Abstract
We review the rationale for seeking inhibitors of homologous recombination (HR) repair for use in cancer therapy. Cells use HR as one way to repair DNA double-strand breaks that arise directly from treatments such as radiotherapy, or indirectly during replication when forks encounter other damage. HR occurs during the S and G2 phases of the cell cycle and is therefore more significant in dividing cancer cells than in non-dividing cells of healthy tissue, giving a potential therapeutic advantage to inhibiting the process. Also, some tumors consist of cells that are defective in other DNA repair pathways, and such cells may be sensitive to HR repair inhibitors because of synthetic lethality, in which blocking two alternative pathways that a cell can use to reach a needed end-point has a much bigger impact than blocking either pathway alone. We review strategies for identifying HR inhibitors and discuss current progress.
Why is Homologous Recombination Repair Important for Cancer Therapy?
Despite the advent of modern molecularly targeted therapy, the mainstay of non-surgical cancer treatment is still radiotherapy and chemotherapy. The cellular target for both of these modalities is DNA, and a wide variety of DNA lesions that lead to cell death are produced by these treatments. Some, such as radiation and alkylating agents, target DNA directly, while others, such as topoisomerase I and II inhibitors, inhibit enzymes critical for DNA metabolism, leading to highly damaging lesions such as DNA double strand breaks. It is not hard to understand why DNA is the most important cellular target for cytotoxic agents. Unlike all other molecules in the cell, DNA is critical for survival as each molecule contains unique genetic material required for cellular reproduction. Further, DNA damage, even if repaired, can lead to inappropriately joined chromosomes that are incompatible with reproductive integrity.
Because of the critical importance of the integrity of DNA for their survival, cells have evolved elaborate mechanisms for the repair of lesions introduced into the DNA, whether they be base damage, single and double strand DNA breaks, or DNA adducts. Each of these lesions has one or more specific repair systems, often involving multiple proteins that function to remove or to repair the DNA lesion. It is these repair mechanisms that also markedly attenuate the efficacy of DNA damaging cytotoxic agents (see ref. Citation1 for an excellent review of the DNA lesions introduced by anticancer agents and their respective repair mechanisms). In recent years considerable attention has been focused on one of these repair mechanisms—homologous recombination (HR) repair—not only because it is involved in the repair of lesions produced by many classes of anticancer agents, but also because of the growing realization that many cancers are to some extent deficient in HR repair and this can be exploited by a process known as synthetic lethality (see below).
Potentially the most lethal of all DNA damages is the double strand break (DSB), for such a break, if not repaired, can lead to chromosome breakage and major loss of genetic material. DSBs can be repaired both by non-homologous end joining (NHEJ) and by HR repair, but for HR to be effective there is an absolute requirement for a nearby undamaged identical copy of the damaged DNA duplex (). In mammalian cells, this primarily involves the replicated chromatids of the same chromosome during the S and G2 phases of the cell cycle.Citation2,Citation3 Thus, HR repair is absent or only minimally involved during G1 when replicated chromatids are not present. In diploid yeast, repair of HO-induced DSBs has been shown to occur preferentially in G2,Citation4,Citation5 however, radiation-induced breaks can be repaired either in G2 or G1, in which case homologous chromosomes are involved.Citation6 In contrast to the two-ended DSBs produced directly by agents such as radiation (), many anticancer agents produce lesions that, if unrepaired prior to DNA synthesis, will stall and collapse the replication forks, resulting in a single-ended DSB at the fork (). Lesions that lead to DSBs at replication forks include base damage, DNA adducts and DNA intra- and inter-strand crosslinks. These single-ended DSBs are repaired by the HR machinery thereby allowing restart of replication. In addition, HR is crucial in repairing the DNA DSBs produced by topoisomerase inhibitors that block the ligating action of the topoisomerase enzymes (topoisomerase I and II) during their action in unwinding DNA during replication. In both situations of a DSB produced by a stalled replication fork or one produced by blocking of topoisomerase II in its action of passing one DNA molecule through another, an unbroken identical DNA molecule is in the immediate vicinity thereby allowing HR repair to occur.
Because of the important role of HR in repairing the lesions produced by most anticancer drugs, it follows that cells with defects in this repair mechanism will be sensitive to these drugs. For example, cells with mutations in BRCA1 are sensitive to mitomycin C,Citation7 and cells with various defects in HR are sensitive to tirapazamine, a hypoxia-activated drug producing DNA damage that is repaired by HR.Citation8 In many cases, cells defective in HR are also extremely sensitive to inhibition of base excision repair or single strand breaks.Citation9,Citation10 Such lesions occur frequently in DNA and are repaired rapidly and efficiently by the base excision and single strand break repair pathways. If either of these pathways is inhibited the base damage or single strand break will produce a stalled and collapsed replication fork, but this can be resolved by the HR pathway. If, however, HR repair is defective, such as in tumors with BRCA1 or BRCA2 inactivation, then inhibitors of base or single strand break repair are extremely toxic. This reflects the phenomenon of synthetic lethality (discussed later in this review), which occurs when there is a strong and lethal synergism between two otherwise non-lethal effects. It is exemplified in this case by specific killing of HR-deficient cells by inhibitors of poly(ADP-ribose) polymerase (PARP),Citation9,Citation10 a protein that facilitates both base excision and single strand break repair. Importantly, the same logic can be applied for an inhibitor of HR. If tumors could be identified with specific lesions in base excision or single strand break repair, then these tumors should be sensitive to an agent that inhibits HR. This would provide tumor specific targeting. Do such tumors exist with defects in base excision or single strand break repair? The answer is “yes,” as has been demonstrated in multiple studies (reviewed in ref. Citation11). Further elucidation of these issues is given in the succeeding sections.
Non-homologous End Joining (NHEJ) and Homologous Recombination (HR): The Two Main Pathways for the Repair of DNA Double Strad Breaks and the Choice of Repair Pathway
Double-stranded DNA breaks can arise in many ways and the exact nature of the broken ends and the circumstances by which breaks arise impact the way cells react to breaks. Nevertheless, cellular methods for rejoining DNA broken in both strands can be classified into two main types, depending on whether or not a separate undamaged template molecule is used in the repair process (). The conceptually simpler process is non-homologous end-joining (NHEJ), in which the broken ends of the same molecule are rejoined by a multi-step enzymatic process that does not involve another DNA molecule. Recent studies suggest that NHEJ is more complex than a simple ligation pathway, and is regulated by the full DNA damage response involving the key serine-threonine protein kinase ATM, especially when DSBs occur in heterochromatin.Citation12 NHEJ is not always precise in that short additions or deletions in the original DNA sequence are commonly introduced at the break-site during the repair.
In contrast, homologous recombination (HR) repair usually leads to an accurately restored molecule, as it relies on a separate undamaged molecule with homologous sequence to help repair the break. In principle, there are two major sources of homologous donor sequence: the homologous chromosome, available throughout the cell cycle, and the sister chromatid of the broken molecule, which is only available after the DNA is replicated. When a sister chromatid is not available, for example in the G1 phase, NHEJ may be more appropriate for DSB repair, since HR using the homologous chromosome as a template could lead to loss of heterozygosity. Therefore, multiple mechanisms evolved to ensure that the risk of genetic changes due to repair is minimal. In general, the choice of repair pathways, and the onset and regulation of HR depend on DNA damage signaling pathways, the nature of the break, chromatin remodeling, transcription of HR proteins, and cyclin-dependent kinase activities present in later phases of the cell cycleCitation13–Citation18 (). This may explain why in mammalian cells HR mainly involves only the sister chromatid, and is usually restricted to the S and G2 phases of the cell cycle.Citation2,Citation3 However, in yeast, NHEJ is not effective for repairing the complex DSBs induced by radiation leaving HR as the main mechanism for IR repair throughout the cell cycle.Citation19
The key first step of HR is resection of double-stranded ends (for more detailed overview of HR see refs. Citation20–Citation23). End resection is a two-step process, where initial limited resection mediated by CtIP and MRE11/RAD50/NBS (MRN) is followed by extensive resection involving EXO1 (or DNA2) and BLM. CtIP is the major target of phosphorylation by CDK limiting HR to S and G2 phases of the cell cycle and making DSB resection the major point where the decision is made as to which pathway, NHEJ or HR, will process the DSB.Citation24 Resection generates single-stranded DNA (ssDNA), which then can anneal to the homologous sequence. The exposed ssDNA is coated by the single strand binding protein RPA, activating ATR and as a result, HR. RPA is further replaced by RAD51, which catalyzes the key reactions of synapsis: homology search and D-loop formation, when strands from the broken molecule pair with the unbroken homologous molecule, which acts as a template (). Efficient formation of RAD51 filaments requires several factors, involving Rad51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3), RAD52 and BRCA2. A series of reactions then occur that involve extension of the broken ends followed by isomerization and ligation. In addition, depending on how the individual strands are cut and rejoined (the resolution stage of the D-loop in ) recombination can occur between the parts of the initially broken and unbroken molecules on either side of the break, or alternatively the molecules may return to their original configuration, leaving only a short region around the break-site itself influenced by transfer of information from one strand to the other (see refs. Citation23 and Citation25 for comprehensive review of the DSBR (classical double-strand break repair), SDSA (synthesis-dependent strand annealing), and BIR (break-induced replication) sub-pathways of HR). 5′ to 3′ strand resection can extend for many kilobases until homologous sequence is found. If resection uncovers direct repeat sequences, both single-stranded DNA ends can be annealed together to repair the break through a process called single-strand annealing (SSA) (). SSA is RAD51-independent and involves reannealing of RPA-covered ssDNA by the RAD52 protein. SSA results in the deletion of the repeat and the loss of the sequence between the repeats. An alternative DSB repair pathway, microhomology-mediated end joining (MMEJ) anneals short regions of homology revealed by limited resection and results in larger sequence deletions than the ones occurring after NHEJ. lists key proteins participating in DSB repair via the HR pathway. Estimates suggest that there are more than 200 proteins involved at different stages of HR,Citation26 providing the necessary variety of context-specific factors and explaining the extraordinary adaptability of HR to repair DSBs, DNA interstrand crosslinks (ICLs), and resolution of stalled/collapsed replication forks. The fact that in human cells HR repair acts preferentially in S phase and G2 cells suggests that inhibiting HR repair offers a potential advantage for cancer therapy by selectively targeting cells that are actively dividing.
Synthetic Lethality and Synergistic Interactions
The phrase “synthetic lethality” (SL) was originally used by geneticists to describe a situation with model organisms, such as bacteria, yeast and flies, in which two mutations, neither of which by itself is required for viability, nevertheless confer a lethal phenotype when they are combined together in the same cell.Citation27–Citation29 Usually, this situation arises when each gene provides an alternative method or route for the cell to accomplish the same goal, so that both must be knocked out to prevent this from being accomplished, thus killing the cell. The term “synthetic lethality” is now used more loosely in anticancer drug discovery to denote interactions between genes when loss of function is achieved not only by mutating a gene, but also by inhibition of its product by a drug. A concept related to SL is that of “synergistic interaction.” In this case, combining two mutations that confer a similar but moderate phenotype, such as radiation sensitivity, may lead to a non-lethal but very strong phenotype, much greater than expected from simply the additive effects of two independent phenotypes. This is the case, for example, when yeast cells with genetic defects in different repair pathways are treated with UV radiation (UV).Citation30 The primary process for repairing UV damage in yeast is nucleotide excision repair (NER) (see ref. Citation31 for review) and mutant strains lacking only NER are highly sensitive to UV radiation. In contrast, mutants lacking only HR repair are only slightly sensitive to UV radiation.Citation32 However, when a mutation in HR (such as deletion of the RAD51 gene) is added to a strain already defective in NER, the same dose of UV radiation now causes much greater cell killing. In short, the HR mutation has a much greater relative effect in cells lacking NER than in cells proficient in NER. The synergistic effect is thought to arise because, in wild-type cells, the same lesions can be channeled into either pathway, so that when only one pathway is blocked, the other pathway can at least partially compensate by accepting more of the damage. When both pathways are blocked, this compensatory effect is abolished, since neither pathway is able to compensate for loss of the other, and the full effect of the damage is seen (reviewed in ref. Citation30).
The recent demonstration of SL between PARP and HRCitation9,Citation10,Citation33 when these pathways are inhibited has drawn much interest to the SL approach from the anticancer therapy community. The current explanation for the extreme sensitivity of HR-deficient cells to PARP inhibitors is based on a central role of PARP in the base excision repair (BER) pathway that processes DNA base damage and SSBs. Unrepaired SSBs stall and collapse replication forks, and given the major role of HR in resolving stalled/collapsed replication fork structures, tumors with defects in BRCA1 or BRCA2, and consequently in HR, are sensitive to inhibitors of PARP. Furthermore, we should expect that SL relationships exist between HR and other proteins involved in BER. This idea is supported by findings of increased sister chromatid exchanges (SCEs) in SSB repair-defective cells,Citation34,Citation35 as well as by our previous finding of hypersensitivity of HR-defective cells to tirapazamine (TPZ), a hypoxia-activated drug that produces base damage and SSBs.Citation8 We observed an increased formation of secondary DSBs during replication after treatment of wild-type cells with TPZ and we found an increase in SCE in the TPZ-treated cells, indicating that unrepaired base damage and SSBs were converted into DSBs during replication and that HR was involved in the repair of those DSBs.Citation8 In addition, the number of replication-associated DSBs and SCE frequencies were increased many fold in the XRCC1-deficient EM9 cells after TPZ treatment compared with normal cells, confirming that elevated levels of replication-associated lesions resulting from unrepaired base damage and SSBs in EM9 cells were repaired through HR (ref. Citation8 and unpublished observations by S.B. Chernikova and J.M. Brown).
The synthetic lethality between HR and BER could be exploited in two ways: (1) by using BER inhibitors in HR-deficient tumors, and (2) using the expectation that tumors with impaired BER should be sensitive to HR inhibitors. The first strategy is best represented by the archetypal example of SL between PARP and HR. The validity of the second approach is demonstrated by the findingCitation36 that survival of cells expressing a truncated polβ variant similar to the variants found in tumors is strongly dependent on HR after ionizing radiation. These strategies have their limitations, as not every protein involved in BER when defective would be synthetically lethal with an HR defect. For example, knockdown of XRCC1, a protein essential in BER, failed to show SL with BRCA2 deficiency.Citation37 HelledayCitation26 pointed out that the success of applicability of PARP inhibitors to treatment of BRCA-defective tumors could be attributed to dual roles of PARP in both BER and HR, and he concluded that interactions such as the one between PARP and BRCA might be rare. The question then remains if the SL approach is more widely applicable or if it is restricted to a few specific proteins that may have functions in both the pathways we are trying to inactivate.
Nevertheless, the finding of a SL interaction between BRCA defects and PARP inhibition provided a proof-of-principle that SL interactions targeting HR and other DNA repair pathways could be identified. The recent discovery of SL interaction of BRCA2 and Rad52 within the HR pathway alone points to a magnitude of possibilities that exist.Citation38 Mammalian BRCA2 and yeast Rad52 share similar activities, thus it is likely that human Rad52 mediates an HR pathway similar to the BRCA2-dependent pathway. Targeting alternative proteins, like Rad52 in BRCA2-deficient tumors, may therefore become an effective monotherapy strategy in targeting tumor cells. We expect to find more SL interactions if we expand our approaches to combination therapies. The synergistic effects seen in yeast when both HR and NER are genetically defective supports the idea that inhibiting HR enzymatically in human cells that are defective in NER may dramatically increase their sensitivity to the effects of these inhibitors when a cytotoxic drug is used that induces NER-repairable damage (e.g., cisplatin). Although the evidence for SL between HR and NER in humans is scarce,Citation39 we expect that many new SL interactions between NER and HR that are useful for anticancer therapy will be uncovered in the future. Both BER, where PARP plays a central role, and NER, when defective, fail to repair damages that stall and collapse replication forks requiring resolution via HR, therefore most findings that apply to SL between BER and HR should also be true for SL interactions between NER and HR. Hence, searching for drugs that specifically inhibit HR repair is currently a rational objective in cancer therapy.
Inhibition of Homologous Recombination as a Tumor Specific Strategy with Radiation Therapy
As discussed earlier, repair of radiation damage in mammalian cells by HR occurs in late S and G2 because of the requirement for a nearby non-damaged homologous DNA duplex, which is efficiently provided by the two chromatids in late S and G 2. Cells in the G1 phase rely preferentially on NHEJ for repair, a process that occurs equally throughout the cell cycle. This is illustrated in . To date the focus of academia and industry in finding inhibitors of DNA repair that could be used with radiotherapy has been on NHEJ inhibitors because mutants in NHEJ appear to be much more sensitive than mutants in HR to killing by ionizing radiation. However, choosing an inhibitor of NHEJ as a sensitizer for radiotherapy has the obvious disadvantage that NHEJ inhibitors radiosensitize tumor and normal tissues equally well. At the present time there is no convincing strategy for selective tumor radiosensitization by an inhibitor of NHEJ, other than the elusive possibility of selective delivery of the inhibitor to the tumor. However, as we discuss below there are compelling reasons why an inhibitor of HR repair could be used for tumor-specific radiosensitization. The following two strategies are likely to be most effective for the combination of an HR repair inhibitor with different radiation fractionation schemes—the first with conventional fractionated irradiation, the second with large doses as currently employed in stereotactic ablative radiotherapy (SABR) (also known as stereotactic body radiotherapy, SBRT).
The first strategy for selective tumor radiosensitization by an HR repair inhibitor is based on the finding that mutants of HR show radiation sensitivity comparable to NHEJ mutants when synchronized in late S/G2.Citation3 The reason why this radiosensitivity of mutants in HR repair has been overlooked is because in experimental systems, whether it is cells in vitro or tumors in vivo, there are always cells in G1, which are not very radiosensitive, so the population as a whole in the HR mutants is only minimally sensitive. This is not the case for mutants in NHEJ which are sensitive throughout the cell cycle. In addition, mutations that completely block HR repair do not kill yeast cells, but are lethal to mammalian cells, probably because of the much larger genome size. This initially made it hard to find HR repair mutants in mammalian cells, leading to an early impression that HR repair might not be important in mammals. The reason why inhibitors of HR could be effective with radiotherapy is that the dose-limiting normal tissues in radiotherapy are those that show so-called “late effects” and these occur in very slowly or non-dividing normal tissues (such as normal brain). The cells in these tissues are all in G0 or G1, and so cannot undergo HR following irradiation. Thus an inhibitor of HR would not be expected to change the radiosensitivity of the cells in these tissues. On the other hand, as tumor cells are dividing they are in all phases of the cell cycle, so those in S and G2 would be radiosensitized by an HR inhibitor. The fact that radiotherapy is typically given in many small doses (on average 30 doses over 6 weeks), would allow the tumor cells in G1 at the time of irradiation to enter the S and G2 phases and be sensitized to subsequent doses provided the inhibitor were given with each radiation dose fraction. Thus an inhibitor of HR could be used to preferentially sensitize tumor cells to irradiation without sensitizing the cells of the critical normal tissues surrounding the tumor. This hypothesis has not been tested yet in preclinical models because of the lack of a drug that can specifically inhibit HR repair and is sufficiently nontoxic to be given with all, or most, of the radiation fractions. As indicated above this strategy would be expected to be useful with conventional fractionated irradiation.
The second potential mechanism by which an inhibitor of HR repair could be a preferential radiosensitizer of tumor vs. normal tissue is based on the fact that a characteristic property of tumor cells is their lack of a G1 arrest following irradiation as a result of inactivation of the p53 pathway in tumor cells. Thus, following irradiation, the majority of tumor cells fail to arrest in G1 but arrest in S and G2 where HR can occur, whereas the majority of the normal cells arrest at the G1/S boundary.
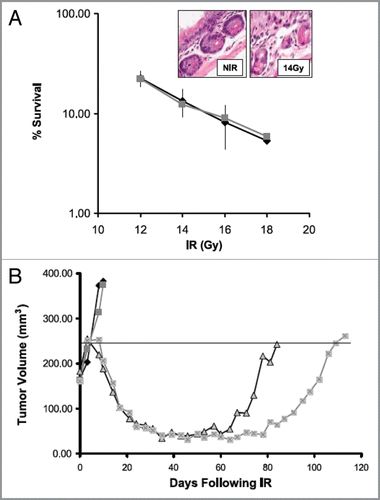
The validity of above strategies is supported by the work of several authors who have demonstrated that various drugs that inhibit HR sensitize tumor but not normal cells to irradiation. For example Noguchi and colleaguesCitation40 demonstrated that the drug 17-AAG, which inhibits heat shock protein 90 (HSP90), radiosensitized two human cancer cell lines (DU145 and SQ-5) in vitro but did not radiosensitize the HFL III normal cell line. They showed further that a 24 h exposure to 17-AAG decreased the levels of the HR proteins BRCA2 and Rad51 but had no effect on the protein levels of the NHEJ proteins DNA-PKcs, Ku80 and Ku70. They also demonstrated that the radiosensitization could be attributed to a decrease in DNA DSB repair in the tumor cell lines. Preferential radiosensitization in vivo of tumor vs. normal tissue has also been demonstrated by inhibition of HR through reduction in Rad51 levels using the clinically used drug imatinib,Citation41 as shown in (data from Choudhury et al.Citation41). The preferential radiosensitization of tumor cells by imatinib is likely to arise from the fact that tumor cells lack the G1/S block and therefore rely more on HR than normal cells due to the expected preferential accumulation of the tumor cells in S and G2 following irradiation. Because the accumulation of cells in the S and G2 phases of the cycle would be most evident following relatively large doses this strategy would be expected to be most useful when such doses are delivered as in SABR.
Current Inhibitors of HR
Despite many attempts to find specific inhibitors of HR, no direct inhibitors of proteins catalyzing the HR reactions are available to date. Nevertheless, many classes of novel anticancer drugs that are currently in clinical trials have recently been shown to target HR, and although they were not developed as inhibitors of HR their activity may in fact be related to their ability to inhibit HR.
The many potential ways that HR may be inhibited can be appreciated by considering the large and growing number of proteins known to be involved in the various aspects of HR. Compounds known to modulate HR activity interfere with the expression, nuclear localization, recruitment, and/or activation of HR. For example, inhibitors of the tyrosine kinases c-Abl (Imatinib or Gleevec) and EGFR (Erlotinib) affect nuclear localization of Rad51 and BRCA1, while inhibitors of histone deacetylation (HDAC) (e.g., valproic acid, PCI-24781), and of HSP90 protein (e.g., 17-AAG) decrease the levels of active HR components, affecting either expression or maturation of key HR gene products,Citation40,Citation42 like Rad51 and BRCA1. On the other hand, proteasome inhibitors (β-lactacystin, MG132, bortezomib, nelfinavir) do not interfere with expression of Rad51, but impair HR through inhibition of proteasome-dependent degradation of mediator proteins, such as MDC1,Citation43 acting downstream of the DSB sensors ATM and MRN, but before the formation of a ssDNA/RPA recombinogenic intermediate.Citation44 Inhibition of the more upstream targets has become possible with the identification of mirin, a compound affecting the Mre11-associated exonuclease activity.Citation45
In light of the recent finding that Rad51 is phosphorylated in a Mec (an ATR homolog)-dependent manner in response to DNA damage in yeast,Citation46 it seems attractive to give closer consideration to compounds that inhibit kinases that directly regulate the HR process. NU6027, originally developed as a CDK2 inhibitor, has recently been shown to inhibit ATR.Citation47 Consistent with a key function of ATR resulting in resolution of stalled replication forks through signaling to the HR pathway, NU6027 has been shown to inhibit Rad51 foci formation. The downstream target of ATR, the CHK1 protein, has also been identified as a regulator of HR,Citation14 and the inhibitor of CHK1, AZD7762, has been shown to hamper assembly of Rad51 foci, as well as to inhibit gene conversion.Citation48 BRCA1 has been recently identified as a phosphorylation target of the CDK1 kinase,Citation49 and this phosphorylation has been shown to be essential for the formation of BRCA1 and Rad51 foci, and for gene conversion.Citation49,Citation50 Consistent with impaired HR, CDK1 inhibition (by RO-3306 and AG024322) decreased formation of Rad51 and BRCA1 foci and gene conversion.Citation50 As expected of cells with defects in HR, all groups of the aforementioned inhibitors were found to sensitize cells to radiation, chemotherapy drugs, and PARP inhibitors.Citation47,Citation48,Citation50
In principle, screens aimed at identifying HR inhibitors can either be target-based, i.e., they search for inhibitors of a specific enzyme known to be involved in HR, or they can be phenotypic, i.e., based on observing inhibition of a cellular phenotype that is dependent on HR. Because of the great complexity of HR, most high-throughput chemical screens are justifiably based on the phenotypic approach. This enables them to detect compounds that may have unknown or multiple enzymatic targets, rather than requiring the screen to identify only inhibitors of one or more chosen targets. Other reasons contributing to the success of the phenotypic approach are in the utilization of cell-based assays relevant to the human disease. Examples of successful phenotypic screens that resulted in identification of small molecule inhibitors of HR include a screen based on phosphorylation of a peptide derived from histone H2AX in cell-free extracts derived from Xenopus laevis eggs.Citation45 Compounds from this screen, which potentially target any step leading to H2AX phosphorylation, have been narrowed down to the compound mirin, which has been shown to interfere with the nuclease activity of Mre11 and subsequently with the MRN-dependent activation of ATM, impairing HR repair.Citation45 Another fruitful approach was based on inhibition of the assembly of FANCD2 foci in response to cross-linking drugs and ionizing radiation.Citation51 The FANCD2 foci formation happens after FANCD2 is monoubiquitinated by a complex of Fanconi Anemia proteins, and the foci are believed to be the sites of HR repair. Among new compounds inhibiting formation of FANCD2 foci were protein kinase inhibitors (wortmannin, H-9 and alsterpaulone) and a natural compound, curcumin.Citation51 Further expansion of the screening library identified more compounds, which included proteasome inhibitors, CDK inhibitors, and an HSP90 inhibitor, well representing the main categories of compounds known to inhibit HR (see above). Significant overlap in terms of proteins involved must exist among the different subpathways of HR, as compounds known to sensitize cells to interstrand crosslinking (ICL) agents often impair Rad51 foci formation and/or gene conversion, and vice versa (refs. Citation41, Citation47, Citation51 and our observations). Our own strategy to screen for inhibitors of HR was based on increased sensitivity to the ICL-inducing agent chlorambucil (Chernikova and Brown, unpublished observations). To screen against compounds that would cause sensitivity to ICL by inhibiting other pathways involved in repair of ICL, i.e., nucleotide-excision repair (NER) and/or translesion synthesis, we re-screened compounds testing positive for sensitization using a gene conversion assay and a Rad51 foci formation assay. Both the gene conversion assay and Rad51 foci formation assay are amenable to the high-throughput format, therefore other approaches may include screening compound libraries using these assays initially. As more HR proteins are characterized and more compounds identified that impair each subpathway of HR, we will have a better overview of the complex HR system.
Although, as mentioned, no inhibitors of proteins directly catalyzing the HR reactions are yet available, the situation might change soon as several potential inhibitors of the Rad51 recombinase activity have recently been identified in an in vitro target-based screen.Citation52 Still at the initial stages of characterization, these compounds have been shown to inhibit IR-induced formation of Rad51 foci and gene conversion (A. V. Mazin, personal communication).
Conclusions
Finding compounds that inhibit HR repair is currently an attractive option in cancer research. Although complete inhibition of recombination for any extended time period is likely to be lethal to all dividing cells, partial or temporary inhibition is likely to be useful in radiotherapy, since, such compounds are expected to have a greater relative impact on radiation sensitivity in dividing cancer cells than on nearby non-dividing healthy cells. Also, HR inhibitors should be especially lethal to cancers that contain mutations in certain genes in other branches of HR repair or in some cases in BER or NER, through the phenomenon of synthetic lethality. Success in finding inhibitors of HR has so far been limited, but several strategies are available for screening libraries of compounds, and promising leads are already on the horizon.
Abbreviations
| HR | = | homologous recombination |
| NHEJ | = | non-homologous end joining |
| DSB | = | double-strand break |
| SSB | = | single-strand break |
Figures and Tables
Figure 1 Homologous recombination participates in resolution of two main types of double-strand breaks (DSBs) from endogenous and exogenous sources: (A) directly produced two-ended DSBs (like the ones produced by ionizing radiation) and (B) one-ended DSBs that result from single-strand breaks (SSBs) encountering replication forks. SSBs are common intermediates in the repair of many DNA lesions.
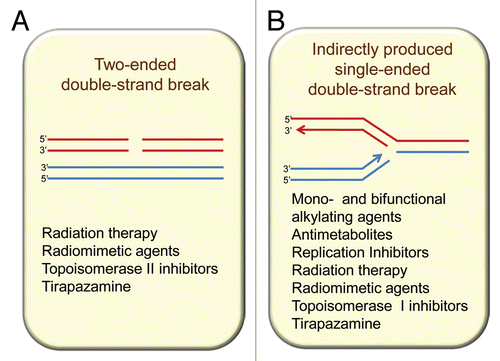
Figure 2 Double-strand break repair in mammalian cells. NHEJ, HR, MMEJ and SSA pathways compete for the repair of DSBs. Proteins involved in various stages of DSB repair are shown in teal. References for proteins involved at different stages can be found in references Citation7 and Citation21–Citation23. NHEJ is involved throughout the cell cycle, and HR is involved after replication, when the sister chromatid is available. A simple DSB occurring within euchromatin will be most likely repaired by NHEJ, while more complex DSBs or those in heterochromatin will initiate the full DNA damage response involving MRE11/RAD50/NBS1 (MRN) complex and ATM protein. MRN at the DSB recruits ATM, which phosphorylates BRE1A/B complex leading to relaxation of chromatin, facilitating repair by either NHEJ or HR. MRN recruits CtIP to initiate resection. Extensive resection is performed upon recruitment of EXO1 or DNA2 and the resulting single-stranded DNA (ssDNA) 3′-overhang is rapidly coated by RPA. The ssDNA-RPA recruits ATRIP leading to activation of ATR. The RPA then is displaced by Rad51 to form a nucleoprotein filament and initiate synapsis, a search for homologous template and DNA strand invasion, leading to formation of a D-loop intermediate. Synapsis is central to all HR reactions, and subsequent resolution of the D-loop is accomplished by at least three different pathways: double-strand break repair (DSBR), synthesis-dependent strand annealing (SDSA), and break-induced replication (BIR). An alternative pathway, microhomology-mediated end joining (MMEJ), which is active throughout the cell cycle, anneals short homologous sequences revealed by limited resection. Single-strand annealing (SSA) mediates repair when perfect repeats are revealed during end resection.
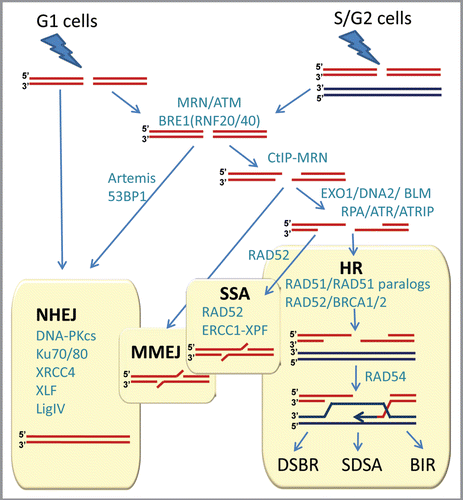
Figure 3 Key proteins involved in homologous recombination (HR). The main engine of HR is Rad51, which catalyzes the key reactions of synapsis-homology search and DNA strand invasion. Altogether, there are more than 200 proteins involved in different aspects of HR, providing a sufficient number of context-specific factors to explain the extraordinary versatility of HR, i.e., its functions in the repair of double-strand breaks (DSBs), DNA interstrand crosslinks (ICLs), and in resolution of stalled/collapsed replication forks. Any of the proteins are potentially targetable for cancer therapy.

Figure 4 Imatinib increases radiotherapy-induced tumor growth delay without increasing intestinal toxicity in a PC-3 prostate cancer xenograft. (A) Intestinal crypt cell survival in BALB/c mice following 1–18 Gy in vivo in the presence of 50 mg/kg/d for 5 d. Surviving crypts were scored 3 d after irradiation based on three mice per treatment group. Points, mean survival; bars, SD. Inset, H&E-stained section of intestinal crypts before and after 14 Gy. (B) Plot of growth delay of PC3 xenografts treated with PBS alone, imatinib (50 mg/kg/d × 8 d) alone, 4 Gy × 5 plus PBS, or 4 Gy × 5 plus imatinib. Irradiated mice were either pretreated with 3 d PBS (sham) or 3 d imatinib (50 mg/kg/d i.p.) before irradiation and received daily PBS or imatinib i.p. dosing during 4 Gy daily radiation fractions for a total treatment period of 8 d similar to drug alone. For clarity, the median animal per group is shown. The calculated mean growth delay based on 9–10 mice per group was 100 ± 7 d for the combined treatment group, compared with 74 ± 14 d for the radiotherapy alone group (p = 0.003). There was no difference in growth delay between the imatinib alone and control groups. (Adapted and reprinted with permission from the American Association for Cancer Research: Choudhury et al., “Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity,” Mol Cancer Ther 2009; 8:203–13).
Acknowledgments
This work was supported by the NIH grant P01 CA-067166 to J.M.B.
References
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8:193 - 204; PMID: 18256616; http://dx.doi.org/10.1038/nrc2342
- Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res 2004; 32:3683 - 3688; PMID: 15252152; http://dx.doi.org/10.1093/nar/gkh703
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706 - 5715; PMID: 12897142; http://dx.doi.org/10.1128/MCB.23.16.5706-5715.2003
- Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 2004; 23:4868 - 4875; PMID: 15549137; http://dx.doi.org/10.1038/sj.emboj.7600469
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011 - 1017; PMID: 15496928; http://dx.doi.org/10.1038/nature02964
- Game JC. Spencer JFT, Spencer D, Smith ARW. Radiation-sensitive mutants and repair in yeast. Yeast Genetics, Fundamental and Applied Aspects 1983; New York Springer-Verlag 109 - 137
- Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res 2001; 61:4842 - 4850; PMID: 11406561
- Evans JW, Chernikova SB, Kachnic LA, Banath JP, Sordet O, Delahoussaye YM, et al. Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res 2008; 68:257 - 265; PMID: 18172318; http://dx.doi.org/10.1158/0008-5472.CAN-06-4497
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913 - 917; PMID: 15829966; http://dx.doi.org/10.1038/nature03443
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 921; PMID: 15829967; http://dx.doi.org/10.1038/nature03445
- Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer?. Cell Cycle 2004; 3:998 - 1001; PMID: 15280658; http://dx.doi.org/10.4161/cc.3.8.1062
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 2008; 31:167 - 177; PMID: 18657500; http://dx.doi.org/10.1016/j.molcel.2008.05.017
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009; 28:3413 - 3427; PMID: 19779458; http://dx.doi.org/10.1038/emboj.2009.276
- Sørensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005; 7:195 - 201; PMID: 15665856; http://dx.doi.org/10.1038/ncb1212
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37 - 45; PMID: 16327781; http://dx.doi.org/10.1038/ncb1337
- Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008; 455:689 - 692; PMID: 18716619; http://dx.doi.org/10.1038/nature07215
- Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell 2011; 41:529 - 542; PMID: 21362549; http://dx.doi.org/10.1016/j.molcel.2011.02.015
- Chernikova SB, Dorth JA, Razorenova OV, Game JC, Brown JM. Deficiency in Bre1 impairs homologous recombination repair and cell cycle checkpoint response to radiation damage in mammalian cells. Radiat Res 2010; 174:558 - 565; PMID: 20738173; http://dx.doi.org/10.1667/RR2184.1
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet 2011; 45:247 - 271; PMID: 21910633; http://dx.doi.org/10.1146/annurev-genet-110410-132435
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 2010; 11:196 - 207; PMID: 20177395; http://dx.doi.org/10.1038/nrm2851
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229 - 257; PMID: 18275380; http://dx.doi.org/10.1146/annurev.biochem.77.061306.125255
- Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 2010; 44:113 - 139; PMID: 20690856; http://dx.doi.org/10.1146/annurev-genet-051710-150955
- Pardo B, Gomez-Gonzalez B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci 2009; 66:1039 - 1056; PMID: 19153654; http://dx.doi.org/10.1007/s00018-009-8740-3
- Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 2009; 284:9558 - 9565; PMID: 19202191; http://dx.doi.org/10.1074/jbc.M808906200
- Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 2008; 18:99 - 113; PMID: 18166982; http://dx.doi.org/10.1038/cr.2008.1
- Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010; 31:955 - 960; PMID: 20351092; http://dx.doi.org/10.1093/carcin/bgq064
- Dobzhansky T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946; 31:269 - 290
- Hartman JL 4th, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science 2001; 291:1001 - 1004; PMID: 11232561; http://dx.doi.org/10.1126/science.291.5506.1001
- Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett 2011; 585:1 - 6; PMID: 21094158; http://dx.doi.org/10.1016/j.febslet.2010.11.024
- Game JC, Cox BS. Synergistic interactions between rad mutations in yeast. Mutat Res 1973; 20:35 - 44; PMID: 4586553; http://dx.doi.org/10.1016/0027-5107(73)90095-X
- Friedberg EC. Deoxyribonucleic acid repair in the yeast Saccharomyces cerevisiae. Microbiol Rev 1988; 52:70 - 102; PMID: 3280967
- Game JC, Mortimer RK. A genetic study of x-ray sensitive mutants in yeast. Mutat Res 1974; 24:281 - 292; PMID: 4606119; http://dx.doi.org/10.1016/0027-5107(74)90176-6
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123 - 134; PMID: 19553641; http://dx.doi.org/10.1056/NEJMoa0900212
- Thompson LH, Brookman KW, Dillehay LE, Carrano AV, Mazrimas JA, Mooney CL, et al. A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair, and an extraordinary baseline frequency of sister-chromatid exchange. Mutat Res 1982; 95:427 - 440; PMID: 6889677; http://dx.doi.org/10.1016/0027-5107(82)90276-7
- Oikawa A, Tohda H, Kanai M, Miwa M, Sugimura T. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem Biophys Res Commun 1980; 97:1311 - 1316; PMID: 6260088; http://dx.doi.org/10.1016/S0006-291X(80)80009-X
- Neijenhuis S, Verwijs-Janssen M, van den Broek LJ, Begg AC, Vens C. Targeted radiosensitization of cells expressing truncated DNA polymerase {beta}. Cancer Res 2010; 70:8706 - 8714; PMID: 20978197; http://dx.doi.org/10.1158/0008-5472.CAN-09-3901
- Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA 2011; 108:3406 - 3411; PMID: 21300883; http://dx.doi.org/10.1073/pnas.1013715108
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA 2011; 108:686 - 691; PMID: 21148102; http://dx.doi.org/10.1073/pnas.1010959107
- Herrero AB, Martin-Castellanos C, Marco E, Gago F, Moreno S. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res 2006; 66:8155 - 8162; PMID: 16912194; http://dx.doi.org/10.1158/0008-5472.CAN-06-0179
- Noguchi M, Yu D, Hirayama R, Ninomiya Y, Sekine E, Kubota N, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun 2006; 351:658 - 663; PMID: 17083915; http://dx.doi.org/10.1016/j.bbrc.2006.10.094
- Choudhury A, Zhao H, Jalali F, Al Rashid S, Ran J, Supiot S, et al. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol Cancer Ther 2009; 8:203 - 213; PMID: 19139130; http://dx.doi.org/10.1158/1535-7163.MCT-08-0959
- Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA 2007; 104:19482 - 19487; PMID: 18042714; http://dx.doi.org/10.1073/pnas.0707828104
- Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, et al. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J Biol Chem 2008; 283:31608 - 31616; PMID: 18757370; http://dx.doi.org/10.1074/jbc.M801082200
- Murakawa Y, Sonoda E, Barber LJ, Zeng W, Yokomori K, Kimura H, et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res 2007; 67:8536 - 8543; PMID: 17875693; http://dx.doi.org/10.1158/0008-5472.CAN-07-1166
- Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol 2008; 4:119 - 125; PMID: 18176557; http://dx.doi.org/10.1038/nchembio.63
- Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP. Regulation of Rad51 function by phosphorylation. EMBO Rep 2011; 12:833 - 839; PMID: 21738226; http://dx.doi.org/10.1038/embor.2011.127
- Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 2011; 105:372 - 381; PMID: 21730979; http://dx.doi.org/10.1038/bjc.2011.243
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972 - 4981; PMID: 20501833; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573
- Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009; 35:327 - 339; PMID: 19683496; http://dx.doi.org/10.1016/j.molcel.2009.06.036
- Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med 2011; 17:875 - 882; PMID: 21706030; http://dx.doi.org/10.1038/nm.2377
- Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther 2006; 5:952 - 961; PMID: 16648566; http://dx.doi.org/10.1158/1535-7163.MCT-05-0493
- Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 2011; 6:628 - 635; PMID: 21428443; http://dx.doi.org/10.1021/cb100428c