targeted cationic compounds exacerbated the anti-proliferative effects of 2-DG in pancreatic cancer cells.Citation47 We propose a similar type of mechanism for the cationic antioxidant, Mito-CP. Blocking tumor cell mitochondrial function with targeted lipophilic cations hypersensitized tumor cells to glycolytic inhibitors.Citation47 As L-NAME did not have any effect on Mito-CP11/fluvastatin-enhanced breast cancer cell cytotoxicity, we can rule out nitric oxide and/or peroxynitrite involvement.
Another caveat is that the half-life of fluvastatin in humans is nearly 3 h, and can reach the steady-state concentration in the plasma up to 102 ng/ml (0.24 µM) with administration of extended release capsules (80 mg). Although the clinical relevance of the combined therapeutic efficacy of MTAs (0.5 µM) and statins (1 µM) remains uncertain, additional investigation in a preclinical animal model using modified dosing schedules is necessary.
Finally, MTAs may enhance the efficacy of conventional chemotherapeutics and act as radioprotectors in radiation therapy.Citation48 With most conventional chemotherapy, the cumulative dose of an antitumor agent that causes tumor cell killing often stimulates toxic side effects. The combination chemotherapy typically involves the use of two or more chemotherapeutic agents, all of which exhibit toxic side effects in normal cells. MTAs can potentially decrease the levels of the conventional chemotherapeutic agent used in cancer treatment. Another potential application for MTA-mediated adjuvant chemotherapy may be related to targeting and killing breast cancer with erbB2 expression that is associated with nearly 30% of breast cancers.
In summary, the present study demonstrates that mitochondria-targeted nitroxides induce antiproliferative effects in breast cancer cells but not in normal mammary epithelial cells. Mito-CP11 exacerbated the cytotoxic effects of fluvastatin in breast cancer cells. Unlike fluvastatin, the effects due to Mito-CP11 were not dependent on mevalonate. Mito-CP11 significantly inhibits NFκB activity in MCF-7 cells, which might contribute to the antiproliferative effects of Mito-CP11 in these cells. Future studies will investigate the chemotherapeutic effects of Mito-CP11 alone and in combination with fluvastatin in a xenograft animal model of breast carcinogenesis.
Materials and Methods
Cell culture.
MCF-7, MDA-MB-231 and MCF-10A cell lines were obtained from ATCC. Cell lines were grown at 37°C in 5% CO2. MCF-7 cells were maintained in MEMα (Invitrogen) containing 10% fetal bovine serum, bovine insulin (10 µg/ml), penicillin (100 U/ml) and streptomycin (100 µg/ml). MDA-MB-231 cells were cultured in DMEM, 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 µg/ml). MCF-10A cells were cultured in DMEM/F12 media (1:1) (Invitrogen) supplemented with 5% horse serum, bovine insulin (10 µg/ml), epidermal growth factor (20 ng/ml), cholera toxin (100 ng/ml) and hydrocortisone (0.5 µg/ml), penicillin (100 U/ml) and streptomycin (100 µg/ml).
Reagents.
Mito-CP11 was synthesized as previously published in reference Citation27. Carboxy-proxyl (CP), Me-TPP+were purchased from Sigma-Aldrich. Fluvastatin was obtained from Calbiochem.
Cell viability and proliferation assays.
Cell viability was determined using the MTT assay. Cells were cultured at a density of 0.8–1 × 104 cells per well of 48-well plates with complete medium overnight prior to treatment. Cells were treated for 24–48 h before MTT was added and the absorbance read at 565 nm. For cell counts, cells were grown in the corresponding media plus treatments for up to 48 h, harvested, diluted in trypan blue dye and counted with an automated cell counter (Invitrogen).
[3H]-thymidine uptake.
DNA synthesis was measured by monitoring the uptake of tritiated thymidine, [3H]TdR (Perkin-Elmer) as previously described in reference Citation1. Cells (0.8–1 × 104/mL) were cultured with different concentrations of Mito-CP11 (0–1 µM). Cells were pulse-chased with [3H]TdR [0.5 µCi (0.185 MBq)/well] during the last 3 h of a 24 h culture, harvested onto glass filters with an automatic cell harvester (Cambridge Technology), and radioactivity on the filters was measured by β-scintillation counting.
SYTOX® green cell death assay.
SYTOX® Green (Invitrogen) cell death assay was performed by plating cells at a density of 5–10 × 103 cells per well of 96-well plates with complete medium overnight prior to treatment. Cells were treated for 48 h with Mito-CP11 and then incubated with 1 µM SYTOX Green for 30 min. Total fluorescence intensities were acquired using a plate reader (Beckman Coulter, DTX-880) equipped with the 485 nm excitation and 535 nm emission filters. Fluorescence from wells containing Sytox Green in the medium without cells was considered as background. Digitonin (120 µM) was used as a permeabilizing agent and Sytox fluorescence in digitonin-treated cells was taken as 100%.Citation49 Samples were imaged using a Nikon Eclipse TE2000U microscope equipped with FITC filters and MetaMorph imaging software (Molecular Devices).
Colony formation assay.
Growth inhibition was monitored using a colony formation assay.Citation43,Citation50 Briefly, the cells were seeded in 6-well plates at a density of 100 cells per well. After overnight attachment of the cells to the plate, the medium was removed and the fresh medium containing Mito-CP11 or CP was added. After a 48 h treatment the medium was removed and the cells were allowed to grow without Mito-CP11 or CP for another 5–7 d. For colony counting, cells were washed with PBS, fixed and stained by treating with PBS containing 6% glutaraldehyde and 0.5% crystal violet for 30 min. Subsequently, cells were washed with tap water and dried at room temperature. The pictures of the plates were taken using an Alphaimager (Alpha Innotech Corporation), and the colonies counted manually using the software provided with the imaging instrument. The surviving fraction (SF) and the plating efficiency (PE) were calculated as follows:
Electron paramagnetic resonance (EPR).
EPR was used to determine the distribution of CP and Mito-CP11 in breast cancer cells. MCF-7 cells were grown to pre-confluency in complete media before treatment with Mito-CP11 or CP (5 µM) for 2 h. Cells were harvested and samples from the media, cell lysate, mitochondria and cytosol were analyzed at room temperature using a Bruker EMX spectrometer at 9.5 GHz (X-band) employing a 100 kHz field modulation.Citation27
Measurements of NFκB activity.
NFκB activity was measured as previously described in reference Citation30. Briefly, the cells were co-transfected with the pNifty-Luc NFκB reporter plasmid (Invivogen), and the β-GAL reporter plasmid and cultured for 24 h. The cells were then incubated in the absence or presence of Mito-CP11 (0.5 µM) and/or fluvastatin (1 µM) for 48 h, then incubated in the absence or presence of TNFα (100 ng/ml, 3 h). Cells were incubated with D-Luciferin (150 µg/ml, 2 min) and luminescence in the living cells was measured using a FLUOstar Omega plate reader (BMG LABTECH). Luminescence was normalized to detection of β-galactosidase expression in the transfected cells, as determined by spectrophotometry.
Vertebrate animals.
All experimental procedures used in the work were in keeping with the recommendations of the Institutional Animal Care and Use Committee and the established guidelines for the humane care of laboratory animals. Female salt-sensitive (SS) rats (obtained from Physiogenics, MCW) were given all DMBA treatments in the biocontainment suite, taking into consideration that DMBA is a carcinogen. Animals were kept in biocontainment for the duration of the experiment. Animals received daily treatments of fluvastatin.
Rats were administered 65 mg/kg of DMBA by oral gavage. Fluvastatin treatment by oral gavage commenced immediately for 15 weeks. In Group 1, rats (n = 5) were given 1 ml of 1 mg/ml fluvastatin in sterile distilled water by oral gavage. In Group 2, rats (n = 4) were given 1 ml of 4 mg/ml of fluvastatin by oral gavage. In Group 3, rats (n = 5) were given sterile water alone (control) by oral gavage. Animals were palpated for tumor formation and weighed on a weekly basis. Tumor latency (the week when the tumor was first palpable) and tumor multiplicity (number of tumors/rat) were calculated. Tumor length (L) and width (W) were measured with a micrometer caliper and tumor volume was calculated using the formula [L/2*W/2*H/2*(4/3)*π] and expressed as mm3.
Statistics.
Statistical significance was determined by two-sample Student's t-tests (p = 0.05). All statistics including mean values and standard deviations were calculated using Microsoft Excel software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Structures of CP, Me-TPP+ and Mito-CP11.
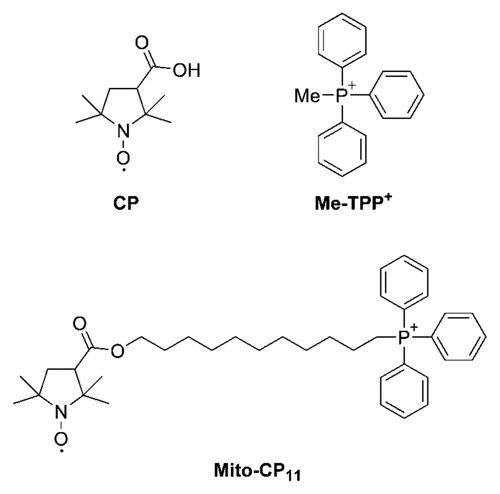
Figure 2 Antiproliferative effects of Mito-CP11 and CP in MCF-7 and MCF-10A cells. (A) MCF-7 and MCF-10A cells were treated with Mito-CP11 and CP (0.5 and 1 µM) for 48 h, and colonies formed were counted after 5 d (for MCF-7 cells) and 7 d (for MCF-10A cells). (B) The cell survival fractions calculated as described in the Materials and Methods section in Mito-CP11- and CP-treated MCF-7 and MCF-10A cells are shown. The plating efficiency calculated for MCF-7 and MCF-10A cells was 65 ± 13 and 38 ± 5, respectively. The error bars represent standard deviation and * symbol indicates a p value of less than 0.02 as determined by Student's t-test (n = 5).
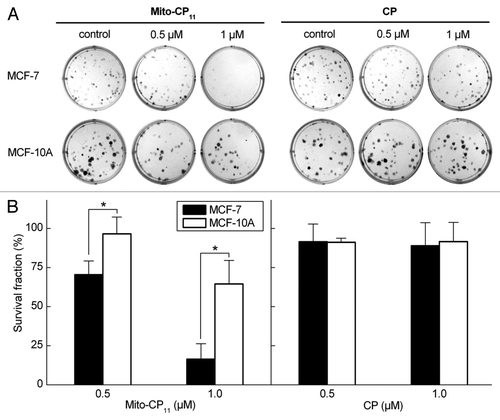
Figure 3 Antiproliferative effects of fluvastatin and Mito-CP11 in MCF-7 cells: Effect of mevalonate. (A) MCF-7 cells were treated with Mito-CP11 (0.5 µM) and fluvastatin (1 or 2.5 µM) in the presence and absence of mevalonate (20 µM) for 48 h, and the colonies counted after an additional 5 d. (B) Cell survival was measured under the same conditions as in (A). The survival fraction was calculated as described in the Materials and Methods section. The error bars represent standard deviation and * symbol indicates p value of less than 0.005 as determined by the Student's t-test (n = 9).
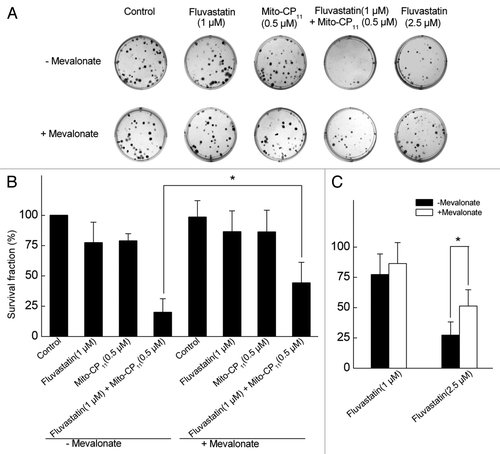
Figure 4 The effect of fluvastatin and Mito-CP11 on the extent of cell death in MCF-7 and MCF-10A cells. (A) MCF-7 and MCF-10A cells were treated with Mito-CP11 (0.5 µM) with or without fluvastatin (1 µM) and mevalonate (20 µM) for 48 h, and dead cells were monitored by staining with SYTOX Green. To measure the total cell number, cells were treated with digitonin (120 µM) when staining with SYTOX Green. Fluorescence intensity from cells grown in 96-well plate was measured using a plate reader (excitation wavelength, 485 nm and the emission wavelength 535 nm). (B) Fluorescence microscopy pictures were obtained using FITC filters under the same experimental cell culture conditions as in (A). Asterisks above a column indicate a statistical comparison between the indicated treatment and the control (*p < 0.05).
Figure 5 Uptake of Mito-CP11 into mitochondria. MCF-7 cells were treated with CP (5 µM) in the presence of Me-TPP+ and Mito-CP11 (5 µM) for 48 h. After treatment, cells were fractionated to isolate mitochondria and cytosol. CP and Mito-CP11 distributions in media, cytosol, mitochondria and cell lysate were determined using EPR. The EPR signals from different samples were normalized to the same concentration of protein.
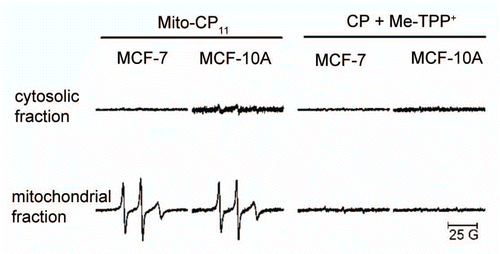
Figure 6 The effect of Mito-CP11 and fluvastatin on NFκB levels in MCF-7 and MCF-10A cells. The activity of NFκB in MCF-7 (A) and MCF-10A (B) cells was measured after incubating the cells for 48 h in the absence or presence of Mito-CP11 (0.5 µM) or fluvastatin (1 µM), followed by incubation in the absence or presence of TNFα (100 ng/ml, 3 h). NFκB activity was normalized to values in control cells that were incubated in the absence of Mito-CP11, fluvastatin and TNFα. Results are the mean ± SEM from three independent experiments conducted with triplicate samples. **p < 0.001 vs. control without TNFα; #p < 0.01, ##p < 0.001 vs. control with TNFα.
Additional material
Download Zip (8.6 MB)Acknowledgments
This work was supported by National Institutes of Health grants R01CA125112 (B.K.), R01CA152810 (B.K.) and R01CA136799 (C.W.). Additional funding was supplied by the Rock River Cancer Research Foundation (C.W.).
References
- Kotamraju S, Williams CL, Kalyanaraman B. Statin-induced breast cancer cell death: role of inducible nitric oxide and arginase-dependent pathways. Cancer Res 2007; 67:8973; PMID: 17671209; http://dx.doi.org/10.1158/0008-5472.CAN-07-0993
- Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, et al. Breast cancer growth prevention by statins. Cancer Res 2006; 66:8707 - 8714; PMID: 16951186; http://dx.doi.org/10.1158/0008-5472.CAN05-4061
- Mück A, Seeger H, Wallwiener D. Inhibitory effects of statins on the proliferation of human breast cancer cells. Int J Clin Pharmacol Ther 2004; 42:695 - 700; PMID: 15624286
- Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteosome, independent of hydroxy glutaryl-CoA reductase. Proc Natl Acad Sci USA 1999; 96:7797 - 7802
- Cafforio P, Dammacco F, Gerone A, Sivestris F. Statins activate the mitochondrial pathway of apoptosis in human lympoblasts and myeloma cells. Carcinogenesis 2005; 26:883 - 891; PMID: 15705602; http://dx.doi.org/10.1093/carcin/bgi036
- Rosenson RS. Pluripotent mechanisms of cardioprotection with HMG-CoA reductase inhibitor therapy. Am J Cardiovasc Drugs 2001; 1:411 - 420; PMID: 14728000; http://dx.doi.org/10.2165/00129784-200101060-00001
- Laezza C, Malfitano AM, Proto MC, Esposito I, Gazzero P, Formisano P. Inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity and of Ras farnesylation mediate antitumor effects of anandamide in human breast cancer cells. Endocr Relat Cancer 2010; 17:495 - 503; PMID: 20304978; http://dx.doi.org/10.1677/ERC-10-0009
- Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basanez G, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res 2008; 68:5246 - 5256; PMID: 18593925; http://dx.doi.org/10.1158/0008-5472.CAN-07-6161
- Laezza C, Fiorentino L, Pisanti S, Gazzerro P, Caragilia M, Portella G, et al. Lovastatin induces apoptosis of k-ras-transformed thyroid cells via inhibition of ras farnesylation and by modulating redox state. J Mol Med 2008; 86:1341 - 1351; PMID: 18779944; http://dx.doi.org/10.1007/s00109-008-0396-1
- McAnally JA, Gupta J, Sodhani S, Bravo L, Mo H. Tocotrienols potentiate lovastatin-mediated growth suppression in vitro and in vivo. Exp Biol Med (Maywood) 2007; 232:523 - 531; PMID: 17392488
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 2002; 82:47 - 95; PMID: 11773609
- Doroshow JH. Redox modulation of chemotherapy-induced tumor cell killing and normal tissue toxicity. J Natl Cancer Inst 2006; 98:223 - 225; PMID: 16478735; http://dx.doi.org/10.1093/jnci/djj065
- Zhang Y, Chen F. Reactive oxygen species (ROS), troublemakers between nuclear factor-kappaB (NFkappaB) and c-Jun NH(2)-terminal kinase (JNK). Cancer Res 2004; 64:1902 - 1905; PMID: 15026320; http://dx.doi.org/10.1158/0008-5472.CAN-03-3361
- Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal 2006; 8:1791 - 1806; PMID: 16987032; http://dx.doi.org/10.1089/ars.2006.8.1791
- Kim DM, Koo SY, Jeon K, Kim MH, Lee J, Hong CY, et al. Rapid induction of apoptosis by combination of flavopiridol and tumor necrosis factor (TNF) alpha or TNF-related apoptosis-inducing ligand in human cancer cell lines. Cancer Res 2003; 63:621 - 626; PMID: 12566305
- Liu M, Ju X, Willmarth NE, Casimiro MC, Ojeifo J, Sakamaki T, et al. Nuclear factor-kappaB enhances ErbB2-induced mammary tumorigenesis and neoangiogenesis in vivo. Am J Pathol 2009; 174:1910 - 1920; PMID: 19349372; http://dx.doi.org/10.2353/ajpath.2009.080706
- Benoit V, Chariot A, Delacroix L, Deregowski V, Jacobs N, Merville MP, et al. Caspase-8-dependent HER-2 cleavage in response to tumor necrosis factor alpha stimulation is counteracted by nuclear factor kappaB through c-FLIP-L expression. Cancer Res 2004; 64:2684 - 2691; PMID: 15087380; http://dx.doi.org/10.1158/0008-5472.CAN-03-2914
- Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. TKs5-dependent, Nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal 2009; 2:53; PMID: 19318623; http://dx.doi.org/10.1126/scisignal.2000368
- Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 2006; 25:695 - 705; PMID: 17160708; http://dx.doi.org/10.1007/s10555-006-9037-8
- Zhang Y, Zhao W, Zhang HJ, Domann FE, Oberley LW. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res 2002; 62:1205 - 1212; PMID: 11861405
- Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res 2004; 64:3580 - 3585; PMID: 15150115; http://dx.doi.org/10.1158/0008-5472.CAN-03-3909
- Arnold RS, He J, Remo A, Ritsick D, Yin-Goen Q, Lambeth JD, et al. Nox1 expression determines reactive oxygen and modulates c-fos-induced growth factor, interleukin-8 and Cav-1. Am J Pathol 2007; 171:2021 - 2032; PMID: 18055552; http://dx.doi.org/10.2353/ajpath.2007.061144
- Hu Y, Rosen DG, Zhao Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese superoxide dismutase expression in ovarian cancer: Role in proliferation and response to oxidative stress. J Biol Chem 2005; 280:39485 - 39492; PMID: 16179351; http://dx.doi.org/10.1074/jbc.M503296200
- Desouki MM, Kulawiec M, Bansal S, Das GM, Singh KK. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol Ther 2005; 4:1367 - 1373; PMID: 16294028; http://dx.doi.org/10.4161/cbt.4.12.2233
- Modica-Napolitano JS, Singh KK. Mitochondria as targets for detection and treatment of cancer. Expert Rev Mol Med 2002; 4:1 - 19; PMID: 14987393
- Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2 or as SOD mimics?. J Biol Chem 1996; 271:26026 - 26031; PMID: 8824242; http://dx.doi.org/10.1074/jbc.271.42.26026
- Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S, Joseph J, et al. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic Biol Med 2005; 39:567 - 583; PMID: 16085176; http://dx.doi.org/10.1016/j.freeradbiomed.2005.04.016
- Suy S, Mitchell JB, Ehleiter D, Haimovitz-Friedman A, Kasid U. Nitroxides tempol and tempo induce divergent signal transduction pathways in MDA-MB 231 breast cancer cells. J Biol Chem 1998; 273:17871 - 17878; PMID: 9651392; http://dx.doi.org/10.1074/jbc.273.28.17871
- Adamovic T, McAllister D, Rowe JJ, Wang T, Jacob HJ, Sugg SL. Genetic mapping of mammary tumor traits to rat chromosome 10 using a novel panel of consomic rats. Cancer Genet Cytogenet 2008; 186:41 - 48; PMID: 18786441; http://dx.doi.org/10.1016/j.cancergencyto.2008.05.014
- Tew GW, Lorimer EL, Berg TJ, Zhi H, Li R, Williams CL. SmgGDS regulates cell proliferation, migration, and NFkappaB transcriptional activity in non-small cell lung carcinoma. J Biol Chem 2008; 283:963 - 976; PMID: 17951244; http://dx.doi.org/10.1074/jbc.M707526200
- Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta 2008; 1777:1028 - 1031
- Mollinedo F, de la Iglesia-Vicente J, Gajate C, Estella-Hermoso de Mendoza A, Villa-Pulgarin JA, de Frias M, et al. In vitro and in vivo selective antitumor activity of edelfosine against mantle cell lymphoma and chronic lymphocytic leukemia involving lipid rafts. Clin Cancer Res 2010; 16:2046 - 2054; PMID: 20233887; http://dx.doi.org/10.1158/1078-0432.CCR-09-2456
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, et al. Mitochondrial dysfunction in SOD1G93 A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci 2008; 28:4115 - 4122; PMID: 18417691; http://dx.doi.org/10.1523/JNEUROSCI.5308-07.2008
- Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci USA 2005; 102:13604 - 13609; PMID: 16157892; http://dx.doi.org/10.1073/pnas.0506390102
- Cauley JA, McTiernan A, Rodabough RJ, LaCroix A, Bauer DC, Margolis KL, et al. Statin use and breast cancer: Prospective results from the Women's Health Initiative. J Natl Cancer Inst 2006; 98:700 - 707; PMID: 16705124; http://dx.doi.org/10.1093/jnci/djj188
- Prowell TM, Stearns V, Trock B. Lipophilic statins merit additional study for breast cancer chemoprevention. J Clin Oncol 2006; 24:2128 - 2129; PMID: 16648517; http://dx.doi.org/10.1200/JCO.2005.05.1649
- Bonovas S, Filioussi K, Tasvaris N, Sitaras NM. Use of statins and breast cancer: A meta-analysis of seven randomized clinical trials and nine observational studies. J Clin Oncol 2005; 23:8606 - 8612; PMID: 16260694; http://dx.doi.org/10.1200/JCO.2005.02.7045
- Lubet RA, Boring D, Steele VE, Ruppert JM, Juliana MM, Grubbs CJ. Lack of efficacy of the statins atorvastatin and lovastatin in rodent mammary carcinogenesis. Cancer Prev Res (Phila) 2009; 2:161 - 167; PMID: 19196723; http://dx.doi.org/10.1158/1940-6207.CAPR08-0134
- Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Traish AM, Mercurio F, Sonenshein GE. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res 2001; 61:3810 - 3818; PMID: 11325857
- El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM. Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol 2003; 23:5516 - 5525; PMID: 12897127; http://dx.doi.org/10.1128/MCB.23.16.551625.2003
- Sanyal U, Chatterjee RS, Das SK, Chakraborti SK. Alkylphosphonium salts as a new class of antitumor agents. Neoplasma 1984; 31:149 - 155; PMID: 6717684
- Manetta A, Gamboa G, Nasseri A, Podnos YD, Emma D, Dorion G, et al. Novel phosphonium salts display in vitro and in vivo cytotoxic activity against human ovarian cancer cell lines. Gynecol Oncol 1996; 60:203 - 212; PMID: 8631539; http://dx.doi.org/10.1006/gyno.1996.0026
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010; 107:8788 - 8793; PMID: 20421486; http://dx.doi.org/10.1073/pnas.1003428107
- Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007; 26:3227 - 3239; PMID: 17496918; http://dx.doi.org/10.1038/sj.onc.1210414
- Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, et al. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinine. J Biol Chem 2010; 285:34447 - 34459; PMID: 20805228; http://dx.doi.org/10.1074/jbc.M110.133579
- Klawitter J, Shokati T, Moll V, Christians U, Klawitter J. Effects of lovastatin on breast cancer cells: a proteo-metabonomic study. Breast Cancer Res 2010; 12:16; PMID: 20205716; http://dx.doi.org/10.1186/bcr2485
- Kurtoglu M, Lampidis TJ. From delocalized lipophilic cations to hypoxia: blocking tumor cell mitochondrial function leads to therapeutic gain with glycolytic inhibitors. Mol Nutr Food Res 2009; 53:68 - 75; PMID: 19072739; http://dx.doi.org/10.1002/mnfr.200700457
- Citrin D, Cotrim AP, Hyodo F, Baum BJ, Krishna MC, Mitchell JB. Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist 2010; 15:360 - 371; PMID: 20413641; http://dx.doi.org/10.1634/theoncologist.2009-S104
- Azzolin L, Basso E, Argenton F, Bernardi P. Mitochondrial Ca2+ transport and permeability transition in zebra fish (Danio rerio). Biochim Biophys Acta 2010; 1797:1775 - 1779
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1:2315 - 2319; PMID: 17406473; http://dx.doi.org/10.1038/nprot.2006.339