Abstract
Phosphoinositide-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitors are an emerging class of anti-cancer agents. Here, we tested the hypothesis that the dual PI3K/mTOR inhibitor, PI103, could synergize with the chemotherapeutic agent, 5-fluorouracil (5-FU) by inhibiting E2F1, thymidylate synthase (TS) and enhancing DNA damage. Drug combination effects were assessed in gastric cancer cells using the median-effect equation. The specific effects of inhibition of E2F1 and PIK3CA were examined by siRNA, and mTOR by rapamycin exposure. Protein expression and apoptosis pre- and post-treatment was measured using standard methods. PI103 and 5-FU was synergistic in 3 out of 5 gastric cancer cell lines tested. Synergy was associated with PI3KCA mutation, reduced TS and E2F1 protein levels, increased H2AX phosphorylation and apoptosis. E2F1 siRNA enhanced sensitivity to 5-FU only in cells displaying synergy. Excess thymidine exposure converted synergism to antagonism in all cells. Inhibition of PI3K and mTOR alone enhanced 5-FU cytotoxicity in only 2 out of 3 cell lines that displayed synergy each. In AGS cells, PI3K inhibition alone enhanced 5-FU sensitivity as much as dual PI3K/mTOR inhibition. In HGC27 cells, dual inhibition increased 5-FU sensitivity more than single PI3K or mTOR inhibition. Combined PI103 and 5-FU treatment reduced in vivo tumor growth more than treatment with single agents. PI3K/mTOR inhibitors can enhance 5-FU cytotoxicity in vitro and in vivo, especially in PIK3CA mutant tumor cells. Dual, rather than single, PI3K/mTOR inhibitors may combine better with 5-FU due to cellular heterogeneity in sensitivity to PI3K and mTOR inhibition.
Introduction
The phosphoinositide-3-kinase (PI3K)-AKT-mTOR pathway mediates numerous signals in cell growth, motility and survival.Citation1,Citation2 The pathway is aberrantly activated in many human cancers, making it an attractive target for pharmacologic intervention.Citation3 Indeed, many PI3K pathway inhibitors have shown promising anti-tumor activity, with some agents progressing to investigation in clinical trials.Citation3,Citation4
The anti-metabolite, 5-Fluorouracil (5-FU) is currently a mainstay of chemotherapy for many cancer types, including gastric cancer.Citation5 Although treatment with 5-FU provides significant survival benefit,Citation6 large inter-patient variability in response has left much room for improvement in its application.
A major mechanism of 5-FU cytotoxicity is its inhibition of thymidylate synthase (TS),Citation7 an enzyme which generates thymidylate precursors for DNA synthesis. The PI3K pathway activates E2F1 and S6, which upregulate the transcription and translation of TS, respectively.Citation8,Citation9 By suppressing E2F1 and S6 and thereby enhancing TS inhibition, PI3K inhibitors could be predicted to be synergistic with 5-FU. Inhibition of PI3K could also repress its activation of DNA-PK,Citation10 a kinase involved in the repair of DNA damage caused by cytotoxic drugs including 5-FU. The aim of this study was to test the hypothesis that PI3K pathway inhibitors could synergize with 5-FU through their complementary pharmacologic activities.
Results
Synergy between PI103 and 5-FU in gastric cancer cells, especially in those with PIK3CA mutations
The IC50 values of 5-FU and PI103 as single agents, and CI index of drug combinations with 5-FU in the five gastric cancer cell lines studied are summarized in . The PIK3CA mutation and PTEN deletion status of the cell lines according to the COSMIC database (www.sanger.ac.uk) is also summarized. Synergy between 5-FU and PI103 was observed in three cell lines (AGS, IM95, HGC27), additivity in one (MKN45) and antagonism in one (NUGC4). The synergy and antagonism observed in all cases was statistically different from additivity (p < 0.05 compared with 1). The three lines in which synergy were observed had mutations in PIK3CA, while the other two did not. In comparison, there was less of an association between single agent drug sensitivity and mutation/deletion status.
Table 1. IC50 values of 5-FU, PI103, and rapamycin, and combination index values of PI103 and 5-FU in gastric cancer cells
Synergy is associated with an increase in apoptosis
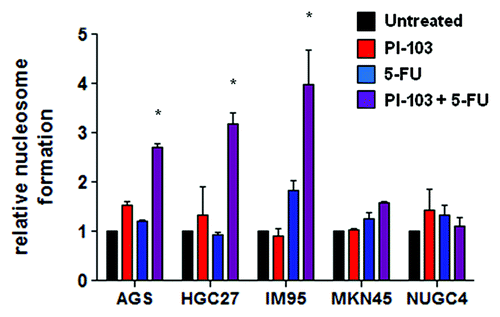
To test whether the observed synergy between PI103 with 5-FU was accompanied by increased apoptosis induction, cells were exposed to IC50 concentrations of either PI103 and 5-FU individually or in combination and measured for nucleosome formation after 24 h. A statistically significant (p < 0.05) increase in cell death was observed with the combination compared with the single agents in all cell lines in which synergy was observed (). Consistent with this, treatment with the combination in NUGC4 cells in which antagonism was observed led to a decrease in apoptosis compared with single agent treatment. In MKN45 cells in which additivity was observed, treatment with the combination led to a minor increase in cell death over that of single agents, consistent with PI103 and 5-FU acting independently.
Figure 1. Effect of PI103, 5-FU and combination on apoptosis induction in gastric cancer cell lines. Nucleosome formation values are displayed as mean ± SD (n = 3) relative to values in untreated control cells. *p < 0.01 compared with controls.
Synergy is associated with decreased E2F1 and TS, and increased pH2AX
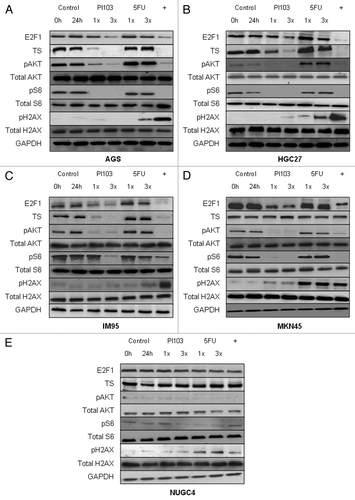
To test whether synergy was associated with reductions in E2F1, TS and increased DNA damage (indicated by pH2AX),Citation11,Citation12 gastric cancer cells were exposed to IC50 and 3xIC50 concentrations of PI103 and 5-FU as single agents or in combination (at IC50 concentrations of both drugs) for 24 h, and the effects on relevant proteins were evaluated by western immunoblotting (). Consistent with our hypothesis, combination treatment led to greater reductions in E2F1 and TS, and elevations in pH2AX when comparing cells in which synergy was observed (AGS, IM95, HGC27) to those in which additivity (MKN45) or antagonism (NUGC4) was observed. These effects were evident whether comparing levels in cells treated with the combination to untreated cells, or to single agent PI103 (for E2F1 and TS) and single agent 5-FU (for TS and pH2AX) treatment. Interestingly, the pattern of H2AX phosphorylation matched the effect of combination in the different cells: highly elevated to 5-FU treatment levels in cells in which synergy was observed (AGS, IM95, NUGC4), similar in cells with additivity (MKN45) and reduced in cells with antagonism (HGC27). In contrast, pAKT and pS6 were similarly reduced by combination and PI103 treatment in all cell lines, suggesting that reductions in pAKT and pS6 may not be a major factor in the observed synergism between PI103 and 5-FU. However, NUGC4 cells displaying antagonism had distinctly low levels of pAKT and pS6 pre- and post-treatment, suggesting that high pAKT and pS6 levels may be pre-requisite for combination synergism or additivity.
Figure 2. Effect of PI103, 5-FU and the combination on protein expression of E2F1, TS, the PI3K/mTOR pathway, and DNA damage in AGS (A), HGC27 (B), IM95 (C), MKN45 (D) and NUGC4 (E) gastric cancer cells. Cells were exposed to 1x and 3x IC50 concentrations of PI103 and 5-FU, and the combination of PI103 and 5-FU (at IC50 levels, denoted with +) for 24 h, and lysates immunoblotted as described in the Methods section. GAPDH was used as a loading control. A representative image of two independent blots, each using independently prepared cell lysates, is displayed.
E2F1 silencing enhances 5-FU sensitivity
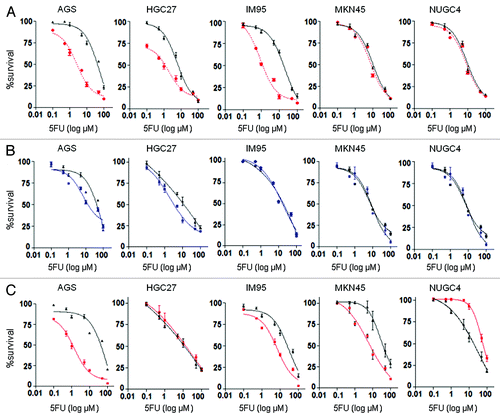
To further test our hypothesis that reduction of E2F1 was a mechanism of combination synergy, concentration-response curves of 5-FU were generated in the presence of E2F1 siRNA or control siRNA. siRNA markedly reduced E2F1 protein levels 24–96 h post-transfection in all cells (data not shown), hence cells were measured 96 h post-transfection and 72 h post-treatment. E2F1 siRNA compared with control siRNA significantly increased the sensitivity of cells to 5-FU in which synergy was observed (5-FU IC50 in AGS after E2F1 vs. control siRNA treatment was 4.1 ± 0.5 µM vs. 61 ± 2 µM respectively; HGC27 1.2 ± 0.3 µM vs. 11 ± 0.5 µM; and IM95 0.9 ± 0.02 µM vs. 21 ± 1.5 µM, all p < 0.05) (), further highlighting the importance of E2F1 inhibition to synergy. In contrast, silencing E2F1 did not have any effect on 5-FU sensitivity in MKN45 (11 ± 0.8 µM vs. 8.0 ± 1.2 µM) and NUGC4 (9.0 ± 1.0 µM vs. 8.6 ± 0.8 µM) cells (). Consistent with these observations, E2F1 siRNA reduced TS levels in cells in which synergy, but not additivity or antagonism, was observed (Fig. S1).
Figure 3. (A) Effect of E2F1 siRNA (red) on 5-FU cytotoxicity in gastric cancer cells. (B) Effect of non-growth inhibitory concentration of rapamycin (blue) on 5-FU cytotoxicity in gastric cancer cells. (C) Effect of PIK3CA siRNA (red) on 5-FU cytotoxicity in gastric cancer cells. Charts display mean (± SD) proportion of cells in three independent experiments.
Excess thymidine reduces synergy between 5-FU and PI103
To test the role of TS in combination synergy, we exposed cells to increasing concentrations of extracellular thymidine and examined its effect on CI values. The use of extracellular thymidine in culture medium for reversing 5-FU cytotoxicty has been previously demonstrated.Citation13 Consistent with TS inhibition being a major factor in combination synergy, the addition of increasing thymidine concentrations increased CI values in all cell lines (). The addition of 10 µM thymidine was sufficient to convert the observed synergy (AGS, HGC27, IM95) and additivity (MKN45) between PI103 and 5-FU to antagonism. In contrast, only the highest concentration of thymidine (1 mM) led to a significant change in 5-FU IC50 values, while PI103 IC50 values were not affected by any of the concentrations of thymidine (data not shown).
Table 2. Effect of exogenous thymidine on PI103 and 5-FU combination
mTOR inhibition enhances 5-FU sensitivity in only 2 out of 3 cell lines in which synergy between 5-FU and PI103 was observed
As PI103 inhibits both PI3K and mTOR,Citation14 we also tested the effect of combining the selective mTOR inhibitor, rapamycin, with 5-FU in the gastric cancer cell lines. The combination of rapamycin and 5-FU was synergistic in AGS and HGC27 cells with CI values at fu0.5 of 0.53 ± 0.02 and 0.25 ± 0.01 respectively (). CI calculations and fixed-ratio experiments could not be performed in IM95, MKN45 and NUGC4 cells as their insensitivity to rapamycin as a single agent prevented determination of their IC50 values.
Consequently, we tested combination effects by measuring changes to 5-FU IC50 values in the presence of a low-fixed dose of 1/5 x IC50 of rapamycin in all cell lines. For the cell lines for which IC50 values could not be calculated (> 100 µM), 1 µM rapamycin was used. This concentration inhibited mTOR signaling (data not shown) and TS without reducing E2F1 (Fig. S2) nor inhibiting proliferation (data not shown). Treatment with rapamycin significantly reduced the IC50 of 5-FU in 2 out of 3 cell lines in which synergy between PI103 and 5-FU was observed (5-FU IC50 in AGS cells treated vs. untreated with rapamycin: 1.4 ± 0.03 µM vs. 81 ± 0.9 µM, p = 0.01; HGC27: 1.8 ± 0.03 µM vs. 21 ± 0.1 µM; p = 0.03) (). Treatment with rapamycin did not change the IC50 of 5-FU of the third cell line displaying synergy (IM95: 29 ± 1.4 µM vs. 30 ± 1.8 µM), nor the cell lines displaying additivity (MKN45: 19 ± 1.2 µM vs. 17 ± 0.98 µM) or antagonism (NUGC4: 15 ± 2 µM vs. 17 ± 1.6 µM).
PIK3CA silencing enhances 5-FU sensitivity in only 2 out of 3 cell lines in which synergy between 5-FU and PI103 was observed
We also transfected siRNA against PIK3CA into cells and measured the IC50 of 5-FU to assess the contribution of PI3K inhibition to the synergy between PI103 and 5-FU. Of the three cell lines in which synergy between PI103 and 5-FU was observed, silencing of PIK3CA significantly reduced the IC50 of 5-FU in AGS (PIK3CA vs. control siRNA: 1.4 ± 0.1 µM vs. 86 ± 1.2 µM, p = 0.01) and IM95 (9.83 ± 0.02 µM vs. 35 ± 0.1 µM, p = 0.02) but not HGC27 cells (11 ± 0.09 µM vs. 11 ± 0.05 µM; p = 0.17) (). The lack of enhancement in HGC27 cells could possibly be explained by the PI3K-p110α selectivity of the PIK3CA siRNA used and the PTEN null status of HGC27 cells (). Cells that are PTEN null are considered to signal more through PI3K-p110β instead of PI3K-p110α.Citation15 Hence, the lack of enhancement of 5-FU sensitivity could be due to a lack of appropriate inhibition of PI3K. Interestingly, the IC50 of 5-FU was also significantly reduced by PIK3CA siRNA in MKN45 cells (7.2 ± 1.8 µM vs. 30.2 ± 1.9 µM, p = 0.01), while it was significantly increased in NUGC4 cells (87 ± 0.10 µM vs. 14 ± 0.03 µM, p = 0.03). Consistent with the above findings, TS levels were reduced following treatment with PIK3CA siRNA in AGS and IM95 cells, but not HGC27 and NUGC4 cells (Fig. S1). However, TS levels were unchanged following PIK3CA siRNA treatment in MKN45 cells, suggesting TS inhibition may not be the only mechanism through which PI3K inhibition may enhance 5-FU sensitivity.
The superiority of dual PI3K/mTOR to single mTOR inhibition is dependent on cell type
To assess the additional value of PI3K inhibition to mTOR inhibition further, we examined the effects of combining PIK3CA siRNA with 1 µM rapamycin in cells in which rapamycin enhanced 5-FU sensitivity (AGS and HGC27). In AGS cells, PIK3CA siRNA alone (5-FU IC50 of treated vs. control treated cells = 1.4 ± 0.1 µM vs. 86 ± 1.2 µM, p = 0.01) and mTOR inhibition alone (1.4 ± 0.03 µM vs. 81 ± 0.9 µM, p = 0.01) both significantly enhanced 5-FU sensitivity (). Treatment with the combination of rapamycin and PIK3CA siRNA did not increase 5-FU sensitivity over PIK3CA siRNA alone (1.4 ± 0.1 µM vs. 1.4 ± 0.03 µM, p = 0.05), indicating PI3K inhibition alone to be as effective as dual inhibition in these cells. This result is consistent with mTOR inhibition being a downstream effect of PI3K inhibition,Citation4 making specific mTOR inhibition redundant.
Figure 4. Evaluation of non-growth inhibitory concentration of rapamycin (rapa) and PIK3CA siRNA on 5-FU sensitivity in AGS (A) and HGC27 (B) cells. Charts display mean (± SD) proportion of cells in three independent experiments. NTC, non-treated control.
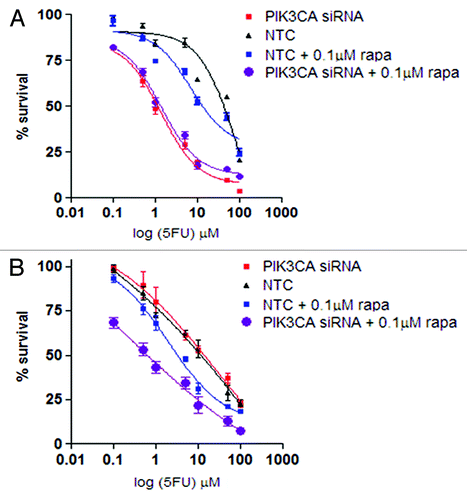
In HGC27 cells, mTOR inhibition alone (1.8 ± 0.03 µM vs. 21 ± 0.1 µM in untreated cells, p = 0.03), but not PI3K inhibition alone (11 ± 0.09 µM vs. 11 ± 0.05 µM in untreated cells, p = 0.17), significantly increased 5-FU sensitivity (), consistent with the findings of earlier experiments (). Nevertheless, the addition of PI3K inhibition to mTOR inhibition further increased the sensitivity of 5-FU compared with mTOR inhibition alone (0.18 ± 0.02 µM vs. 1.8 ± 0.03 µM, p = 0.002), indicating that dual inhibition may be more effective than single inhibition in enhancing 5-FU cytotoxicity in these cells. This is consistent with observations that PI3K inhibition may enhance mTOR inhibition by reducing feedback upregulation of AKT following mTOR inhibition.Citation16 The possibility that this feedback involves upregulation of PI3K-p110α and not PI3K-p110β may explain why PIK3CA siRNA was ineffective as a single treatment, but was effective in combination with mTOR inhibition in these cells.
PI103 and 5-FU in combination but not as single agents reduces tumor burden in mouse tumor xenografts
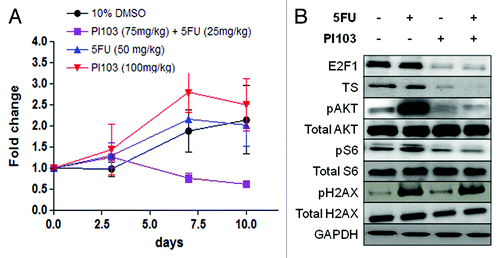
To determine whether PI103 could sensitize tumor cells in vivo to 5-FU, HGC27 xenografts were established in NOD/SCID mice and treated with PI103 and 5-FU in combination or as single agents, or 10% DMSO as a control. Consistent with the in vitro findings, the combination substantially reduced HGC27 xenograft tumor growth (). In contrast, no apparent effect on tumor xenograft growth was observed with PI103 or 5-FU treatment as a single agent compared with control. This is despite the dosage of the single agents (PI103 at 100 mg/kg, 5-FU at 50 mg/kg) being administered at a higher dosage than the combination (PI103 and 5-FU at 75 mg/kg and 25 mg/kg respectively). Also consistent was the reduction of E2F1, TS, pAKT and pS6 levels as well as an increase in pH2AX following treatment with the combination ().
Figure 5. (A) Tumor sizes of mouse xenograft models of HGC27 cells treated with PI103, 5-FU, the combination and control (10% DMSO). The chart displays mean (± SD) size of tumors (n = 3). (B) Western immunoblot showing effect of PI103, 5-FU and the combination on levels of E2F1, TS, the PI3K/mTOR pathway proteins and H2AX in HGC27 xenograft tumor samples. A representative of two independent blots from independently prepared tissue homogenates is displayed.
Discussion
The frequency of aberrant PI3K activation in oncogenesis provides a rational basis for combining PI3K pathway inhibitors with other anti-cancer therapies.Citation3,Citation17 Recent studies have reported synergy between PI103 and doxorubicin in glioblastoma cells,Citation18 and with radiotherapy in colorectal cancer cells.Citation19 Other studies have shown that mTOR inhibitors are synergistic with 5-FU in hepatocellularCitation20 and gastricCitation21,Citation22 cancer cells. The results of the present study have shown for the first time that dual PI3K/mTOR inhibition can synergize with 5-FU both in vitro () and in vivo () in gastric cancer cells. These findings add to the growing body of evidence indicating that PI3K pathway inhibitors could be useful agents for enhancing current treatment strategies.
Consistent with our hypothesis, we found synergy was associated with reductions in E2F1 and TS levels and increased H2AX phosporylation (). Indeed, combination treatment enhanced phosphorylation of H2AX following 5-FU treatment and reduction of E2F1 by PI103 (), in line with the synergy being more than a simple complement of drug activities. Synergy was also associated with increased apoptosis, consistent with the involvement of the PI3K pathway in pro-survival signaling,Citation4 and the cytotoxic activity of 5-FU.Citation5 These results support the notion that the combination could function by reducing E2F1 (through reduction of PI3K activation of E2F1Citation8 by PI103) and TS activity (through reduction of TS transcription by E2F1,Citation9 and 5-FU binding),Citation5 enhancing DNA damage (through incorporation of 5-FU into DNA,Citation5 and inhibition of DNA-PK and DNA repair by PI103Citation10) and inducing apoptosis. This notion is further supported by the more direct evidence of enhancement of 5-FU sensitivity () and reduction of TS (Fig. S1) by E2F1 silencing in only cells with synergy, and the conversion of synergistic combination effects to antagonism through excess thymidine treatment ().
In comparison, AKT and S6 phosphorylation appeared to have a lesser role in mediating synergy, as their reduction appeared to be similar between cell lines displaying synergy and additivity (). Nevertheless, NUGC4 cells displaying antagonism had a distinct relative lack of AKT and S6 phosphorylation pre- and post-treatment, suggesting that elevated basal levels of these phosphorylation events could be at least a pre-requisite for combination additivity and synergy. This may be related to the association between PIK3CA mutation and synergy observed in this study. The PIK3CA mutations observed in these cell lines constitutively activate the PI3K pathway,Citation23 and this type of activation is commonly associated with a mutation-“addicted” cell phenotype.Citation4 Cellular “addiction” to mutations has been implicated in the enhanced potency of their respective targeted agents in many cases, with the sensitivity of EGFR-mutated cells to EGFR inhibitors being a prominent example.Citation24,Citation25 Taken together, these results highlight the possibility that cellular addiction to PI3K signaling could also be a significant determinant of combination synergy.
A link between PIK3CA mutations and combination synergy would present PIK3CA mutations as a useful biomarker for predicting sensitivity to the combination of PI3K pathway inhibitors and 5-FU. The role of PIK3CA mutations in predicting sensitivity to PI3K pathway inhibitors is currently controversial, with most studies focused on single-agent inhibitory activity.Citation4 In the current study, PIK3CA mutations were more closely associated with combination synergy than single agent PI103, 5-FU or rapamycin activity (), suggesting that 5-FU may accentuate signaling or “addiction” differences between PIK3CA mutant and wild-type cells. The possibility that these differences could derive from the DNA-damaging activity of 5-FU, presents the enticing prospect that the predictive significance of PIK3CA mutations could extend to combinations of PI3K inhibitors and cytotoxic agents other than 5-FU. The result also highlights the concept that predicting combination synergy may be a more viable goal than predicting single agent activity for PI3K pathway inhibitors.
To better understand the roles of PI3K and mTOR inhibition in synergy between PI103 and 5-FU, and assess the merits of single and dual inhibition, we also assessed the individual () and combined effects () of PIK3CA siRNA and rapamycin treatment on 5-FU sensitivity. Our results revealed variability in the abilities of PI3K and mTOR inhibition alone (), as well the sufficiency of single and dual inhibition (), to enhance 5-FU sensitivity between cell types. The variability in single PI3K and mTOR inhibition effects supports that dual inhibition may be the optimal approach for achieving synergy with 5-FU in the widest range of cells. The conclusion is consistent with the logic that inhibition of two frequently activated proteins would be more effective than the inhibition of one. The conclusion is also consistent with the superior activity of PI103 to rapamycin observed in glioblastoma cells.Citation26,Citation27 In this study, superiority of PI103 was attributed to the reduction of the feedback induction of PI3K from mTOR inhibition.
Nevertheless, PI3K inhibition alone was able to increase 5-FU sensitivity in MKN45 cells () in which a lack of enhancement by rapamycin, and only additivity between PI103 and 5-FU was observed (). Reduction of TS did not accompany the enhancement of 5-FU sensitivity in MKN45 cells (Fig. S2), highlighting the possibility that PI3K inhibition could enhance 5-FU sensitivity through mechanisms other than TS inhibition. Moreover, the inability of PI3K inhibition to enhance 5-FU sensitivity in HGC27 cells () could potentially be accounted for by the PTEN null status of the cells (), the preference for PTEN null cells for p110β signaling,Citation15 and the selectivity of the PI3K siRNA used for p110α. Hence, it remains possible that PI3K inhibition alone could be at least as effective in combining with 5-FU than PI103. This would be of interest to test once selective PI3K inhibitors become more readily available.
In summary, the results of this study have suggested that PI3K pathway inhibition may be a promising, rationally-based approach for enhancing 5-FU chemotherapy. In addition, PIK3CA mutations were identified as potentially useful biomarkers for predicting synergy. Results indicated that dual PI3K/mTOR inhibition may provide a greater scope of synergism with 5-FU to single PI3K or mTOR inhibition, although this conclusion awaits further testing with isoform selective PI3K inhibitors. Additional work is required in a wider range of cancer types and models to further characterize the potential scope of synergy, to further optimize the combination effects by different scheduling, and to test the potential of PI3K pathway inhibitors to sensitize other current treatments.
Materials and Methods
Cell culture
Gastric cancer cells were obtained from American Type Culture Collection (ATCC), or Japanese Collection of Research Bioresources (JCRB). Cells were routinely cultured in DMEM or RPMI1740 supplemented with 10% fetal bovine serum (Invitrogen), Fifty-thousand units penicillin and 50 mg streptomycin (Sigma) at 37°C in a humidified atmosphere containing 5% CO2.
Drug sensitivity analysis
PI103 and rapamycin were obtained from Cayman Chemicals, and 5-FU from Sigma. All stock solutions were prepared in DMSO (MP Biomedicals) at a final concentration in culture media of 0.25% (v/v). Cells in 90 µl medium were seeded (3,000 cells/well) onto 96-well microtiter plates (Nunc). After 24 h, 10 µl of medium containing compounds in graded concentrations ranging from 0.1–1,000 µM was added to the wells. The effect on cell numbers was assessed using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega) (MTS assay) at 72 h post-treatment. The IC50 was calculated as the drug concentration that inhibited cell proliferation by 50% compared with vehicle controls as previously described.Citation28
Drug combination analysis
The effect of combining compounds was evaluated using the median-effect equation and combination index (CI) method of Chou and Talalay.Citation29 Two compounds were combined at a fixed 1:1 ratio of their IC50 simultaneously for 72 h. For each level of fraction unaffected (fu), a CI was calculated as follows: CI = (D)1/(Df)1 + (D)2/(Df)2 + [(D)1(Df)2/(Df)1(Df)2], where (D)1 and (D)2 are the concentrations of the combination required to produce fu, and (Df)1 and (Df)2 are the concentrations of the individual drugs required to produce fu. Data giving linear regression coefficients (r2) of median-effect plots < 0.95 were excluded. CI values of < 1, 1 and > 1 were considered to indicate synergy, additivity and antagonism respectively. CI values with the non-exclusive assumption have been reported.
Apoptosis measurement
Apoptosis was measured using the Cell Death ELISA (Roche) kit. Cells were plated in 96-well plates (3,000 cells/well) and in the following day were treated with drug or solvent in a volume adjusted to 200 μL with 10% FCS/RPMI. After 24 h, nucleosomes were quantified according to the manufacturer's instructions.
Western blotting
Cells were harvested and lysed in 1× denaturing lysis buffer (10 mM TRIS-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin) (New England Biolabs). Samples were then centrifuged at 1,500 g for 10 min at 4°C and assayed for protein concentration using the BCA protein estimation kit (Pierce Biotechnology). Equal amount of protein (50 µg) were separated by electrophoresis through a 4–12% Tris-Glycine gel (Invitrogen). Proteins were then transferred onto 0.45 µm nitrocellulose pore membranes and probed with antibodies. The following antibodies were used: sheep polyclonal antibodies against TS (Abcam) and GAPDH (Millipore), mouse monoclonal antibodies against pAKT(Ser473), AKT, pS6 (Ser235/236), S6, and p-Histone H2AX (Ser139), rabbit polyclonal antibodies against Histone H2AX and E2F1 (New England Biolabs) and appropriate horseradish peroxidase-conjugated secondary antibodies (New England Biolabs and Abcam).
E2F1 and PIK3CA siRNA treatment
Cells were transfected with siGENOME SMARTpool siRNA or siGENOME Non-Targeting pool siRNA (Thermo Fisher Scientific) against E2F1 or PIK3CA respectively at concentrations of 20 nM, and exposed to a range of concentrations of 5-FU 24 h post-transfection. The effect on cell numbers was measured at 72 h post-transfection by the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega).
Thymidine exposure
Exponentially growing cells were seeded into 96-well plates at 3,000 cells per/well and allowed to attach for 24 h before the addition of 10 µM, 100 µM or 1 mM thymidine (Sigma) for 24 h. The media was changed after 24 h before 5-FU addition, and then assessment of cell numbers using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega) at 72 h.
Rapamycin treatment
Cells were either exposed to one-fifth of the IC50 or 1 µM (for cells in which the rapamycin IC50 was greater than 100 µM) of rapamycin, along with graded concentrations of 5-FU, before cell numbers were measured by the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega) at 72 h post-treatment.
Tumour xenograft drug response
Twelve female NOD/SCID mice (5–6 weeks old) were obtained from Animal Resources Center. The animals were housed under a 12:12 h light-dark regimen, and were fed a standard pellet diet ad libitum and provided free access to water. The mice were subcutaneously injected with approximately 4 million HGC27 cells in 0.1 ml of media on the right flank. When tumors reached 100 mm3, mice were randomly assigned to 2 × 2 factorial design treatment or control groups. All drugs were dissolved in 100% DMSO to attain the following concentrations: PI103 (100 mg/kg), 5-FU (50 mg/kg), PI103 and 5-FU (75 mg/kg and 25 mg/kg respectively) and a 10% DMSO control. Caliper measurements of tumors were made twice weekly and tumor volumes were calculated according to the following formula: (L × W)2/2, where L and W are the length and width of the tumor respectively. Animals were euthanized by CO2 asphyxiation when their tumors reached 1,500 mm3. Tumor xenografts were collected at the final treatment day from euthanized animals. The samples were snap frozen using liquid nitrogen and stored at -80°C until processed. All animal experiments were performed according to the guidelines on the care and use of animals for scientific purposes, and experimental protocols approved by the animal ethics committee of the National University of Singapore.
Statistics
A two-tailed, one-sample t-test was used to compare experimental CI at fu0.5 values with the predicted value for additivity of 1. For apoptosis assays, one-way ANOVA, followed by Dunn’s Multiple Comparison test, was performed to compare for differences between control and treatment arms. Differences in 5-FU IC50 between siRNA and non-targeting controls were assessed using a two-tailed paired sample t-test. All statistical analyses were performed using GraphPad Prism 4.00 software (GraphPad Software Inc.). Statistical significance was considered if p < 0.05.
| Abbreviations: | ||
| 5-FU | = | 5-fluorouracil |
| BCA | = | bicinchoninic acid |
| CI | = | combination index |
| DNA-PK | = | DNA-dependent protein kinase |
| E2F1 | = | E2 transcription factor 1 |
| fu | = | fraction unaffected |
| IC50 | = | inhibitory concentration 50 |
| mTOR | = | mammalian target of rapamycin |
| PI3K | = | phosphatidylinositol 3 kinase |
| siRNA | = | short interfering RNA |
| TS | = | thymidylate synthase |
Additional material
Download Zip (177.9 KB)Acknowledgments
We thank Ms. Marie Loh for her assistance with statistics, and Dr. Barry Iacopetta for his review of the manuscript. This work was supported by a research grant from the National Medical Research Council of Singapore (NMRC/1123/2007).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Michl P, Downward J. Mechanisms of disease: PI3K/AKT signaling in gastrointestinal cancers. Z Gastroenterol 2005; 43:1133 - 9; http://dx.doi.org/10.1055/s-2005-858638; PMID: 16220453
- Arkenau HT. Gastric cancer in the era of molecularly targeted agents: current drug development strategies. J Cancer Res Clin Oncol 2009; 135:855 - 66; http://dx.doi.org/10.1007/s00432-009-0583-7; PMID: 19363621
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010; 28:1075 - 83; http://dx.doi.org/10.1200/JCO.2009.25.3641; PMID: 20085938
- Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res 2010; 70:2146 - 57; http://dx.doi.org/10.1158/0008-5472.CAN-09-4355; PMID: 20179189
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3:330 - 8; http://dx.doi.org/10.1038/nrc1074; PMID: 12724731
- Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV, et al. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer Res 1995; 55:1407 - 12; PMID: 7882343
- Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta 2002; 1587:194 - 205; PMID: 12084461
- Dynlacht BD. Live or let die: E2F1 and PI3K pathways intersect to make life or death decisions. Cancer Cell 2008; 13:1 - 2; http://dx.doi.org/10.1016/j.ccr.2007.12.017; PMID: 18167332
- Kasahara M, Takahashi Y, Nagata T, Asai S, Eguchi T, Ishii Y, et al. Thymidylate synthase expression correlates closely with E2F1 expression in colon cancer. Clin Cancer Res 2000; 6:2707 - 11; PMID: 10914714
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006; 125:733 - 47; http://dx.doi.org/10.1016/j.cell.2006.03.035; PMID: 16647110
- Clingen PH, Wu JY, Miller J, Mistry N, Chin F, Wynne P, et al. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem Pharmacol 2008; 76:19 - 27; http://dx.doi.org/10.1016/j.bcp.2008.03.025; PMID: 18508035
- Ikeda M, Kurose A, Takatori E, Sugiyama T, Traganos F, Darzynkiewicz Z, et al. DNA damage detected with gammaH2AX in endometrioid adenocarcinoma cell lines. Int J Oncol 2010; 36:1081 - 8; PMID: 20372780
- Peters GJ, Laurensse E, Leyva A, Pinedo HM. Purine nucleosides as cell-specific modulators of 5-fluorouracil metabolism and cytotoxicity. Eur J Cancer Clin Oncol 1987; 23:1869 - 81; http://dx.doi.org/10.1016/0277-5379(87)90053-8; PMID: 3436351
- Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 2007; 67:5840 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-06-4615; PMID: 17575152
- Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J 2008; 415:97 - 110; http://dx.doi.org/10.1042/BJ20080639; PMID: 18498248
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26:1932 - 40; http://dx.doi.org/10.1038/sj.onc.1209990; PMID: 17001314
- Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol 2008; 8:393 - 412; http://dx.doi.org/10.1016/j.coph.2008.08.004; PMID: 18721898
- Westhoff MA, Kandenwein JA, Karl S, Vellanki SH, Braun V, Eramo A, et al. The pyridinylfuranopyrimidine inhibitor, PI-103, chemosensitizes glioblastoma cells for apoptosis by inhibiting DNA repair. Oncogene 2009; 28:3586 - 96; http://dx.doi.org/10.1038/onc.2009.215; PMID: 19633683
- Prevo R, Deutsch E, Sampson O, Diplexcito J, Cengel K, Harper J, et al. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI-103 enhances tumor radiosensitivity. Cancer Res 2008; 68:5915 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-08-0757; PMID: 18632646
- Bu X, Le C, Jia F, Guo X, Zhang L, Zhang B, et al. Synergistic effect of mTOR inhibitor rapamycin and fluorouracil in inducing apoptosis and cell senescence in hepatocarcinoma cells. Cancer Biol Ther 2008; 7:392 - 6; http://dx.doi.org/10.4161/cbt.7.3.5366; PMID: 18075305
- Lee KH, Hur HS, Im SA, Lee J, Kim HP, Yoon YK, et al. RAD001 shows activity against gastric cancer cells and overcomes 5-FU resistance by downregulating thymidylate synthase. Cancer Lett 2010; 299:22 - 8; http://dx.doi.org/10.1016/j.canlet.2010.07.020; PMID: 20727673
- Matsuzaki T, Yashiro M, Kaizaki R, Yasuda K, Doi Y, Sawada T, et al. Synergistic antiproliferative effect of mTOR inhibitors in combination with 5-fluorouracil in scirrhous gastric cancer. Cancer Sci 2009; 100:2402 - 10; http://dx.doi.org/10.1111/j.1349-7006.2009.01315.x; PMID: 19764996
- Liu Z, Roberts TM. Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle 2006; 5:675 - 7; http://dx.doi.org/10.4161/cc.5.7.2605; PMID: 16627990
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 500; http://dx.doi.org/10.1126/science.1099314; PMID: 15118125
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 39; http://dx.doi.org/10.1056/NEJMoa040938; PMID: 15118073
- Guillard S, Clarke PA, Te Poele R, Mohri Z, Bjerke L, Valenti M, et al. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle 2009; 8:443 - 53; http://dx.doi.org/10.4161/cc.8.3.7643; PMID: 19177002
- Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res 2007; 67:7960 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-07-2154; PMID: 17804702
- Tan WL, Bhattacharya B, Loh M, Balasubramanian I, Akram M, Dong D, et al. Low cytosine triphosphate synthase 2 expression renders resistance to 5-fluorouracil in colorectal cancer. Cancer Biol Ther 2011; 11:599 - 608; http://dx.doi.org/10.4161/cbt.11.6.14670; PMID: 21378502
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27 - 55; http://dx.doi.org/10.1016/0065-2571(84)90007-4; PMID: 6382953