Abstract
Activation of the epidermal growth factor receptor (EGFR) regulates cellular proliferation, survival, and migration of breast cancer cells. In particular, EGFR recruits signaling proteins to the cell membrane leading to their phosphorylation and activation. However, EGFR also localizes to other cellular structures, including endosomes, mitochondrion, and nuclei. Recently, we demonstrated that lipid raft localization of EGFR in triple-negative breast cancer cell lines promotes EGFR protein-dependent, EGFR kinase-independent activation of Akt. Here, we further define the mechanism by which lipid rafts regulate EGFR signaling to Akt. Specifically, we show that the non-receptor tyrosine kinase c-Src co-localizes and co-associates with EGFR and lipid rafts. Breast cancer cells resistant to treatment with EGFR inhibitors, were also resistant to treatment with Src family kinase (SFK) inhibitors; however, the combination of EGFR and SFK inhibitors synergistically decreases cell viability. We found that this decrease in cell viability observed with EGFR and SFK inhibitor co-treatment correlates with loss of Akt phosphorylation. In addition, we found that in breast cancer cell lines with EGFR and c-Src co-localized to lipid rafts, phospho-inositide 3 kinase (PI3K) was also associated with lipid rafts. Together, the data herein suggest that lipid rafts provide a platform for the interaction of EGFR, c-Src, and PI3K, leading to activation of cellular survival signaling in breast cancer cells.
Introduction
Epidermal growth factor receptor (EGFR) is a mitogenic receptor tyrosine kinase expressed on the membrane as asymmetric dimers that upon ligand binding undergo a conformational change to induce autophosphorylation.Citation1 Autophosphorylation of tyrosine residues provides docking sites for mediators of downstream signaling including phospho-inositide-3-kinase (PI3K), Shc, Grb2, phospholipase C γ (PLCγ), and signal transducers and activators of transcription (STATs).Citation2 Recruitment and subsequent phosphorylation of these proteins results in activation of downstream signaling leading to cellular proliferation, migration, differentiation and survival (reviewed in ref. Citation2). While the most well known localization of EGFR is at the plasma membrane, evidence demonstrates that EGFR is active within other cellular membrane bound organelles, including endosomes, the nucleus and mitochondria.Citation3–Citation5 In addition, EGFR localizes to discrete membrane microdomains, referred to as lipid rafts.Citation6 Within these lipid rafts, EGFR signaling has been shown to be stimulated as well as inhibited, depending on the cellular context.Citation7–Citation10
High concentrations of signaling molecules can be found within lipid rafts.Citation11–Citation13 Such molecules include a variety of GPI-anchored proteins, lipid modified proteins and transmembrane proteins.Citation14,Citation15 Lipid rafts have both positive and negative effects on cellular signaling. Specifically, rafts may act as platforms to localize components of a signaling pathway in close proximity, or may facilitate crosstalk between signaling pathways by keeping components of both pathways near each other. However, lipid rafts may also act as sequestering regions in the membrane to prevent association of pathway components, leading to downregulation of signaling events (reviewed in ref. Citation16). As mentioned above, EGFR is capable of lipid raft localization, which can regulate EGFR-dependent signaling pathways. Another protein family known be present within lipid rafts is the Src family of tyrosine kinases.Citation17,Citation18 Lipid modification, as well as basic residues within the unique domain of the protein, facilitates association of c-Src, the prototypical member of this family, to lipid rafts.Citation18 Such localization has been described in neuronal, hematopoietic, and cervical and lung cancer cell lines.Citation17–Citation21 Like EGFR, lipid rafts also mediate c-Src signaling, including c-Src-dependent activation of PI3K/Akt.Citation17
c-Src interacts with a number of receptor tyrosine kinases including EGFR.Citation22 Activation of EGFR increases c-Src catalytic activity.Citation23 c-Src, in turn, phosphorylates novel sites on EGFR, which promotes signaling downstream of the receptor.Citation24–Citation26 Phosphorylation of EGFR by c-Src has been shown to regulate EGF-induced mitogenesis.Citation27,Citation28 Specifically, phosphorylation of EGFR by c-Src mediates the binding of PI3K to EGFR leading to Akt phosphorylation and cellular survival signaling.Citation26 The interaction between these two proteins enhances transformation of mouse fibroblasts and human mammary epithelial cell lines, and tumor formation in nude mice.Citation27,Citation29
We have previously shown that EGFR is localized to plasma membrane lipid rafts in breast cancer cells that are resistant to EGFR tyrosine kinase inhibitors.Citation30 Interestingly, inhibiting lipid raft structure with cholesterol biosynthesis inhibitors sensitizes these resistant cells to EGFR inhibitors.Citation30 Mechanistically, we found that inhibiting lipid raft structure decreased signaling through the Akt pathway, as previously reported by others.Citation30,Citation31 Here, we demonstrate co-localization and co-association between c-Src and EGFR within plasma membrane lipid rafts of one such breast cancer cell line, SUM159. While inhibition of either of these proteins alone was insufficient to reduce the viability of breast cancer cells, co-inhibition of c-Src and EGFR kinase activities resulted in a synergistic decrease in cell viability. Additionally, when EGFR and c-Src colocalized within lipid rafts, c-Src kinase inhibition abrogated EGFR-kinase independent Akt phosphorylation.
Results
c-Src localizes to lipid rafts in SUM159 cells.
Previously, we have shown that EGFR localizes to lipid rafts in four EGFR TKI resistant breast cancer cell lines.Citation30 This localization of EGFR to lipid rafts was required for Akt phosphorylation and EGFR disassociation with lipid rafts subsequently sensitized these cells EGFR kinase inhibition.Citation30 However, the signaling mechanism by which lipid raft localization of EGFR mediates Akt phosphorylation remained unknown. Interestingly, other cell types have shown that c-Src, an EGFR interacting kinase, may also associate with lipid rafts.Citation18 Using biochemical raft isolation techniques, density fractions were collected from the EGFR TKI resistant SUM159 cells and the EGFR TKI sensitive SUM149 cells. Fractions 1–7 were determined to be lipid raft fractions due to absence of transferrin receptor, presence of flotillin protein (), and the presence of the lipid raft specific ganglioside GM1 (data not shown).Citation30,Citation32 Similar to EGFR localization, significantly more c-Src was present in fractions 1–7 from SUM159 cells than from SUM149 cells (; p value = 0.0492). To determine if c-Src localizes to plasma membrane lipid rafts, SUM159 cells were co-immunostained with cholera toxin subunit B (red) which binds to GM1, and with c-Src antibodies (green, ). Co-localization between GM1 and c-Src occurred along the membrane of SUM159 cells (yellow, arrows), while staining of c-Src alone (green) was mostly intracellular. No co-localization was observed between GM1 and c-Src in SUM149 cells. Taken together, these data further confirm the association of c-Src with lipid rafts and define the localization of this association to be at the plasma membrane.
EGFR and c-Src co-localize in SUM159 cells.
Although EGFR and c-Src proteins both localize to lipid rafts in SUM159 cells, it was unknown if the proteins co-associate within the same lipid raft domain. Thus, to determine if c-Src and EGFR co-localize, SUM159 and SUM149 breast cancer cells were immunostained with Alexafluor labeled EGFR (green) and c-Src (red) antibodies (). The merged image shows co-localization of EGFR and c-Src (yellow) intracellularly in both cell lines, however; colocalization at the plasma membrane (yellow, arrows) occurs solely in SUM159 cells. These results suggest that c-Src and EGFR co-localize in both SUM159 and SUM149 cells, but only co-localize at the plasma membrane in SUM159 cells.
Physical interaction between EGFR and c-Src has been described in a number of cell types.Citation27,Citation28 In order to determine if EGFR and c-Src physically interact in our breast cancer cell lines, co-immunoprecipitation was performed. Whole cell lysate (WCL) was utilized as a positive immunoblotting control (, lane 1), while mouse immunoglobulin immunoprecipitation was performed as a negative immunoprecipitation control (IgG, , lane 2). We have previously shown that c-Src does not co-immunoprecipitate with EGFR from SUM149 cells.Citation33 However, in SUM159 cells, when EGFR was pulled down from whole cell lysates, c-Src co-immunoprecipitated (, lane 3). When the reverse immunoprecipitation was performed, EGFR co-immunoprecipitated with c-Src (, lane 3). These data support the co-localization results demonstrating that EGFR and c-Src are localized to similar regions in SUM159 cells.
The immunofluoresence data from the SUM159 cells suggests that EGFR and c-Src co-associate at the plasma membrane, and even more specifically within lipid rafts in SUM159 cells. Therefore, biochemical raft isolation was performed, equal sample volumes from fractions 1–7 [lipid raft (LR) fractions] and 8–14 [non-lipid raft (NR) fractions] were pooled, and EGFR or c-Src was immunoprecipitated. Samples were then subjected to SDS-PAGE followed by immunoblotting for EGFR and c-Src. When EGFR was pulled down from lipid raft fractions of SUM159 cells, c-Src co-associated (, lane 3). Low levels of EGFR were also precipitated from non-lipid raft fractions (, lane 4). When the reverse precipitation was performed, EGFR was found to co-associate with c-Src immunoprecipitates from lipid raft fractions (, lane 5). Similar results were also obtained when immunoprecipitation was performed in the presence of detergent (data not shown). These data suggest that EGFR and c-Src may functionally interact within the lipid rafts of SUM159 cells.
The co-localization data suggested that EGFR and c-Src were in close proximity intracellularly as well as at the membrane. Therefore, to determine if EGFR and c-Src co-immunoprecipitate independent of lipid-raft membranes, cells were treated with cholesterol biosynthesis inhibitor methyl-β-cyclodextran (MBCD), lysed and immunoprecipitated with antibodies against EGFR (). The immunoprecipitates were separated by SDS-PAGE and immunoblotted using EGFR and c-Src antibodies. As expected, EGFR and c-Src remain associated in the absence of lipid rafts, potentially as an intracellular complex. These data demonstrate that EGFR and c-Src associate within lipid raft membranes, but that this association is not exclusive to lipid rafts.
EGFR and Src family kinase inhibitors synergistically inhibit cell viability when both EGFR and c-Src are localized to lipid rafts.
Src family kinase (SFK) inhibitors are currently approved and in use for cancer treatment. Specifically, dasatinib, a SFK TKI, was approved in 2006 for the treatment of leukemia, and is currently in clinical trials in a number of solid tumor types including breast cancer.Citation34 Tyrosine phosphorylation of c-Src on tyrosine 419 is required for activation of c-Src. Thus, to determine if dasatinib inhibits c-Src kinase activity in our model system, phosphorylation of c-Src on tyrosine 419 was measured. Cells were treated with 0.5 µM dasatinib for 2 h and lysates were immunoblotted with phospho-tyrosine 419 specific antibodies. As shown in , phosphorylation of c-Src on tyrosine 419 was abrogated with dasatinib treatment.
Preclinical studies demonstrate that dasatinib treatment decreases cell growth in select breast cancer cell lines.Citation34 To determine if inhibition of SFKs was sufficient to decrease SUM159 cell growth, cellular proliferation assays were performed. SUM159 cells were treated with increasing doses of dasatinib and proliferation was measured by cell counting on days 1, 4 and 8. SUM159 cells continued to proliferate in the presence of all doses of dasatinib tested (). Similar results were observed with the SFK inhibitors SU6656 and AZD0530 (data not shown). We have previously shown that SUM159 cells also continue to proliferate in the presence of the EGFR TKI gefitinib.Citation30 Therefore, to determine if concomitant treatment with dasatinib and gefitinib could alter viability of SUM159 cells, cell viability analyses were performed. Cells were treated with dasatinib and gefitinib alone, or in combination, for 72 h. Gefitinib IC50 values were calculated for each dose of dasatinib tested and isobolograms were generated to determine drug synergy (). The IC50 of gefitinib alone was plotted on the y-axis and the IC50 of dasatinib alone was plotted on the x-axis. A line was drawn between them. This line represents an additive relationship between two drugs. IC50 points that fall on the line are considered additive, while points that fall below the line are considered synergistic (). In the SUM159 cells, the IC50 of gefitinib at each dose of dasatinib fell below the line of additivity indicating that the effects of gefitinib and dasatinib were synergistic. Combination index (CI) values were also determined for co-treatment of dasatinib and gefitinib. At all doses of dasatinib, the CI-value was significantly below 1 (p < 0.05), which indicates that gefitinib and dasatinib have a synergistic interaction in SUM159 breast cancer cells.
Inhibition of c-Src kinase activity and depletion of lipid rafts is additive.
We have previously shown that by decreasing cholesterol biosynthesis, we could sensitize cell lines in which EGFR localizes to lipid rafts to gefitinib.Citation30 Therefore, to determine if decreasing cholesterol content and subsequent lipid raft formation could sensitize cells to dasatinib, SUM159 cells were treated with dasatinib in combination with lovastatin, a cholesterol biosynthesis inhibitor, and cell viability analyses were performed. Interestingly, increasing doses of lovastatin did not synergistically decrease the IC50 for dasatinib (). In fact, the data points fall very close to the line suggesting an additive interaction between the two drugs. Similarly, the CI-values for the combination of dasatinib and lovastatin were not significantly different from one, indicating that these two drugs are not synergistic ().
c-Src kinase activity mediates EGFR kinase-independent Akt phosphorylation.
In 2007, Arcaro and colleagues found that lipid raft associated c-Src was critical for the activation of the PI3K/Akt signaling pathway in small cell lung cancer (SCLC) cell lines.Citation17 Their results suggested that lipid rafts provided a platform for interaction between c-Src and PI3K. To determine if lipid rafts may also provide such a platform in SUM159 breast cancer cells, biochemical raft isolation was performed followed by immunoblotting for the catalytic subunit of PI3K, p110α (). While the SUM149 cells contained little p110α in lipid rafts, p110α was increased in the lipid raft fractions of SUM159 cells (36 vs. 45% of total c-Src protein). Thus, it is possible that PI3K signaling mediates EGFR/c-Src synergy in lipid rafts.
We have previously shown that depletion of lipid rafts results in decreased EGFR-kinase independent Akt phosphorylation in SUM159 cells.Citation30 c-Src is known to be a downstream mediator of EGFR signaling pathways, including Akt and MAPK.Citation24,Citation26,Citation28 Thus, to determine if c-Src mediates EGFR kinase-independent signaling in SUM159 cells, lysates were immunoblotted for phosphorylation of Akt and MAPK following treatment with gefitinib or dasatinib alone, or in combination (). As shown previously, gefitinib effectively abrogates MAPK phosphorylation in SUM159 cells while Akt phosphorylation persists (, lane 2, and 30). SFK inhibition by dasatinib treatment resulted in a small decrease of MAPK phosphorylation, and was sufficient to abrogate Akt phosphorylation in SUM159 cells (, lane 3). Co-treatment with gefitinib and dasatinib resulted in no additional decrease in MAPK or Akt phosphorylation. These data suggest that the activity of SFKs regulate Akt phosphorylation in SUM159 cells.
Discussion
This study provides evidence that c-Src plays a role in lipid raft-dependent, EGFR kinase-independent, signaling in SUM159 cells. Specifically, we have shown that c-Src co-localized and co-associated with EGFR in lipid rafts. When EGFR and c-Src are co-localized within lipid rafts, the cells are sensitive to co-treatment with EGFR and SFK inhibitors: gefitinib and dasatinib. In fact, we found this inhibition to be synergistic. Interestingly, in contrast to our previous results demonstrating that gefitinib and the cholesterol biosynthesis inhibitors were synergistic, dasatinib and lovastatin inhibitors lacked synergy. Lastly, we have shown that a catalytic subunit of PI3K (p110α) co-localizes with EGFR and c-Src within lipid rafts in SUM159 cells and that inhibition of SFK activity decreased Akt phosphorylation. Taken together, these data have further characterized a mechanism for lipid raft-dependent EGFR TKI resistance by showing that c-Src is active and present in lipid rafts where it has the ability to regulate PI3K/Akt survival signals.
SFKs previously have been localized to lipid rafts.Citation18 While other Src family members are likely to associate preferentially to lipid rafts through their double lipid modification, c-Src is only singly lipid modified.Citation35,Citation36 c-Src localization to lipid rafts is thought to occur through interactions between membrane lipids and basic residues within the unique domain of c-Src.Citation18 Localization of c-Src to lipid rafts has been demonstrated in neuronal,Citation19,Citation20 hematopoietic,Citation37 and madin-darby canin kidney cells,Citation36 as well as skeletal myoblasts,Citation38 murine fibroblasts,Citation35 and cervical and lung cancer cell lines.Citation17,Citation36 Here we have shown that c-Src localizes to lipid rafts in SUM159 breast cancer cells and that this localization may be important for cell proliferation and survival ().
We have identified an interaction between EGFR and c-Src within lipid raft membrane microdomains in breast cancer cells ( and ). Co-precipitation between these two proteins has been noted previously. Specifically, Wu and colleagues have shown that zinc is capable of inducing an association between EGFR and c-Src leading to Ras activation in human fibroblasts.Citation6 Maa and colleagues demonstrated EGFR and c-Src coimmunoprecipitation in a murine fibroblast model.Citation27 In this model system, a physical association was noted between c-Src and EGFR, resulting in the novel phosphorylation of tyrosine residues on EGFR, as well as other downstream targets, leading to increased cellular transformation. Nair and colleagues have shown co-immunoprecipitation between EGFR and c-Src in PC12 neuronal cells where this interaction promotes Akt signaling.Citation39 c-Src was also co-immunoprecipitated as part of a complex between EGFR and c-Met in SUM229 breast cancer cells.Citation33,Citation40 This interaction was described to mediate EGFR TKI resistance of SUM229 cells. Our data indicate that EGFR and c-Src associate in SUM159 breast cancer cells (). More specifically, coimmunoprecipitation occurs between these two proteins within lipid raft fractions of SUM159 cells (). While it is possible that immunoprecipitation from lipid raft fractionation may bring down larger sheets of membrane, resulting in a falsely positive interaction, we have treated the lysate with detergent including triton x-100 and have seen similar co-immunoprecipitation results (data not shown). While previous studies have revealed that these two proteins are both capable of localizing to lipid rafts in the same cell line,Citation19,Citation41,Citation42 the actual co-association within these lipid raft domains has not been previously shown.
Our data indicate the concomitant inhibition of SFK and EGFR kinase activities results in decreased viability in breast cancer cells. In particular, we have shown that while SUM159 cells are resistant to EGFRCitation30 and SFK inhibitors (), cotreatment with dasatinib and gefitinib resulted in synergistic decreases in cell viability (). These data are supported by the knowledge that c-Src is overexpressed in many of the same cancer types as EGFR, and has been shown to be co-overexpressed with EGFR in a subset of breast tumors.Citation27 Additionally, EGFR and c-Src co-overexpression in mouse fibroblasts and human mammary epithelial cells results in synergistic increases in tumorigenesis.Citation24,Citation27,Citation29,Citation43 In other models of cancer, dual inhibition of EGFR and c-Src kinase activities decreases cancer cell viability. In particular, Johns and colleagues have demonstrated that downregulation of c-Src expression improved the response of gliomas to EGFR-directed monoclonal antibodies.Citation44 Decreased c-Src kinase activity resulted in increased sensitivity to EGFR inhibitors in head and neck cancer and epidermoid carcinoma cell lines.Citation45,Citation46 A recent Phase I/II study has also been published utilizing dasatinib in combination with the EGFR TKI erlotinib in non-small cell lung cancer.Citation47 Not only was this combination tolerable in patients, but disease control was also observed. Thus, our data suggest that combinatorial inhibition of EGFR and c-Src kinase activities may result in a decrease of the viability of breast cancer cells.
Our data are specifically analyzing the interaction of EGFR and c-Src in breast cancer cells. However, because we used of the SFK dasatinib for most of our studies, the influence of other SFKs in SUM159 cell viability cannot be excluded. Specifically, the IC50 values for dasatinib for all SFK, as well as Abl, are less than 1 nM, indicating that at the doses used in our study we are also inhibiting other SFKs.Citation48 SUM159 cells express c-Src, Yes, Lyn and Abl (data not shown). To determine if Yes, Lyn, and/or Abl kinase activities are critical to the viability of SUM159 cells in the presence of an EGFR TKI, further studies would need to be done using more selective inhibitors and dominant-negative or siRNA methods for inhibiting SFK activity. That being said, these data provide strong evidence for a role of SFKs in EGFR TKI resistance.
Localization of c-Src to lipid rafts has been shown to regulate c-Src signaling.
Specifically, lipid rafts are important in mediation of the c-Src dependent activation of PI3K/Akt signaling in human SCLC cell linesCitation17 and in the c-Src dependent activation of FAK to promote early contact signaling in cells.Citation49 Also, c-Src trafficking and co-localization with EGFR promotes EGFRligand independent MAPK signaling.Citation50 We have shown that lipid rafts promote c-Src-dependent signaling in SUM159 breast cancer cells. In particular, we suggest that lipid raft localized c-Src plays a role in EGFR-kinase independent signaling seen in these cells, as inhibition of c-Src kinase activity with dasatinib results in abrogation of EGFR-kinase independent Akt phosphorylation ().
The results described here suggest a model for the activation of EGFR-dependent signaling pathways independent of EGFR-kinase activity. We have described a role for the non-receptor tyrosine kinase c-Src in the activation of EGFR-kinase independent Akt signaling in the EGFR TKI resistant SUM159 breast cancer cell line. Specifically, c-Src co-localized and co-associated with EGFR in plasma membrane lipid rafts in SUM159 cells. Combined treatment with dasatinib, a small molecule SFK inhibitor, and gefitinib, an EGFR TKI, resulted in a synergistic decrease in cell viability when both EGFR and c-Src localized to lipid rafts, however lipid raft depletion and c-Src kinase inhibition were not synergistic. Furthermore, EGFR kinase-independent Akt phosphorylation was c-Src dependent in SUM159 cells. We therefore hypothesize that lipid rafts provide a platform whereby EGFR, c-Src and PI3K interact to promote EGFR kinase-independent cellular survival signaling in breast cancer cells.
Materials and Methods
Reagents.
Gefitinib was provided by AstraZeneca. All other reagents were purchased from Sigma or VWR unless otherwise noted.
Cell lines.
The SUM series of cell lines were obtained from Dr. Stephen Ethier (Wayne State University/Karmanos Cancer Institute). SUM159 and SUM149 cell lines are grown in 5% IH media (Ham's F-12 media, supplemented with 5% FBS, 1 µg/ml hydrocortisone and 5 µg/ml insulin) and cultured in 10% CO2. All media are supplemented with 2.5 µg/ml amphotericin B and 25 µg/ml gentamicin.
Biochemical raft isolation.
Biochemical lipid raft isolation was performed following established protocols.Citation51 Briefly, cells were plated at a density of 0.5 × 106 cells in six-100 mm plates and grown in normal growth media for 72 h. Cells were scraped in base buffer (20 mM Tris, pH 7.8, 250 mM Sucrose, 1 mM MgCl2, 1 mM CaCl2, 100 µM sodium orthovanadate) and then lysed in base buffer containing 1x protease inhibitor cocktail (EMD Biosciences) by passing through a 22 gauge × 1.5″ needle 40 times. Lysates were centrifuged as described and the first and second post-nuclear supernatants were combined and frozen at −20°C. Samples were thawed and combined with equal volume of 50% Opti-Prep (Greiner Bio One) and 0–20% Opti-Prep gradient was applied. Gradients were centrifuged for 90 min at 52,000x g and then fractionated into 16-0.56 mL fractions. Fractions were separated via SDS-PAGE, transferred to Immobolin-P (Millipore), and immunoblotted utilizing c-Src (2–17, S. Parsons, University of Virginia), p110α, transferrin (Invitrogen), or flotillin (BD Biosciences) antibodies. Fractions were dot blotted with Cholera Toxin Subunit B-HRP (Invitrogen) to determine GM-1 expression. Incubation with enhanced chemiluminescence (GE Healthcare) was followed by exposure to film. Experiments were repeated at least three independent times and quantified using densitometry (NIH Image).
Immunostaining.
Anti-EGFR (mab108, M. Weber, University of Virginia) was labeled with Alexa-fluor-488 (Invitrogen) and Anti-Src (2–17) was labeled with Alexa-flour-594 (Invitrogen). Cells were plated on coverslips at a density of 1.5 × 105 cells per 35 mm dish and grown in normal growth media for 48 h. For lipid raft staining, cells were incubated with Alexa-fluor-594 labeled cholera toxin subunit B (Invitrogen,) at 1 µg/ml for 10 min on ice prior to fixation.Citation9,Citation52 Cells were then fixed with formalin, permeabilized with 0.1% triton-x 100, blocked in 20% goat serum for 1 h, then incubated with EGFR (Alexa-fluor labeled) or c-Src (2–17 unlabeled for lipid raft staining, 594 labeled for EGFR costaining) antibody for 1 h (followed by corresponding Alexa-fluor 488 secondary antibody if necessary), washed, and mounted onto slides with Prolong Gold containing DAPI (Invitrogen). Imaging was performed via confocal microscopy using a Zeiss Axioplan2 apotome microscope fitted with a 63x 1.25 oil immersion lens at the Microscopy and Imaging Resources Laboratory (Wayne State University). Images are representative of at least three independent experiments.
Immunoprecipitation.
Whole cell lysates or two hundred microliters of each lipid raft fractions (1–7) or non-lipid raft fractions (8–14) were pooled and immunoprecipitated with 5 µL of EGFR antibody (mab108) or 5 µL c-Src antibody (2–17) with or without the addition of CHAPs lysis buffer for 1 h at 4°C. Protein A agarose beads were added for 30 min. Samples were then pelleted and washed three times in CHAPs lysis buffer [10 mmol/L CHAPs, 50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl and 2 mmol/L EDTA with 10 µmol/L sodium orthovanadate and 1x protease inhibitor cocktail], then 25 µL 2x lamelli buffer was added, and samples were boiled for 5 min. Samples were subjected to SDS-PAGE followed by immunoblotting for EGFR and c-Src. Whole cell lysates were utilized as a migration control. Immunoprecipitation with 1.75 µL mouse IgG antibody served as a negative control.
Immunoblotting.
Breast cancer cell lines were plated at a density of 1 × 106 cells per 100-mm dish and grown in normal growth media for 48 h. Cells were treated with indicated reagents [1.0 µM gefitinib for 30 min and/or 1.0 µM dasatinib (LC Laboratories; 2 h)] then lysed in CHAPs lysis buffer. For immunoblotting, 10 to 100 µg of protein lysate were separated by SDS-PAGE and transferred to Immobilon P. Membranes were blocked in either 5% nonfat dry milk for 1 h at 25°C or overnight at 4°C [phosphomitogen activated protein kinase (MAPK)] or 5% BSA overnight at 4°C (phospho-SrcY419, Cell Signaling Technology, Danvers, MA). Membranes were probed with EGFR (Cell Signaling Technology), Akt (Cell Signaling Technology), MAPK (Cell Signaling Technology), phospho-Akt (Ser473; Cell Signaling Technology,), phospho-ERK1/2 (MAPK) (Invitrogen), phospho-SrcY416 and c-Src (Cell Signaling Technology) antibodies. All antibodies were incubated overnight at 4°C, except for phos-pho-MAPK and phospho-SrcY416 (2 h at room temperature). Membranes were washed with TBS + 0.1% Tween 20 three times for 10 min, followed by incubation with corresponding secondary antibody and another series of three washes. Incubation with enhanced chemiluminescence (GE Healthcare) was followed by exposure to film. Experiments were repeated at least three independent times and quantified using densitometry (NIH Image).
MTS assays.
Breast cancer cells were plated at a density of 1–2 × 103 in 96-well plates, incubated overnight, and then treated with indicated concentrations of lovastatin (Calbiochem) dasatinib, and/or gefitinib for 72 h. Twenty microliters of CellTiter 96 Aqueous One Solution Cell Proliferation Assay reagent (Promega) were added to each well and allowed to incubate at 37°C. Absorbance at 490 nm was detected at 2 h using a OpsysMR microplate reader (Dynex). Absorbance units were normalized to the mean of a single dose to compare between experiments. Dose response curves were generated using nonlinear sigmoidal dose response curve analyses in GraphPad Prism. IC50 values were calculated and plotted on isobolograms. Points in the graph represent a mean of three independent experiments performed in triplicate.
Statistics.
Student's t-test was performed utilizing the statistical software in GraphPad Prism. p values of < 0.05 were considered statistically significant. To perform synergy analyses, the IC50 gefitinib was calculated for each dose of lovastatin. The combination index (CI-value) was calculated as follows: (IC50 gefitinib at × dose lovastatin)/(IC50 gefitinib alone) + (dose of lovastatin)/(IC50 lovastatin alone).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| EGFR | = | epidermal growth factor receptor |
| SFK | = | Src family kinases |
| TKIs | = | tyrosine kinase inhibitors |
Figures and Tables
Figure 1 c-Src localizes within lipid rafts in SUM159 breast cancer cells. (A) SUM159 or SUM149 cells were plated and cultured for 72 h in normal growth media. Lipid rafts were separated by ultracentrifugation and immunoblotting was performed for EGFR, c-Src, transferrin receptor and flotillin. Lanes numbered 1–7 indicate lipid raft fractions (Macdonald and Pike, 2005). (B) Densitometry was performed on c-Src immunoblots from (A). Bars represent the percent of total c-Src in lipid raft fractions (1–7) compared with the total c-Src (fractions 1–14). Fractions 15 and 16 were excluded as pellet fractions. Experiments were repeated at least three independent times. p value was calculated by comparing the percent of c-Src in lipid rafts in SUM159 cells compared with SUM149 cells using the Student's t-test. (C) Two hundred thousand cells were plated onto coverslips and cultured under normal growth conditions for 48 h. Coverslips containing cells were then incubated with Alexa-fluor 594 labeled cholera toxin subunit B (red) to stain lipid rafts, fixed, permeabilized, blocked and then stained for c-Src utilizing 2–17 and an alexa-fluor 488 labeled secondary antibody (green). DAPI was used to stain the nucleus (blue). Imaging was performed using Zeiss Axioplan2 apotome microscope fitted with 63x 1.25 oil immersion lens at the Microscopy and Imaging Resources Laboratory (Wayne State University, Detroit, MI). Arrows represent lipid raft localized c-Src. Three representative images from three distinct experiments are shown.
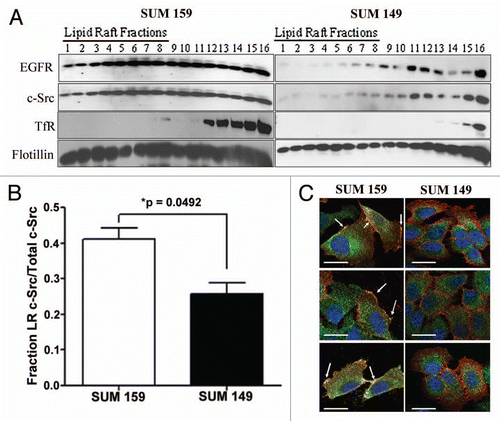
Figure 2 EGFR and c-Src co-localize and co-associate within lipid rafts. (A) Two hundred thousand cells were plated onto coverslips and cultured under normal growth conditions for 48 h. Coverslips containing cells were blocked, and then incubated with fluorphore-labeled antibodies (EGFR-488: green; Src-594: red). The nucleus was stained with DAPI (blue). Imaging was performed using Zeiss Axioplan2 apotome microscope fitted with 63X 1.25 oil immersion lens at the Microscopy and Imaging Resources Laboratory (Wayne State University, Detroit, MI). Arrows indicate areas of co-localization at the plasma membrane (yellow). (B and C) SUM159 cells were plated, allowed to grow for 48 h, and then lysed. Five hundred micrograms of protein was immunoprecipitated with EGFR antibodies (B) or c-Src antibodies (C), then subjected to SDS-PAGE and immunoblotting for EGFR and c-Src. Ten micrograms (2% input) of whole cell lysate (WCL) was utilized as a positive control and mouse IgG immunoprecipitation was performed as a negative control (IgG). (D) Lipid rafts were isolated from SUM159 cells by biochemical raft isolation as described. Two hundred microliters of each fraction [Lipid raft fractions 1–7 (LR) or Non-lipid raft fractions 8–14 (NR)] was pooled (approximately 100 ug), and EGFR or c-Src was immunoprecipitated. Immunoblotting was then performed for EGFR and c-Src. Ten micrograms of whole cell lysate (WCL, 10% input) was utilized as a positive control, while immunoprecipitation with IgG was utilized as a negative control. Arrows indicate the c-Src band. Immunoblots were performed at least three independent times. (E) Cells were treated with 1 mM MBCD for 1 h. Lysates were prepared and 500 µg of protein were immunoprecipitated using EGFR antibodies. Immunoprecipitates were separated by SDS-PAGE and immunoblotted using EGFR and c-Src antibodies.
![Figure 2 EGFR and c-Src co-localize and co-associate within lipid rafts. (A) Two hundred thousand cells were plated onto coverslips and cultured under normal growth conditions for 48 h. Coverslips containing cells were blocked, and then incubated with fluorphore-labeled antibodies (EGFR-488: green; Src-594: red). The nucleus was stained with DAPI (blue). Imaging was performed using Zeiss Axioplan2 apotome microscope fitted with 63X 1.25 oil immersion lens at the Microscopy and Imaging Resources Laboratory (Wayne State University, Detroit, MI). Arrows indicate areas of co-localization at the plasma membrane (yellow). (B and C) SUM159 cells were plated, allowed to grow for 48 h, and then lysed. Five hundred micrograms of protein was immunoprecipitated with EGFR antibodies (B) or c-Src antibodies (C), then subjected to SDS-PAGE and immunoblotting for EGFR and c-Src. Ten micrograms (2% input) of whole cell lysate (WCL) was utilized as a positive control and mouse IgG immunoprecipitation was performed as a negative control (IgG). (D) Lipid rafts were isolated from SUM159 cells by biochemical raft isolation as described. Two hundred microliters of each fraction [Lipid raft fractions 1–7 (LR) or Non-lipid raft fractions 8–14 (NR)] was pooled (approximately 100 ug), and EGFR or c-Src was immunoprecipitated. Immunoblotting was then performed for EGFR and c-Src. Ten micrograms of whole cell lysate (WCL, 10% input) was utilized as a positive control, while immunoprecipitation with IgG was utilized as a negative control. Arrows indicate the c-Src band. Immunoblots were performed at least three independent times. (E) Cells were treated with 1 mM MBCD for 1 h. Lysates were prepared and 500 µg of protein were immunoprecipitated using EGFR antibodies. Immunoprecipitates were separated by SDS-PAGE and immunoblotted using EGFR and c-Src antibodies.](/cms/asset/11dd4278-27ab-4647-9e77-f89c8af262ea/kcbt_a_10916907_f0002.gif)
Figure 3 Dasatinib sensitizes SUM159 cells to gefitinib, but not lovastatin. (A) SUM159 cells were treated with 0.5 uM dasatinib for 2 h and lysates were subjected to SDS-PAGE followed by immunoblotting for EGFR, c-Src and phospho-Y416 c-Src. (B) SUM159 cells were plated in 35 mm plates and grown for eight days. Treatment with the indicated doses of dasatinib occurred every other day and cell counts were performed on days 1, 4 and 8. Error bars represent average of three independent experiments performed in triplicate. (C) SUM159 cells were plated in a 96-well plate and treated with increasing doses of dasatinib and gefitinib for 72 h. Cell viability was measured using MTS assays. The IC50 of gefitinib at each dose of dasatinib was calculated and plotted on an isobologram. (D) The combination index (CI-value) was calculated as follows: (IC50 gefitinib at × dose of dasatinib)/(IC50 gefitinib alone) + (dose of dasatinib)/(IC50 dasatinib alone). (E) Cells were placed in a 96-well plate and treated with variable doses of dasatinib and lovastatin for 72 h. Cell viability was measured using MTS assays. The IC50 of dasatinib at each dose of lovastatin was calculated and plotted on an isobologram. (F) The CI-value was calculated and the p value was determined as compared with 1. All experiments were performed at least three independent times. p values were calculated as a difference between CI-values and one utilizing the Student's t-test.
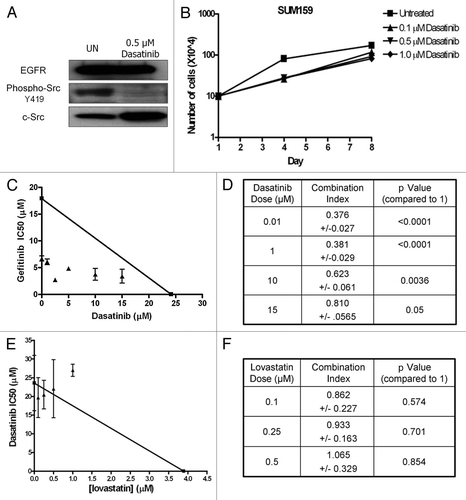
Figure 4 Phosphorylation of Akt is c-Src kinase-dependent in SUM159 cells. (A) Biochemical raft isolation was performed as described on SUM159 and SUM149 cells. Fractions were separated by SDS-PAGE and immunblotting for p110α, transferrin receptor and flotillin was performed. Blots are representative of at least three independent experiments. (B) Whole cell lysates were collected from SUM159 cells treated with 1.0 µM gefitinib and 1.0 µM dasatinib alone or in combination. Expression and phosphorylation of Akt and MAPK, as well as expression of EGFR was determined by immunoblotting. Immunoblots were repeated at least three independent times.
Acknowledgments
We would like to thank Kelly Mueller, B.S. for technical assistance. We are grateful to the Microscopy and Imaging Resources Laboratory Shared Resource facility for providing services. The Microscopy and Imaging Resources Laboratory is supported, in part, by NIH Center grants P30ES06639 to the Institute of Environmental Health Sciences, P30CA22453 to The Karmanos Cancer Institute, Wayne State University, and U54RR020843 to The Center for Proteolytic Pathways, the Burnham Institute, and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University. This work was supported by NCI T32-CA009531 (M.E.I.) and Susan G. Komen for the Cure Career Catalyst Grant (KG081416; J.L.B.).
References
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006; 125:1137 - 1149; PMID: 16777603; http://dx.doi.org/10.1016/j.cell.2006.05.013
- Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 2003; 284:31 - 53; PMID: 12648464; http://dx.doi.org/10.1016/S0014-4827(02)00098-8
- Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol 2004; 24:7059 - 7071; PMID: 15282306; http://dx.doi.org/10.1128/MCB.24.16.7059-71.2004
- Cohen S, Fava RA. Internalization of functional epidermal growth factor:receptor/kinase complexes in A-431 cells. J Biol Chem 1985; 260:12351 - 12358; PMID: 2995372
- Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3:802 - 808; PMID: 11533659; http://dx.doi.org/10.1038/ncb0901-802
- Wu W, Graves LM, Gill GN, Parsons SJ, Samet JM. Src-dependent phosphorylation of the epidermal growth factor receptor on tyrosine 845 is required for zinc-induced Ras activation. J Biol Chem 2002; 277:24252 - 24257; PMID: 11983694; http://dx.doi.org/10.1074/jbc.M200437200
- Chen X, Resh MD. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J Biol Chem 2002; 277:49631 - 49637; PMID: 12397069; http://dx.doi.org/10.1074/jbc.M208327200
- Ringerike T, Blystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J Cell Sci 2002; 115:1331 - 1340; PMID: 11884532
- Roepstorff K, Thomsen P, Sandvig K, van Deurs B. Sequestration of epidermal growth factor receptors in non-caveolar lipid rafts inhibits ligand binding. J Biol Chem 2002; 277:18954 - 18960; PMID: 11886870; http://dx.doi.org/10.1074/jbc.M201422200
- Westover EJ, Covey DF, Brockman HL, Brown RE, Pike LJ. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J Biol Chem 2003; 278:51125 - 51133; PMID: 14530278; http://dx.doi.org/10.1074/jbc.M304332200
- Lisanti MP, Tang Z, Scherer PE, Kubler E, Koleske AJ, Sargiacomo M. Caveolae, transmembrane signalling and cellular transformation. Mol Membr Biol 1995; 12:121 - 124; PMID: 7767370; http://dx.doi.org/10.3109/09687689509038506
- Wu C, Butz S, Ying Y, Anderson RG. Tyrosine kinase receptors concentrated in caveolae-like domains from neuronal plasma membrane. J Biol Chem 1997; 272:3554 - 3559; PMID: 9013605; http://dx.doi.org/10.1074/jbc.272.6.3554
- Hope HR, Pike LJ. Phosphoinositides and phosphoinositide-utilizing enzymes in detergent-insoluble lipid domains. Mol Biol Cell 1996; 7:843 - 851; PMID: 8816992
- Danielsen EM, van Deurs B. A transferrin-like GPI-linked iron-binding protein in detergent-insoluble noncaveolar microdomains at the apical surface of fetal intestinal epithelial cells. J Cell Biol 1995; 131:939 - 950; PMID: 7490295; http://dx.doi.org/10.1083/jcb.131.4.939
- Sargiacomo M, Scherer PE, Tang Z, Kubler E, Song KS, Sanders MC, et al. Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc Natl Acad Sci USA 1995; 92:9407 - 9411; PMID: 7568142; http://dx.doi.org/10.1073/pnas.92.20.9407
- Zajchowski LD, Robbins SM. Lipid rafts and little caves. Compartmentalized signalling in membrane microdomains. Eur J Biochem 2002; 269:737 - 752; PMID: 11846775; http://dx.doi.org/10.1046/j.0014-2956.2001.02715.x
- Arcaro A, Aubert M, Espinosa del Hierro ME, Khanzada UK, Angelidou S, Tetley TD, et al. Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell Signal 2007; 19:1081 - 1092; PMID: 17275257; http://dx.doi.org/10.1016/j.cellsig.2006.12.003
- Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD. Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc Natl Acad Sci USA 1994; 91:12253 - 12257; PMID: 7527558; http://dx.doi.org/10.1073/pnas.91.25.12253
- Kasai A, Shima T, Okada M. Role of Src family tyrosine kinases in the downregulation of epidermal growth factor signaling in PC12 cells. Genes Cells 2005; 10:1175 - 1187; PMID: 16324154; http://dx.doi.org/10.1111/j.1365-2443.2005.00909.x
- Mukherjee A, Arnaud L, Cooper JA. Lipid-dependent recruitment of neuronal Src to lipid rafts in the brain. J Biol Chem 2003; 278:40806 - 40814; PMID: 12912979; http://dx.doi.org/10.1074/jbc.M306440200
- Shenoy-Scaria AM, Gauen LK, Kwong J, Shaw AS, Lublin DM. Palmitylation of an amino-terminal cysteine motif of protein tyrosine kinases p56lck and p59fyn mediates interaction with glycosyl-phosphatidylinositol-anchored proteins. Mol Cell Biol 1993; 13:6385 - 6392; PMID: 8413237
- Luttrell DK, Luttrell LM, Parsons SJ. Augmented mitogenic responsiveness to epidermal growth factor in murine fibroblasts that overexpress pp60c-src. Mol Cell Biol 1988; 8:497 - 501; PMID: 2447487
- Weernink PA, Rijksen G. Activation and translocation of c-Src to the cytoskeleton by both platelet-derived growth factor and epidermal growth factor. J Biol Chem 1995; 270:2264 - 2267; PMID: 7530720; http://dx.doi.org/10.1074/jbc.270.5.2264
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem 1999; 274:8335 - 8343; PMID: 10075741; http://dx.doi.org/10.1074/jbc.274.12.8335
- Sato K, Sato A, Aoto M, Fukami Y. Site-specific association of c-Src with epidermal growth factor receptor in A431 cells. Biochem Biophys Res Commun 1995; 210:844 - 851; PMID: 7539262; http://dx.doi.org/10.1006/bbrc.1995.1735
- Stover DR, Becker M, Liebetanz J, Lydon NB. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85 alpha. J Biol Chem 1995; 270:15591 - 15597; PMID: 7797556; http://dx.doi.org/10.1074/jbc.270.26.15591
- Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA 1995; 92:6981 - 6985; PMID: 7542783; http://dx.doi.org/10.1073/pnas.92.15.6981
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci USA 1999; 96:1415 - 1420; PMID: 9990038; http://dx.doi.org/10.1073/pnas.96.4.1415
- Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res 2007; 67:4164 - 4172; PMID: 17483327; http://dx.doi.org/10.1158/0008-5472.CAN-06-2580
- Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol 2010; PMID: 21140447
- Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS, Hong SJ. Cholesterol level of lipid raft microdomains regulates apoptotic cell death in prostate cancer cells through EGFR-mediated Akt and ERK signal transduction. Prostate 2007; 67:1061 - 1069; PMID: 17469127; http://dx.doi.org/10.1002/pros.20593
- Wolf AA, Jobling MG, Wimer-Mackin S, Ferguson-Maltzman M, Madara JL, Holmes RK, et al. Ganglioside structure dictates signal transduction by cholera toxin and association with caveolae-like membrane domains in polarized epithelia. J Cell Biol 1998; 141:917 - 927; PMID: 9585411; http://dx.doi.org/10.1083/jcb.141.4.917
- Mueller KL, Yang ZQ, Haddad R, Ethier SP, Boerner JL. EGFR/Met association regulates EGFR TKI resistance in breast cancer. J Mol Signal 2010; 5:8; PMID: 20624308; http://dx.doi.org/10.1186/1750-2187-5-8
- Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19:415 - 425; PMID: 20113198; http://dx.doi.org/10.1517/13543781003592097
- Robbins SM, Quintrell NA, Bishop JM. Myristoylation and differential palmitoylation of the HCK protein-tyrosine kinases govern their attachment to membranes and association with caveolae. Mol Cell Biol 1995; 15:3507 - 3515; PMID: 7791757
- Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J Cell Biol 1994; 126:353 - 363; PMID: 7518463; http://dx.doi.org/10.1083/jcb.126.2.353
- Stoddart A, Dykstra ML, Brown BK, Song W, Pierce SK, Brodsky FM. Lipid rafts unite signaling cascades with clathrin to regulate BCR internalization. Immunity 2002; 17:451 - 462; PMID: 12387739; http://dx.doi.org/10.1016/S1074-7613(02)00416-8
- Smythe GM, Eby JC, Disatnik MH, Rando TA. A caveolin-3 mutant that causes limb girdle muscular dystrophy type 1C disrupts Src localization and activity and induces apoptosis in skeletal myotubes. J Cell Sci 2003; 116:4739 - 4749; PMID: 14600260; http://dx.doi.org/10.1242/jcs.00806
- Nair VD, Sealfon SC. Agonist-specific transactivation of phosphoinositide-3-kinase signaling pathway mediated by the dopamine D2 receptor. J Biol Chem 2003; 278:47053 - 47061; PMID: 12970364; http://dx.doi.org/10.1074/jbc.M303364200
- Mueller KL, Hunter LA, Ethier SP, Boerner JL. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res 2008; 68:3314 - 3322; PMID: 18451158; http://dx.doi.org/10.1158/0008-5472.CAN-08-0132
- Hur EM, Park YS, Lee BD, Jang IH, Kim HS, Kim TD, et al. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J Biol Chem 2004; 279:5852 - 5860; PMID: 14630916; http://dx.doi.org/10.1074/jbc.M311687200
- Jeon JH, Kim SK, Kim HJ, Chang J, Ahn CM, Chang YS. Lipid raft modulation inhibits NSCLC cell migration through delocalization of the focal adhesion complex. Lung Cancer 2010; 69:165 - 171; PMID: 19945766; http://dx.doi.org/10.1016/j.lungcan.2009.10.014
- Biscardi JS, Tice DA, Parsons SJ. c-Src, receptor tyrosine kinases and human cancer. Adv Cancer Res 1999; 76:61 - 119; PMID: 10218099; http://dx.doi.org/10.1016/S0065230X(08)60774-5
- Johns TG, Perera RM, Vernes SC, Vitali AA, Cao DX, Cavenee WK, et al. The efficacy of epidermal growth factor receptor-specific antibodies against glioma xenografts is influenced by receptor levels, activation status and heterodimerization. Clin Cancer Res 2007; 13:1911 - 1925; PMID: 17363548; http://dx.doi.org/10.1158/1078-0432.CCR-06-1453
- Andersen P, Villingshoj M, Poulsen HS, Stockhausen MT. Improved response by co-targeting EGFR/EGFRvIII and Src family kinases in human cancer cells. Cancer Invest 2009; 27:178 - 183; PMID: 19235590; http://dx.doi.org/10.1080/07357900802570759
- Koppikar P, Choi SH, Egloff AM, Cai Q, Suzuki S, Freilino M, et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res 2008; 14:4284 - 4291; PMID: 18594011; http://dx.doi.org/10.1158/1078-0432.CCR-07-5226
- Haura EB, Tanvetyanon T, Chiappori A, Williams C, Simon G, Antonia S, et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J Clin Oncol 2010; 28:1387 - 1394; PMID: 20142592; http://dx.doi.org/10.1200/JCO.2009.25.4029
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-Chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a Dual Src/Abl Kinase Inhibitor with Potent Antitumor Activity in Preclinical Assays. J Med Chem 2004; 47:6658 - 6661; PMID: 15615512; http://dx.doi.org/10.1021/jm049486a
- Baillat G, Siret C, Delamarre E, Luis J. Early adhesion induces interaction of FAK and Fyn in lipid domains and activates raft-dependent Akt signaling in SW480 colon cancer cells. Biochem Biophys Acta 2008; 1783:2323 - 2331; PMID: 18805444; http://dx.doi.org/10.1016/j.bbamcr.2008.08.008
- Donepudi M, Resh MD. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligandindependent EGF receptor activation and signaling. Cell Signal 2008; 20:1359 - 1367; PMID: 18448311; http://dx.doi.org/10.1016/j.cellsig.2008.03.007
- Macdonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res 2005; 46:1061 - 1067; PMID: 15722565; http://dx.doi.org/10.1194/jlr.D400041-JLR200
- Liu Y, Sun R, Wan W, Wang J, Oppenheim JJ, Chen L, et al. The involvement of lipid rafts in epidermal growth factor-induced chemotaxis of breast cancer cells. Mol Membr Biol 2007; 24:91 - 101; PMID: 17453416; http://dx.doi.org/10.1080/10929080600990500