Abstract
The development of systemic therapy drug resistance for breast cancer treatment is an ongoing problem, thus, so are the potential molecular mechanisms of it. AZD6244 is a novel ATP-uncompetitive inhibitor to MAP/ERK kinase (MEK) 1/2 which has been demonstrated to be potent, selective and safe in the clinical trials and previous studies. However, the precise role of resistance to AZD6244 is largely unknown. We and other groups have reported that the novel oncogene Metadherin (MTDH) is associated with multiple drug resistance, but there is no report about its role in treatment of AZD6244. Here we report that the resistance to AZD6244 can be reserved by downregulating MTDH in breast cancer cell lines. When the MTDH was downregulated, the breast cancer cells exhibited a significantly increased sensitivity to AZD6244 as measured by MTT assay. After treated with AZD6244 the MTDH-knockdown cells showed more apoptosis rate and growth inhibition. We also showed that knockdown of MTDH cannot only increase expression of FOXO3a but also activate it by promoting its translocation via MTDH/ERK1/2/FOXO3a pathway rather than MTDH/AKT/FOXO3a pathway. In conclusion knockdown MTDH can enhance the breast cancer cells sensitivity to AZD6244 via regulating the expression and activity of FOXO3a. These indicate us that MTDH is a candidate marker to predict the clinical efficacy of AZD6244 and targeting MTDH could overcome the resistance to AZD6244 in breast cancer cells.
Keywords: :
Introduction
Breast cancer is the most common female cancer in the world and has become the first cause of cancer death in women of developing countries.Citation1,Citation2 Despite the recent advances made in breast cancer treatment utilizing improved local and systemic treatments to improve disease-free and overall survival in the general female breast cancer population, about one-third of patients diagnosed with early-stage breast cancer eventually die of recurrent disease due to the development of drug resistance to systemic therapy.Citation3,Citation4 The specific nature in which certain breast cancers develop chemotherapy resistance, the biologic mechanisms of action, and the role of genes involved in the transport of anticancer agents that lead to resistance remain unclear. In order to overcome drug resistance to breast cancer therapies and develop successful therapeutic strategies to overcome drug resistance, a better understanding of the underlying molecular mechanisms is mandatory. The magnitude of the resistance problem has prompted both pre-clinical and clinical efforts at identifying underlying prognostic features for drug resistance and potential mechanisms of action.
The understanding of certain oncogenes and their signal transduction cascades which lead to tumor development, proliferation, cell death, angiogenesis and metastasisCitation5 has provided for the development of targeted therapies for receptors such as estrogen receptor, HER2 (human epidermal growth factor receptor) and VEGF (vascular endothelial growth factor). Currently, a large percentage of breast cancer patients will receive some form of targeted therapy, which have been demonstrated to improve breast cancer outcomes, including local control, distant-disease free survival, and overall survival.Citation6-Citation8
Unfortunately, a significant number of patients develop drug resistance to these agents. The mechanism of drug resistance is likely to be heterogenous and multi-factoral and thus, extensive investigation of individual agents and pathways leading to drug resistance will need to be conducted.
Activated extracellular signal-regulated kinase (ERK1/2) in breast cancer has been reported to be essential for cell proliferation, differentiation, survival, tumor angiogenesis, motility and invasion,Citation10-Citation13 and is activated by MEK1/2 via phosphorylation.Citation9 It has been recently described that Selumetinib (AZD6244, ARRY-142886), a novel potent oral inhibitor of MEK1/2 kinases, is an active inhibitor of the RAF/MEK/ERK pathway, and thus has potential to block cell proliferation, survival, metastasis and/or invasion.Citation14-Citation16 In preliminary clinical trials, AZD6244 has demonstrated activity in various tumors including colorectal cancer, lung cancer, acute myelogenous, breast cancer.Citation14,Citation17-Citation20 AZD6244 selectively targets cancer cells and thus the treatment-related toxicity is reported to be low.Citation21 Some phase I/II clinical trials have reported AZD6244 to be relatively safe, efficacious and promising for solid tumors.Citation22-Citation24 Several preliminary investigations suggest the efficacy of AZD6244 to be comparable to capecitabine with regards to disease-free/progression-free survival.Citation25 Currently, these pilot studies remain investigational.Citation26
Despite the preliminary promising results of AZD6244, development of drug resistance remains an ongoing problem. While several molecular markers such as BRAF, AKT, FOXO3aCitation20,Citation21,Citation27 have been implicated in potentiating this resistance, other markers need to be explored.
Metadherin (MTDH, also known as AEG-1 and Lyric) is a novel multifunctional oncogene which was first cloned by subtraction hybridization in 2002.Citation28 It has been found to be overexpressed in various cancers including squamous cell carcinoma of the esophagus, breast carcinomas, melanomas, hepatocellular carcinoma, and epithelial ovarian cancers.Citation29-Citation31 Data suggest that MTDH plays an important role in promoting cell proliferation, migration, tumor metastasis and angiogenesis via the regulation of multiple molecular pathways including PI3K/AKT, NF-κB, MAPK, Wnt, vascular endothelial growth factor (VEGF), and the transcription factor family (FOXO1, FOXO3a etc.).Citation31-Citation37 In breast cancer specifically, our group has previously demonstrated that greater than 40% of tumors overexpress MTDH, that overexpression of MTDH correlates with worse clinical outcomes,Citation32,Citation38 that MTDH enhance the invasiveness of cells by inducing epithelial to mesenchymal transition (EMT),Citation39 and lastly, that MTDH has the potential to induce multiple drug resistance in breast cancer (doxorubicin, paclitaxel, cisplatin).Citation40 However, the mechanism of MTDH involvement in the treatment of AZD6244 in breast cancer has yet to be been explored. In this study, we utilized a triple negative breast cancer cell line that is insensitive to AZD6244Citation22,Citation41 to demonstrate that knockdown MTDH can distinctly sensitize breast cancer cells to AZD6244 treatment. We also explored a potential mechanism that MTDH may affect the sensitivity to AZD6244: by upregulating the expression of FOXO3a and activating it by promoting translocation.
Results
Knockdown MTDH sensitizes MDA-MB-231 cells to AZD6244
To test the hypothesis that MTDH plays an important role in the sensitivity to AZD6244 in breast cancer, the short hairpin RNA (shRNA) targeting MTDH gene was designed and inserted into the pSUPER.retro.puro plasmid, then transfected into MDA-MB-231 cells. Stable cell lines were selected by puromysin, then MTDH expression was measured using RNA levels (real time PCR) and protein levels (western blot) as shown in . The 231-prpM cells demonstrated significantly less MTDH expression in both mRNA and protein levels compared with the control 231-prpn.
Figure 1. Knockdown of MTDH sensitizes MDA-MB-231 cells to AZD6244. (A) Relative MTDH mRNA and protein levels in the breast cancer cell line MDA-MB-231 transfected with prpn and prpM vector, respectively. Error bars represent means ± SD of triplicate measurements. (B) Cell viability was evaluated using the MTT assay after treated cells with various concentration of AZD6244 for 96 h. Marked difference of survival fraction was observed between 231-prpn and 231-prpM cells (p = 0.005). Data shown are the means ± SD of three independent experiments. (C and D) Percentages of G0/G1 cells of each treated sample were measured using flow cytometric analysis for PI staining for cell cycle after 24 h treatment with AZD6244 0.5uM while the cells without treatment as control. (Data represent mean ± SD and results are representative of three independent experiments, *p < 0.01 vs. respect control). (E and F) 231-prpn and 231-prpM cells were treated with AZD6244 10 μM for 96 h and subjected to sub-G1 analysis using flow cytometric analysis with the cells without treatment as control. Cell death was measured by percentages of sub-G1 cells. Data represent mean ± SD and results are representative of three independent experiments, *p < 0.01 vs. respect control.
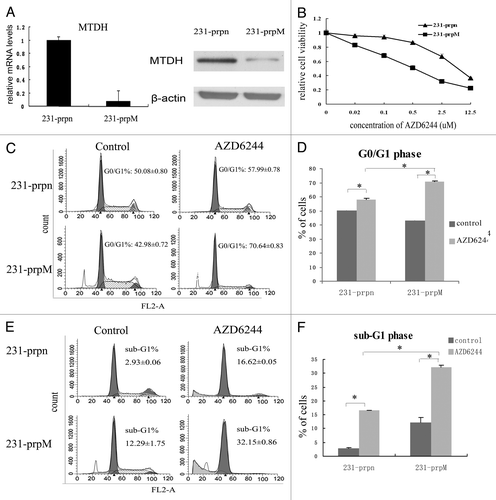
Subsequently, the two selected cell lines were treated with various concentrations of AZD6244 for 96 h, and MTT was used to quantitate changes in viability. The observed changes in sensitivity for the cell lines with MTDH are shown in . While AZD6244 reduced the cell viability in a dose-dependent manner in both cell lines, the 231-prpM cells were significantly more sensitive to AZD6244 growth inhibition (IC50 from 6.04 ± 0.08 μM in 231-prpn to 0.52 ± 0.04 μM in 231-prpM, p = 0.005).
MTDH knockdown enhanced the AZD6244 induced G0/G1 cell cycle arrest and cell apoptosis
To further validate the effect of MTDH on cell sensitivity to AZD6244, we tested whether knockdown MTDH sensitizes cell cycle arrest and apoptosis after exposure to AZD6244. Indeed, the reduced expression of MTDH led to a higher percentage of cells accumulating in G0/G-phase (70.64–42.98 vs. 57.99–50.08%) () and more sub-G1 phase (32.15 vs. 16.62%) (). These results indicated that MTDH knockdown enhanced cells respond to AZD6244 in the G0/G-phase arrest and promoted cell apoptosis.
MTDH knockdown increased expression of FOXO3a and promoted its nuclear translocation through regulating ERK1/2 but not AKT activity
To identify a potential mechanism of action, protein expression of ERK1/2, p-ERK1/2 was measured. The expression of AKT, p-AKT and FOXO3a were also tested with the rationale that expression levels and activation of AKT have been reported to be important in mediating the resistance to AZD6244Citation42 and share with ERK1/2 the same downstream molecular-the transcription factor FOXO3a.Citation43 Furthermore, FOXO3a can be induced by AZD6244 and make cells more sensitive to this drug.Citation21 These findings are shown in : MTDH knockdown (A) inhibited the activity of ERK1/2 (B) activated AKT and (C) upregulated the expression of FOXO3a. It has been reported that activate either ERK1/2 or AKT can make FOXO3a inactive and degradation via phosphorylation.Citation44,Citation45 Based on these findings, we further postulated that the upregulation of FOXO3a was through the reduced activity of ERK.
Figure 2. MTDH knockdown increases expression of FOXO3a and promote its nuclear translocation. (A) The FOXO3a expression in protein level was examined by western blot, both ERK1/2 and AKT activity were measured by the level of p-ERK1/2 and p-AKT. (B) The expression of p-FOXO3a (Ser 294) was measured by western blot. The phosphorylation level of FOXO3a was measured by the ratio of p-FOXO3a/FOXO3a. (Data represent mean ± SD and results are representative of three independent experiments, *p < 0.01 vs. 231-prpn.) (C) The expression of FOXO3a in cytoplasma and nucleus was measured by western blot. (D and E) The efficiency of FOXO3a knockdown by siRNA in 231-prpn and 231-prpM cells was tested by western blot after transfected cells for 24 h. The changes of 231-prpM and 231-prpn cells sensitivity to AZD6244 was measured by MTT after treated cells for 96 h.
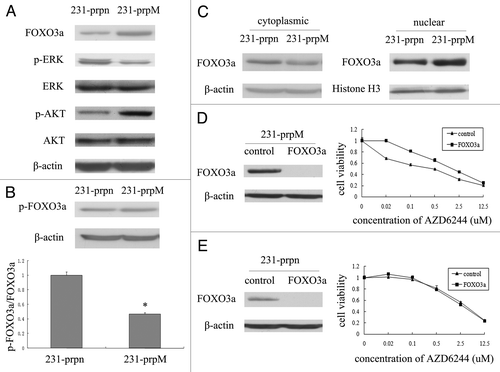
Previous study has shown that ERK interacts with and phosphorylates FOXO3a at Ser 294, Ser 344 and Ser 425 to promote its cytoplasmic translocation and degradation via MDM2.Citation44 To test our hypothesis, we measured the p-FOXO3a (Ser 294) expression. The results demonstrate that while MTDH knockdown did not change the expression level of p-FOXO3a, a reduction in the ratio between p-FOXO3a and total FOXO3a was observed (). To assess whether MTDH knockdown activates FOXO3a via enhancment of its translocation, we tested the expression of FOXO3a in both nuclear and cytoplasma in 231-prpn and 231-prpM cells and found that the expression of FOXO3a was decreased in cytoplasm but increased in the nuclear after MTDH knockdown (). These results indicated the enhanced expression and nuclear translocation of FOXO3a may be achieved by the inactivation of ERK1/2.
FOXO3a knockdown reverse the sensitivity to AZD6244 induced by MTDH knockdown
To demonstrate the role of FOXO3a in MTDH knockdown induced sensitivity to AZD6244, we utilized knockdown FOXO3a in both 231-prpn and 231-prpM cells using siRNA to test the knockdown efficiency by western blot. Additionally, we used MTT assay to measure the changes of sensitivity to AZD6244 with the negative siRNA transfected cells as control. As shown in , in 231-prpM cells, FOXO3a knockdown resulted in more cellular resistance to AZD6244 (p = 0.018), which was not observed in 231-prpn cells ().
Discussion
Previous studies have suggested that AZD6244 is potent, selective and safe in various tumors but the emergence of resistance may limit its clinical applicability.Citation14,Citation17-Citation20 Understanding the mechanism(s) of drug-resistance may allow us to identify methods for overcoming the major limitation of this drug. Several studies have suggested that cell resistance to AZD6244 is associated with wild-type BRAF,Citation20 high level expression and activity of AKT,Citation27,Citation42 reduced FOXO3a activity,Citation21 and upregulation of Wnt pathway.Citation46 We contribute to the existing limited literature with data suggesting that the MTDH gene may also contribute to resistance in AZD6244.
MTDH is involved in regulating cell proliferation, migration, tumor metastasis and angiogenesisCitation32,Citation33 in malignant tumors via multiple pathways including ERK1/2 and AKT pathways. Our group has previously demonstrated the association between MTDH and drug resistance,Citation31,Citation32,Citation47 and found preliminary data to suggest that MTDH may contribute to the drug resistance to AZD6244. In this study, we found MTDH knockdown can significantly sensitize MDA-MB-231 cells to AZD6244. As AZD6244 has been reported to induce cell cycle arrest and apoptosisCitation21 we further demonstrated an increased cell cycle accumulation in G0/G1-phase and increased sub-G1 rate in MTDH knockdown cells after treatment with AZD6244.
FOXO3a is a transcription factor which plays a crucial role in regulation of the cell cycle and cell apoptosis. Furthermore, FOXO3a has been reported to promote cell sensitivity to AZD6244.Citation21 The activation of FOXO3a occurs via nuclear translocation. Cytoplasmic translocation or proteasomal degradation of FOXO3a is through phosphorylation that is regulated by ERK1/2, AKT or IKK activity in response to growth factor and insulin stimulation. All of the ERK1/2 and the AKT pathways are reported to be regulated by MTDH.Citation44,Citation45,Citation48
Our first hypothesis was that MTDH knockdown induced sensitivity to AZD6244 through FOXO3a activation. We tested our hypothesis by measuring the expression and activation change of the AKT, ERK1/2 and FOXO3a and found that knockdown MTDH in MDA-MB-231 can inactivate ERK1/2, activate AKT, upregulate and promote nuclear translocation of FOXO3a. Furthermore, we hypothesized that the increased expression of FOXO3a was induced by its enhanced stability via decreased ERK1/2 activity. Our results supported this hypothesis in that the decreased activity of ERK1/2 by knockdown MTDH did, in fact, reduce the phosphorylation level of FOXO3a at Ser 294 which is the specific phosphorylation site of FOXO3a interacting with ERK1/2. These findings suggest that MTDH knockdown not only increases expression of FOXO3a but also activates it by promoting its nuclear translocation which may occur through the MTDH/ERK1/2/FOXO3a pathway.
Lastly, we hypothesized that the activity of FOXO3a plays a crucial role in the sensitivity to AZD6244 induced by MTDH knockdown. Using siRNA targeted FOXO3a to inhibit its activity, we tested the changes of sensitivity to AZD6244 in both 231-prpn and 231-prpM cells. Our findings suggest that FOXO3a plays an important role in cells sensitivity to AZD6244 in MTDH knockdown cells 231-prpM but not in the 231-prpn cells, which express low levels of FOXO3a. These results indicate that the effect of FOXO3a in MTDH knockdown induced cell sensitivity to AZD6244 is related to MTDH. Whether the MTDH oncogene affects the cellular response to AZD6244 by additional mechanisms remains to be addressed.
In recent years, the identification of specific genes, proteins and biological molecular pathways that are associated with the development and progression of breast cancer have been identified which have ultimately become targets for therapy.Citation6,Citation7 MTDH is an important oncogene in breast cancer which has been found to play a critical role in tumor progression and thus prognosis. In this study, we have identified MTDH as a potential novel target for drug resistance to AZD6244, a finding which has not been previously reported. Our findings suggest that regulation of MTDH pathways may provide a promising approach to sensitizing breast cancer cells to AZD6244.
Materials and Methods
Reagents
AZD6244 (Selleck Chemicals, S1008) was dissolved in dimethyl sulphoxide (DMSO, Sangon Biotech, 0231) to a final concentration of 50 mM and stored at -20°C. Anti-MTDH rabbit polyclone antibody (Invitrogen Carlsbad), rabbit anti-ERK1/2, p-ERK1/2, AKT, p-AKT, FOXO3a and p-FOXO3a (Ser 294) (Cell Signaling Technology), rabbit anti-Histone H3 (Beyotime Institute of Biotechnology), mouse anti-β-actin monoclone antibody (Sigma), and HRP-labeled secondary antibodies (KPL) were utilized.
Cell culture
The cell line MDA-MB-231 (ATCC) was grown in Dulbecco modified Eagle medium (Gibco-BRL, 12800-017) with 10% fetal bovine serum (Haoyang biological manufacture Co. Ltd), 100 U/ml penicillin and 100 ug/ml streptomycin at 37°C 5% CO2 incubator.
siRNA and vector construction
The plasmid construction was performed as described previously.Citation32 Briefly, the 19-nt sequence 5′ ATGAACCAGAATCAGTCAGC 3′ was used to generate MTDH shRNA. The 60-nt oligonucleotides were annealed and cloned into the HindIII and BglII sites of the pSUPER.retro.puro vetor which is also used as control and called prpn. The shRNA interference vetor called prpM was confirmed by sequencing. The small interfering RNA (siRNA) sequence CCUGUCACUGCAUAGUCGAtt for FOXO3a and a negative control (Shanghai Gene Pharma Co. Ltd) were utilized.
Transfection and expression of shRNA
For transfection Lipofectamine 2000 (Invitrogen, 11668-019) was used as per manufacturer’s instructions. The cells were plated at a density of 5 × 104 cells/well, and the cells were washed twice with PBS the following day and transfection mixes added. The cells were incubated for 4 h at 37°C and 5% CO2. Subsequently, the transfection medium was replaced by the complete medium. After 24 h, the cells were plated for additional experimentation for transient transfection while for stable transfection cells were passaged at a 1:10 dilution into fresh growth medium. After 24 h, 0.5 μg/ml puromycin (Invitrogen) was added in order to select the stable cell lines. The empty prpn vector was used to establish the 231-prpn cell line while the shRNA interference vector prpM was used to generate the 231-prpM cell line.
Cytotoxicity assay
The sensitivity of drugs was measured by 3-(4, 5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT, Sigma, M2128). Cells were plated in 96-well culture plates at a density of 2.5 × 103 cells/well. The next day, different concentrations of AZD6244 were added into 100 μl dilutions of the culture medium. The DMEM medium was utilized as the bank control while the cells without reagents were the negative control. After incubation for 96 h at 37°C, 5% CO2, 20 μl MTT were added to each well. Cells were incubated for another 4 h and DMSO were added 100ul/well to dissolve the formazan after removing the culture medium. The reduction of MTT was quantified by absorbance at a wavelength of 490 nm using Microplate Reader (Bio-Rad).
Western blot
Cells were washed with ice-cold PBS and lysed with ice-cold RIPA buffer (Shennengbocai) [1 × PBS, 1% NP40, 0.1% sodium dodecyl sulfate (SDS), 5 mM EDTA, 0.5% sodium deoxycholate] containing protein inhibitor cocktail (SIGMA) and phosphatase inhibitor sodium fluoride (NaF) and sodium vanadate (NaVO3). Nuclear and cytoplasmic extraction reagents (Beyotime Institute of Biotechnology) were used to extract nuclear proteins, which were then quantified by BCA Protein Assay Kit (Merck, 71285-3) and separated by 5–10% SDS-PAGE. These were transferred onto the PVDF membrane (Millipore), which were blocked in 1 × TBST with 5% skim milk for 1 h and subsequently incubated with the corresponding primary antibodies overnight at 4°C. The membranes were subsequently washed three times with TBST and incubated with secondary antibody for 1 h at room temperature. After washing three times in TBST, each protein was detected using an ECL system (Merck). The bands were quantified by densitometry and normalized to the appropriate loading controls and analyzed using the public domain Image J program.
Real-time RT-PCR
Total RNA was isolated using Trizol (Invitrogen, 15596-026) as per manufacturer’s protocol. RNAs were reverse transcripted by using PrimeScript RT reagent Kit (Takara). The resulting cDNA was used for quantitative realtime PCR on a StepOne Plus instrument from Applied Biosystems with SYBR green detection. Primers involved were the MTDH primers (forward 5′-AAATAGCCAGCCTATCAAGACTC-3′, reverse 5′-TTCAGACTTGGTCTGTGAAGGAG-3′) and the GAPDH primers (forward 5′-GGGCTGCTTTTAACTCTGGTAAAG-3′, reverse 5′-CCATGGGTGGAATCATATTGG-3′). StepOne Plus software was used to calculate crossing threshold (Ct) points from the fluorescence curves and the delta/delta-Ct method was applied to quantify the induction of mRNA as compared with the control sample.
Cell cycle analysis by flow cytometry
Cells were harvested by trypsin, washed by PBS and then stained with the buffer including 50 μg/ml propidium iodide (Sigma), for 30 min at room temperature. For fluorescence-activated cell sorting (FACS) analysis, data was collected using a FACSCalibur (BD Bioscience) flow cytometer. The cell-cycle distribution was evaluated by counting > 20,000 cells/samples.
Statistical analysis
All experiments were repeated at least three times independently. All data were presented as means ± SD of the mean and analyzed with Microsoft Excel analysis tools. Differences between individual groups were analyzed by paired t-test. P values of < 0.05 were considered statistically significant.
Acknowledgments
This project is supported by Program for New Century Excellent Talents in University, Key Project of Chinese Ministry of Education (No. 108080), National Natural Science Foundation of China (No. 30772133; No.81072150; No.81172529) and Independent Innovation Foundation of Shandong University (IIFSDU, No. 2009JQ007) to Y.Q.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reference
- Igene H. Global health inequalities and breast cancer: an impending public health problem for developing countries. Breast J 2008; 14:428 - 34; http://dx.doi.org/10.1111/j.1524-4741.2008.00618.x; PMID: 18821930
- Krcova Z, Ehrmann J, Krejci V, Eliopoulos A, Kolar Z. Tpl-2/Cot and COX-2 in breast cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2008; 152:21 - 5; PMID: 18795070
- Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5:591 - 602; http://dx.doi.org/10.1038/nrc1670; PMID: 16056258
- Frieboes HB, Edgerton ME, Fruehauf JP, Rose FR, Worrall LK, Gatenby RA, et al. Prediction of drug response in breast cancer using integrative experimental/computational modeling. Cancer Res 2009; 69:4484 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-08-3740; PMID: 19366802
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist 2004; 9:361 - 77; http://dx.doi.org/10.1634/theoncologist.9-4-361; PMID: 15266090
- Gasparini G. Therapy of breast cancer with molecular targeting agents. Ann Oncol 2005; 16:iv28 - 36; http://dx.doi.org/10.1093/annonc/mdi905; PMID: 15923426
- Schlotter CM, Vogt U, Allgayer H, Brandt B. Molecular targeted therapies for breast cancer treatment. Breast Cancer Res 2008; 10:211; http://dx.doi.org/10.1186/bcr2112; PMID: 18671839
- Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 2008; 65:3525 - 44; http://dx.doi.org/10.1007/s00018-008-8170-7; PMID: 18668205
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22:153 - 83; http://dx.doi.org/10.1210/er.22.2.153; PMID: 11294822
- Pagès G, Milanini J, Richard DE, Berra E, Gothie E, Vinals F, et al. Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann N Y Acad Sci 2000; 902:187 - 200; http://dx.doi.org/10.1111/j.1749-6632.2000.tb06313.x; PMID: 10865838
- Joslin EJ, Opresko LK, Wells A, Wiley HS, Lauffenburger DA. EGF-receptor-mediated mammary epithelial cell migration is driven by sustained ERK signaling from autocrine stimulation. J Cell Sci 2007; 120:3688 - 99; http://dx.doi.org/10.1242/jcs.010488; PMID: 17895366
- Price DJ, Avraham S, Feuerstein J, Fu Y, Avraham HK. The invasive phenotype in HMT-3522 cells requires increased EGF receptor signaling through both PI 3-kinase and ERK 1,2 pathways. Cell Commun Adhes 2002; 9:87 - 102; http://dx.doi.org/10.1080/15419060214147; PMID: 12487410
- Nishioka C, Ikezoe T, Yang J, Yokoyama A. Inhibition of MEK/ERK signaling induces apoptosis of acute myelogenous leukemia cells via inhibition of eukaryotic initiation factor 4E-binding protein 1 and down-regulation of Mcl-1. Apoptosis 2010; 15:795 - 804; http://dx.doi.org/10.1007/s10495-010-0483-y; PMID: 20221697
- Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS ONE 2010; 5:e13026; http://dx.doi.org/10.1371/journal.pone.0013026; PMID: 20885957
- Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res 2008; 68:6145 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-08-1430; PMID: 18676837
- Yoon J, Koo KH, Choi KY. MEK1/2 Inhibitors AS703026 and AZD6244 may be potential therapies for kras mutated colorectal cancer that is resistant to egfr monoclonal antibody therapy. Cancer Res 2011; 71:445 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-10-3058; PMID: 21118963
- Yoon YK, Kim HP, Han SW, Oh do Y, Im SA, Bang YJ, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Mol Carcinog 2010; 49:353 - 62; http://dx.doi.org/10.1002/mc.20607; PMID: 20358631
- Garon EB, Finn RS, Hosmer W, Dering J, Ginther C, Adhami S, et al. Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY-142886) in human breast cancer and non-small cell lung cancer cell lines. Mol Cancer Ther 2010; 9:1985 - 94; http://dx.doi.org/10.1158/1535-7163.MCT-10-0037; PMID: 20587667
- Ball DW, Jin N, Rosen DM, Dackiw A, Sidransky D, Xing M, et al. Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J Clin Endocrinol Metab 2007; 92:4712 - 8; http://dx.doi.org/10.1210/jc.2007-1184; PMID: 17878251
- Yang JY, Chang CJ, Xia W, Wang Y, Wong KK, Engelman JA, et al. Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res 2010; 70:4709 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-09-4524; PMID: 20484037
- Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther 2007; 6:2209 - 19; http://dx.doi.org/10.1158/1535-7163.MCT-07-0231; PMID: 17699718
- Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 2008; 26:2139 - 46; http://dx.doi.org/10.1200/JCO.2007.14.4956; PMID: 18390968
- Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol 2010; 5:1630 - 6; http://dx.doi.org/10.1097/JTO.0b013e3181e8b3a3; PMID: 20802351
- Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs 2011; 29:1021 - 8; http://dx.doi.org/10.1007/s10637-010-9392-8; PMID: 20127139
- Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2010; 55:668 - 77; http://dx.doi.org/10.1002/pbc.22576; PMID: 20806365
- Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res 2010; 70:8736 - 47; http://dx.doi.org/10.1158/0008-5472.CAN-10-0902; PMID: 20959481
- Su ZZ, Kang DC, Chen Y, Pekarskaya O, Chao W, Volsky DJ, et al. Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene 2002; 21:3592 - 602; http://dx.doi.org/10.1038/sj.onc.1205445; PMID: 12032861
- Li C, Liu J, Lu R, Yu G, Wang X, Zhao Y, et al. AEG -1 Overexpression: A novel indicator for peritoneal dissemination and lymph node metastasis in epithelial ovarian cancers. Int J Gynecol Cancer 2011; 21:602 - 8; http://dx.doi.org/10.1097/IGC.0b013e3182145561; PMID: 21543927
- Yu C, Chen K, Zheng H, Guo X, Jia W, Li M, et al. Overexpression of astrocyte elevated gene-1 (AEG-1) is associated with esophageal squamous cell carcinoma (ESCC) progression and pathogenesis. Carcinogenesis 2009; 30:894 - 901; http://dx.doi.org/10.1093/carcin/bgp064; PMID: 19304953
- Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest 2009; 119:465 - 77; http://dx.doi.org/10.1172/JCI36460; PMID: 19221438
- Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 2009; 15:9 - 20; http://dx.doi.org/10.1016/j.ccr.2008.11.013; PMID: 19111877
- Emdad L, Lee SG, Su ZZ, Jeon HY, Boukerche H, Sarkar D, et al. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc Natl Acad Sci USA 2009; 106:21300 - 5; http://dx.doi.org/10.1073/pnas.0910936106; PMID: 19940250
- Song L, Li W, Zhang H, Liao W, Dai T, Yu C, et al. Over-expression of AEG-1 significantly associates with tumour aggressiveness and poor prognosis in human non-small cell lung cancer. J Pathol 2009; 219:317 - 26; http://dx.doi.org/10.1002/path.2595; PMID: 19644957
- Li C, Li R, Song H, Wang D, Feng T, Yu X, et al. Significance of AEG-1 expression in correlation with VEGF, microvessel density and clinicopathological characteristics in triple-negative breast cancer. J Surg Oncol 2011; 103:184 - 92; http://dx.doi.org/10.1002/jso.21788; PMID: 21259255
- Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, et al. Knockdown of astrocyte-elevated gene-1 inhibits prostate cancer progression through upregulation of FOXO3a activity. Oncogene 2007; 26:7647 - 55; http://dx.doi.org/10.1038/sj.onc.1210572; PMID: 17563745
- Li J, Yang L, Song L, Xiong H, Wang L, Yan X, et al. Astrocyte elevated gene-1 is a proliferation promoter in breast cancer via suppressing transcriptional factor FOXO1. Oncogene 2009; 28:3188 - 96; http://dx.doi.org/10.1038/onc.2009.171; PMID: 19633686
- Su P, Zhang Q, Yang Q. Immunohistochemical analysis of Metadherin in proliferative and cancerous breast tissue. Diagn Pathol 2010; 5:38; http://dx.doi.org/10.1186/1746-1596-5-38; PMID: 20565850
- Li X, Kong X, Huo Q, Guo H, Yan S, Yuan C, et al. Metadherin enhances the invasiveness of breast cancer cells by inducing epithelial to mesenchymal transition. Cancer Sci 2011; 102:1151 - 7; http://dx.doi.org/10.1111/j.1349-7006.2011.01919.x; PMID: 21371176
- Khuda II, Koide N, Noman AS, Dagvadorj J, Tumurkhuu G, Naiki Y, et al. Astrocyte elevated gene-1 (AEG-1) is induced by lipopolysaccharide as toll-like receptor 4 (TLR4) ligand and regulates TLR4 signalling. Immunology 2009; 128:e700 - 6; http://dx.doi.org/10.1111/j.1365-2567.2009.03063.x; PMID: 19740331
- Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res 2010; 70:2264 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-09-1577; PMID: 20215513
- Meng J, Peng H, Dai B, Guo W, Wang L, Ji L, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther 2009; 8:2073 - 80; http://dx.doi.org/10.4161/cbt.8.21.9844; PMID: 19783898
- Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal 2010; 5:10; http://dx.doi.org/10.1186/1750-2187-5-10; PMID: 20642839
- Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol 2008; 10:138 - 48; http://dx.doi.org/10.1038/ncb1676; PMID: 18204439
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857 - 68; http://dx.doi.org/10.1016/S0092-8674(00)80595-4; PMID: 10102273
- Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, et al. Identification of Predictive Markers of Response to the MEK1/2 Inhibitor Selumetinib (AZD6244) in K-ras-Mutated Colorectal Cancer. Mol Cancer Ther 2010; 9:3351 - 62; http://dx.doi.org/10.1158/1535-7163.MCT-10-0376; PMID: 20923857
- Liu H, Song X, Liu C, Xie L, Wei L, Sun R. Knockdown of astrocyte elevated gene-1 inhibits proliferation and enhancing chemo-sensitivity to cisplatin or doxorubicin in neuroblastoma cells. J Exp Clin Cancer Res 2009; 28:19; http://dx.doi.org/10.1186/1756-9966-28-19; PMID: 19216799
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004; 117:225 - 37; http://dx.doi.org/10.1016/S0092-8674(04)00302-2; PMID: 15084260