Abstract
Vimentin, a mesenchymal marker, is frequently overexpressed in epithelial carcinomas undergoing epithelial to mesenchymal transition (EMT), a condition correlated with invasiveness and poor prognosis. Therefore, vimentin is a potential molecular target for anticancer therapy. Emerging studies in experimental models underscore the functions of homeodomain-interacting protein kinase 2 (HIPK2) as potential oncosuppressor by acting as transcriptional corepressor or catalytic activator of molecules involved in apoptosis and response to antitumor drugs. However, an involvement of HIPK2 in limiting tumor invasion remains to be elucidated. This study, by starting with a microarray analysis, demonstrates that HIPK2 downregulates vimentin expression in invasive, vimentin-positive, MDA-MB-231 breast cancer cells and in the non-invasive MCF7 breast cancer cells subjected to chemical hypoxia, a drive for mesenchymal shift and tumor invasion. At functional level, vimentin downregulation by HIPK2 correlates with inhibition of breast tumor cell invasion. Together, these data show that vimentin is a novel target for HIPK2 repressor function and that HIPK2-mediated vimentin downregulation can contribute to inhibition of breast cancer cells invasion that might be applied in clinical therapy.
Introduction
Homeodomain-interacting protein kinase-2 (HIPK2) is a transcriptional co-repressor involved in cell development and in tumor regression as a potential oncosuppressor. Previous studies by our group and others’ showed that HIPK2 is able to modulate the transcription of genes by acting on a growing number of transcription factors and their chromatin-remodelling complexes, leading to regulation of cell survival and apoptosis.Citation1,Citation2 Exogenous HIPK2 expression or DNA damage-induced HIPK2 activate the apoptotic function of p53 oncosuppressor;Citation3 however, the induction of p53-independent apoptotic pathways by HIPK2 has been also reported.Citation4-Citation6 Among the regulated molecules, we found that HIPK2 inhibits hypoxia-inducible factor 1 (HIF-1) activity, restraining tumor angiogenesis and chemoresistance.Citation7,Citation8 For these reasons, activation of HIPK2 function or HIPK2 overexpression is an important tool for regulating molecular pathways involved in tumor progression and aggressiveness and the identification of novel molecules regulated by HIPK2 may be beneficial for antitumor therapies.
Vimentin is an intermediate filament protein normally expressed in cells of mesenchimal origin.Citation9 However, vimentin can also be expressed in epithelial cells undergoing epithelial-to-mesenchymal transition (EMT),Citation10 a reversible critical event in the progression toward cancer metastasisCitation11 characterized by reduced expression of epithelial markers such as E-cadherin and upregulation of mesenchymal markers such as N-cadherin and vimentin.Citation12,Citation13 Accumulating evidences demonstrated that epithelial-mesenchymal transition has also been implicated in the onset of drug resistance and tumor relapses, representing an escape mechanism from apoptosis.Citation11 Vimentin expression is induced in invasive epithelial carcinoma cell linesCitation14-Citation16 and is correlated with poor prognosis.Citation17,Citation18 The functional relevance of vimentin overexpression was recently shown by its correlation with upregulation of several genes associated with EMT and the basal-like phenotype, by microarray analysis of clinical samples.Citation19 Therefore, attenuation of the EMT phenotype in cancer cells may enhance classical chemotherapy treatment and improve the clinical management of cancer. By virtue of its overexpression in various epithelial cancers including, prostate, breast, lung, gastrointestinal cancers, and its association with tumor growth and metastasis, vimentin downregulation is as an attractive strategy for anticancer therapy.Citation20 In the present study we investigated the effect of HIPK2 overexpression on invasive vimentin-positive MDA-MB-231 breast cancer cells and in non-invasive MCF7 breast cancer cells undergoing hypoxia-induced invasive phenotype, with the aim of examining a possible link between HIPK2, vimentin and cell migration. Our data show that HIPK2 overexpression leads to downregulation of vimentin that correlates with impairment of breast cancer cell migration, strongly supporting a role of HIPK2 in modulating carcinoma cell invasion.
Results
HIPK2 downregulates vimentin expression in MDA-MB-231 cells
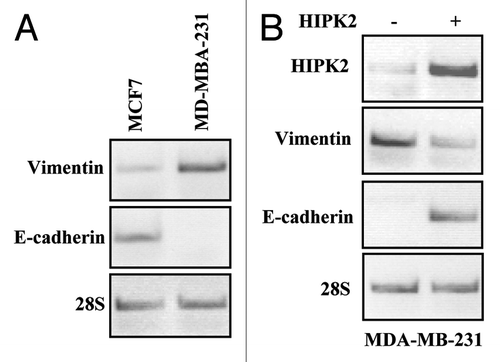
To gain insight into the genes modulated by HIPK2, we compared differential gene expression in Mock and HIPK2-transfected colon cancer RKO cells by microarray technique. Of the 24,746 filtered probesets compared, 178 genes were upregulated (Table S1) and 180 genes were donwregulated by more than 2-fold change (Table S2) as a result of HIPK2 overexpression. Among the group of downregulated genes, several cytoskeleton genes were identified including vimentin (VIM), ARHGEF2, TUBE1, TTLL1, HAUS1, MID1IP1 and TUBB2C. We confirmed the microarray results by semi-quantitative RT-PCR, showing that the vimentin expression levels were indeed downregulated by HIPK2 overexpression in RKO cells (not shown). Therefore, we asked whether HIPK2 might regulate vimentin expression in breast cancer cells as vimentin overexpression is associated with breast cancer invasion.Citation11 To this aim we used the MDA-MB-231 and the MCF7 cells which were, respectively, strong and weak positive for vimentin () and were reported to be, respectively, invasive and non-invasive in Boyden chamber assay.Citation14,Citation21 As shown in , the MDA-MB-231 cells transfected with HIPK2 showed downregulation of vimentin mRNA levels that strongly correlated with re-expression of E-cadherin indicative of a reversion of EMT phenotype. However, the increase of E-cadherin mRNA did not allow E-cadherin protein expression (not shown), in agreement with recent data suggesting that post-translational modifications may take part, in a time-dependent manner, to regulate E-cadherin stabilization and likely to fully promote the reversion of EMT phenotype.Citation22,Citation23
Figure 1. mRNA expression of vimentin and E-cadherin in breast cancer cells after HIPK2 overexpression. (A) Semi-quantitative RT-PCR analysis of vimentin and E-cadherin in MCF7 and MDA-MB-231 cells. 28S mRNA levels were detected as control of cDNA inputs. (B) MDA-MB-231 cells were transfected with HIPK2 expression vector and vimentin and E-cadherin mRNA levels were detected 24 h after transfection. HIPK2 mRNA levels are indicative of HIPK2 overexpression. 28S mRNA levels were detected as control of cDNA inputs.
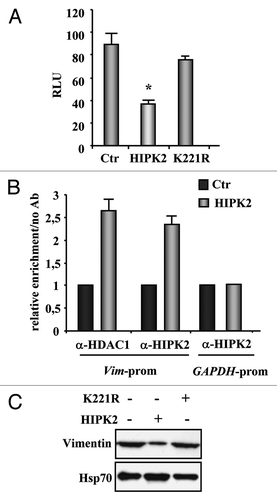
To further evaluate the role of exogenous HIPK2 in vimentin regulation, we performed a luciferase assay by using a reporter plasmid containing the human vimentin promoter,Citation24 in transient transfection experiments. As shown in , HIPK2 overexpression led to a significant repression of the vimentin promoter (about 52% reduction compared with the control cells) while the kinase-deficient K221R mutant failed to do so. Next the HIPK2 recruitment onto the vimentin promoter was evaluated by ChIP assay. MDA-MB-231 cells were transfected with Flag-HIPK2 vector and 24 h after transfection DNA/protein complexes were immunoprecipitated with anti-HIPK2 and anti-HDAC1 antibodies. PCR analysis was performed using specific primers flanking the human vimentin promoter.Citation25 The results show that, following HIPK2 overexpression, both HIPK2 and HDAC1 were efficiently co-recruited onto vimentin promoter (), while the HIPK2 binding to the human GAPDH promoter, used as control of HIPK2 binding specificity, did not show any enrichment (). Finally, western immunoblotting of MDA-MB-231 cells showed that HIPK2 overexpression strongly reduced vimentin protein levels, compared with the kinase-deficient K221R mutant that failed to do so (), suggesting that the HIPK2 catalytic activity is involved in vimentin downregulation.
Figure 2. Regulation of vimentin transcription by HIPK2. (A) H1299 cells were co-transfected with the vimentin-luciferase reporter plasmid together with HIPK2, kinase-deficient K221R mutant, or empty vector (Ctr). Luciferase activity was measured 36 h after transfection. Results, normalized to β-gal activity, are the mean of seven independent experiments, performed in duplicate, ± SD. RLU, relative luciferase units. *p < 0.0001. (B) ChIP analysis with anti-HIPK2 and anti-HDAC1 antibodies or no antibody as control was performed in MDA-MB-231 cells transfected with HIPK2 expression vector or empty vector (Ctr) for 24 h. Co-recruitment of HIPK2 and HDAC1 onto the vimentin promoter was detected by RT-PCR, and analyzed by densitometry. Relative enrichment of HIPK2 and HDAC1 onto vimentin promoter, compared with no antibody, is shown. Amplification of the GAPDH promoter was used as control of HIPK2 binding specificity to the vimentin promoter. The data represent the mean of two independent experiments ± SD (C) MDA-MB-231 cells were transfected with HIPK2 and K22R expression vector and equal amount of total cell extracts were analyzed by protein gel immunoblotting with anti-vimentin antibody. Hsp70 detection was used as protein loading control.
HIPK2 knock-down impairs the ADR-induced vimentin downregulation
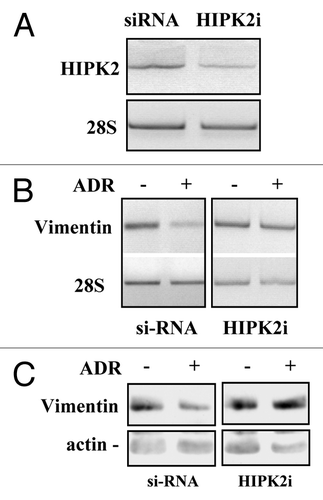
As HIPK2 can be activated by antitumor drugs including adryamicin (ADR),Citation26 to evaluate the role of endogenous HIPK2 in vimentin downregulation, in response to drug, we first knocked down HIPK2 with siRNA interference in MDA-MB-231 cells (). Next, control and HIPK2-interfered cells were treated with ADR for 16 h before analyzing vimentin expression. The results show that ADR treatment downregulated vimentin at both mRNA () and protein () levels only in si-RNA control cells, while, HIPK2 interference (HIPK2i) strongly impaired ADR-induced vimentin downregulation (). Intriguingly, HIPK2 depletion did not per se induced vimentin mRNA expression, highlighting the fact that different systems of gene manipulation, such as siRNA interference or exogenous expression, might modulate different molecular pathways. This result though, is in agreement with our hypothesis that HIPK2 activation is involvement in vimentin downregulation. On the other hand, as resistance to ADR represents a major obstacle to the unsuccessful treatment of patients with breast cancer,Citation27 it is tempting to speculate the use of exogenous HIPK2 as a promising gene therapy tool for antitumor purpose and to overcome the negative side effects due to drug resistance. Alternatively to drug activation, the use of small peptides or targeted micro RNA to specifically activate HIPK2 could be taken in consideration in future studies.
Figure 3. HIPK2 knock-down impairs the ADR-induced vimentin downregulation. (A) MDA-MB-231 cells were transfected with the control (siRNA) or specific HIPK2 interfering vector (HIPK2i) and 36 h after transfection mRNA was extracted for semi-quantitative RT-PCR analysis of HIPK2 levels. 28S mRNA levels were detected as control of cDNA inputs. (B) MDA-MB-231 control or HIPK2-interfered cells as in (A) were treated with ADR (1.5 μg/ml) and for 12 h after treatment mRNA was extracted for semi-quantitative RT-PCR analysis of vimentin levels. 28S mRNA levels were detected as control of cDNA inputs. (C) MDA-MB-231 control or HIPK2-interfered cells were treated as in (B) equal amount of total cell extracts were analyzed by protein gel immunoblotting with anti-vimentin antibody. Hsp70 detection was used as protein loading control.
HIPK2 inhibits MDA-MB-231 cell migration
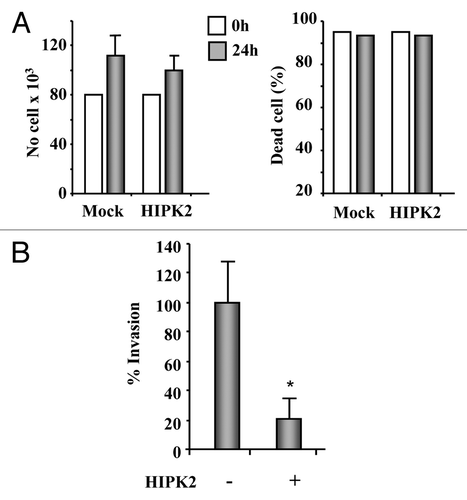
These encouraging results prompted us to further evaluate the potential therapeutic efficacy of exogenous HIPK2 overexpression. Therefore, the invasive potential of HIPK2-transfected MDA-MB-231 cells was tested, by counting the number of cells passing through filters coated with reconstituted basement membrane, in a Boyden chamber assay. Transfection of the GFP-tagged HIPK2 expression vector or co-transfection of the Flag-tagged HIPK2 expression vector with the GFP vector allowed to monitor transfection efficiency by fluorescence microscope that was about 70% in this assay. To exclude an indirect effect on cell migration mediated by HIPK2 role on cell apoptosis, growth curves comparing wild-type with HIPK2 transfected cells were performed. The results showed no difference in proliferation (, left panel) or cell death (, right panel) over the time-course during which the assays took place. Cell migration assay shows that the invasive behavior of MDA-MB-231 cells was significantly reduced by HIPK2 overexpression (reduction of cell invasion of about 79%, p = 0.0001) (), suggesting that HIPK2, by modulating different molecular pathways, might exert additional antitumor effects other than the apoptotic one, particularly in mutant-p53 cells, that are worth to study further.
Figure 4. HIPK2 overexpression inhibits MDA-MB-231 cell invasion. (A) The highly invasive MDA-MB-231 cell line was transfected with HIPK2 expression vector and 24 h after transfection both floating and adherent cells were collected and cell proliferation (left panel) and viability (right panel) were determined by Trypan blue exclusion. The results are the mean of three independent experiment ± S.D. (B) MDA-MB-231 cells were transfected with HIPK2 expression vector (+) or empty vector (-). Sixteen hours after transfection, cells were serum-starved (2% FBS) for 24 h and cell invasion was measured using a Boyden’s chamber invasion assay. Mean invaded cells/microscopic field, ± S.D., is shown. *p < 0.0001.
HIPK2 downregulates hypoxia-induced vimentin expression in MCF7 cells and inhibits cell migration
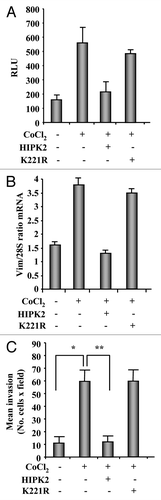
MCF7 cells have been shown to be weak positive for vimentin (see ) and were reported to be non-invasive in Boyden chamber assay,Citation14,Citation21 and hypoxia has been shown to induce a shift in expression of epithelial markers (e.g., E-cadherin) in MCF7 cells to mesenchymal markers (e.g, vimentin) leading to invasive phenotype.Citation28 We have found that HIPK2, but not the K221R kinase-deficient mutant, downregulates the hypoxia-inducible factor 1α, inhibiting the HIF-1 transcriptional activityCitation8,Citation29 with sensitization of hypoxia-treated cells to drug-induced apoptosis, regardless of their p53 status.Citation7 Therefore, in this scenario, we attempted to evaluate whether exogenous HIPK2 expression could affect vimentin expression and cell migration in hypoxia-treated cells. To this aim, we used cobalt chloride (CoCl2), that was shown to mimic hypoxia by stabilizing HIF-1α.Citation30 We first analyzed the vimentin-luciferase reporter activity in MCF7 cells with or without cobalt chloride treatment. As shown in , cobalt induced vimentin reporter activity that was strongly inhibited by HIPK2 overexpression while the K221R mutant did not affect the reporter activity. Then, in vivo analysis of gene expression showed that 24 h of cobalt treatment induced vimentin mRNA expression that was strongly inhibited by HIPK2 overexpression but not by K221R mutant (). Finally, the invasive potential of MCF7 cells, treated with cobalt with or without HIPK2 and K221R overexpression, was evaluated by Boyden chamber assay. As shown in , MCF7 invasion induced by chemical hypoxia was completely counteracted by HIPK2 overexpression while it was not affected by K221R overexpression. To emphasize the effect of HIPK2 on cell migration we performed a viability assay that showed about 20% cell death in cobalt-treated MCF7 cells after HIPK2 overexpression, while K221R mutant did not affect cell viability (not shown). This result is in agreement with our previous results showing that HIPK2 overexpression is able to induce transient cell death in cobalt-treated wtp53-expressing cancer cells,Citation7 although, 20% cell death is not sufficient to explain the complete inhibition of cobalt-induced cell invasion by HIPK2 overexpression. Altogether, these data show that HIPK2-induced vimentin downregulation correlated with inhibition of the hypoxia-induced breast cancer cell invasion.
Figure 5. Chemical hypoxia induces vimentin expression and cell migration in MCF7 cells that may be reverted by HIPK2 overexpression. (A) MCF7 cells were co-transfected with the vimentin-luciferase reporter plasmid together with HIPK2, K221R mutant or empty vector. Sixteen hours after transfection, cells were treated with CoCl2 (200 µM). Luciferase activity was measured 24 h after treatment. Results, normalized to β-gal activity, are the mean of three independent experiments, performed in duplicate, ± S.D. RLU: Realtive Luciferase Units. (B) Semi-quantitative RT-PCR analysis of vimentin mRNA in MCF7 treated as in (A). Results are shown as the ratio between vimentin and 28S mRNA levels, as measured by densitometric analysis. (C) The low invasive MCF7 cell line was transfected with HIPK2 or K221R expression vectors empty vector. Sixteen hours after transfection, cells were treated with CoCl2 (200 µM) and 24 h later serum-starved (2% FBS) for additional 24 h. Cell invasion was measured using a Boyden’s chamber invasion assay. Mean invaded cells/microscopic field, ± S.D., is shown. *p < 0.007, **p < 0.0003.
Discussion
It is now established that vimentin is one of the most important marker of epithelial-to-mesenchymal transition (EMT), a reversible critical event in the progression toward cancer metastasis.Citation9-Citation11 Thus, vimentin overexpression is reported in many sarcoma and in several different carcinomas including breast cancer, and influences cell migration. Therefore, vimentin is considered a potential molecular target for cancer therapy.Citation20 Here, our findings showed a novel target of HIPK2, that is vimentin, whose downregulation correlated with inhibition of the invasive phenotype of MDA-MB-231 breast cancer cells and hypoxia-treated MCF7 cells.
HIPK2 activity is necessary for regulating different molecular pathways, positively or negatively involved in tumor development, to restrain tumor progression. HIPK2 regulates gene expression by phosphorylating transcription factors, or accessory components of the transcription machinery or by recruitment of co-repressor components.Citation1,Citation2 Among the molecules regulated by HIPK2, we demonstrated that HIPK2 by phosphorylating p53 specifically induces its apoptotic activity,Citation3 and that HIPK2 inhibits the β-catenin-induced vascular endothelial growth factor (VEGF)Citation31 expression and inhibits HIF-1 activity.Citation7,Citation8 In agreement with its co-repressor function, here we show that HIPK2 inhibited the vimentin luciferase reporter activity and downregulated its mRNA expression. As a proof of principle, the HIPK2 kinase-deficient K221R mutant did not affect vimentin expression, suggesting that HIPK2 catalytic activity is involved in this regulation. Vimentin function can be regulated either by post-translational modifications or at the gene transcription level.Citation20 In the latter condition, vimentin promoter can be downregulated by the zinc finger transcriptional repressor ZBP-89 via the specific recruitment of histone deacetylase (HDAC1),Citation25 on the contrary, vimentin expression was shown to be transactivated by β-catenin/TCF.Citation24 As HIPK2 has been shown to act as transcriptional co-repressor in complex with HDAC1Citation32 and we found here that both HIPK2 and HDAC1 were recruited onto vimentin promoter, it is conceivable to speculate that HIPK2 may recruit, likely through its catalytic activity, HDAC1 to repress vimentin transcription.
Numerous studies showed that vimentin downregulation impairs in vitro tumor cell invasiveness or migration. Thus, vimentin targeting by vimentin-specific antisense cDNA, oligonucleotide transfection or siRNA impairs in vitro invasiveness or migration of several tumor cells including breast cancer,Citation33-Citation36 emphasizing a functional contribution of vimentin to epithelial cell invasion/migration. In line with these findings, we found a strong correlation between HIPK2-induced vimentin downregulation and inhibition of breast cancer cell migration by Boyden chamber assay. To exclude an indirect effect on cell migration mediated by HIPK2 role on growth arrest or apoptosis, growth curves were performed showing no difference in proliferation or cell death in MDA-MB-231 HIPK2-transfected cells. This is in line with previous observations by us demonstrating that HIPK2 induces apoptosis in wt-p53 but not in mutant-p53 cells,Citation3 suggesting that HIPK2, by modulating different molecular pathways, might exert additional antitumor effects other than the apoptotic one, particularly in mutant-p53 cells such as MDA-MB-231 cells, that are worth to study further.
The use of exogenous HIPK2 is supported also by the finding that HIPK2 is inhibited by hypoxiaCitation37 but this inhibition may be overcome by HIPK2 overexpression.Citation7 Intratumoral hypoxia is one of the most important mechanisms that promotes metastasis, induces chemoresistance and angiogenesis and thereby increases tumor aggressiveness.Citation38 Therefore, targeting hypoxia is a valuable strategy in anticancer therapy.Citation39 In this regard, we recently reported that HIPK2, but not its kinase-deficient K221R mutant, downregulates HIF-1α subunit inhibiting HIF-1 activity with sensitization of hypoxia-treated cells to antitumor drugs.Citation7,Citation8,Citation29 Here we showed that hypoxia-mimicking condition induced vimentin expression in MCF7 cells which correlated with acquisition of their invasive behavior; interestingly, both effects were efficiently counteracted by exogenous HIPK2 but not by the K221R mutant. On the other hand, viability assay showed about 20% cell death in cobalt-treated MCF7 cells after HIPK2 overexpression, which could be explained by HIPK2-dependent p53 reactivation since MCF7 cells carry endogenous wtp53. However, 20% cell death was not sufficient to explain the complete abolishment of hypoxia-treated cell invasion after HIPK2 overexpression, or at least suggests that HIPK2 might act at different molecular levels to impinge on tumor progression.
Endogenous HIPK2 can be activated by several drugs (adriamycin, cisplatin, roscovitin), UV light, and IR irradiation,Citation40 so we asked whether we could parallel the effect obtained with exogenous HIPK2 expression with that obtained by drug activation. Therefore, we treated MDA-MB-231 cells with ADR that reduced vimentin expression, an effect that indeed was impaired by HIPK2 knock-down, as a proof of principle of HIPK2 activation in vimentin downregulation. Thus, HIPK2 knock-down per se did not increase vimentin mRNA expression (); however, it was previously shown that HIPK2 depletion increases colon cancer cell invasion by de-repressing the pro-invasive integrin β4.Citation42 Although ADR is currently considered one of the most effective agents in the treatment of breast cancers, drug resistance represents a major obstacle to successful outcome in many patients.Citation27 This perspective strongly suggests to identify novel therapeutical strategies to overcome drug resistance and inhibit tumor progression. In this regard, the proposed use of exogenous HIPK2 expression as a potential therapeutical tool for antitumor purpose does not appear trivial. Thus, we previously showed that exogenous HIPK2 overexpression circumvents chemoresistant signaling pathways, reactivating the apoptotic response,Citation41 or sensitizes to drug-induced apoptosis tumor cells treated with hypoxia, regardless of their p53 status.Citation7
Altogether, the previous findings and the proposed effect on vimentin expression warrant further studies to develop new therapeutical approaches, such as the use of micro RNA or small peptides to activate HIPK2 to restrain tumor progression and to potentiate antitumor cytotoxic therapies.
Materials and Methods
Cell lines and treatments
Human invasive, vimentin-positive, breast cancer MDA-MB-231 cell line (carrying p53 mutation R280K) and the non-invasive human breast cancer MCF7 cell line (carrying wild-type p53) were maintained in DMEM (Life Technology, Invitrogen), while human colon cancer RKO (carrying wild-type p53) and lung cancer H1299 (p53 null) cell lines were maintained in RPMI-1640 (Life Technology, Invitrogen), all supplemented with 10% heat-inactivated fetal bovine serum (FBS) plus glutamine and antibiotics. For chemical hypoxia treatment, cobalt chloride (CoCl2, 200 μM) was added to sub-confluent cell layer for 24 h. Subconfluent cells were treated with adriamycin (ADR) diluted into the medium to a final concentration of 1.5 μg/ml.
Microarray analysis
Human colon cancer RKO cells were transfected with Flag-HIPK2 expression vector or empty vector as control using the cationic polymer LipofectaminePlus method (Invitrogen) according to manufacturers’ instructions and harvested 24 h after transfection. Total RNA was extracted using TRIzol Reagent (Invitrogen) as indicated by the manufacturer, and hybridized to Affymetrix HU-gene st1.0 microarrays, containing 28,830 probe sets. Experimental samples were processed in duplicates. Iter-Plier normalization algorithm (available at: www.affymetrix.com/support/technical/technotes/pliertechnote.pdf) was applied on the .CEL files, followed by modified Lowess correction algorithm and log2 transformation.Citation43 Only probe sets with expression above threshold (t = 16) in both replicates were included. To gain more statistical power we used a method developed by Zeisel et al.,Citation44 which estimates the intensity dependent variance of the noise, using the distribution of the probe sets having similar expression levels in each pair of replicates. Differential expression between the control and the HIPK2 transfected cells was detected using these estimated variances, by calculating p values using the two tailed z-score test for each probe set. The p values were ranked and FDR of 5% was applied to control the number of false positives. Unique genes with expression ratios above 2 (or below 0.5) were selected for further investigation.
RNA extraction and reverse transcription (RT)-PCR analysis
Cells were harvested in TRIzol Reagent (Invitrogen) and total RNA was isolated following the manufacturer’s instructions. The first strand cDNA was obtained by 2 µg RNA as previously reportedCitation7 and semi-quantitative reverse-transcribed (RT)-PCR was performed by using HotMaster Taq (Eppndorf) and 2 µl cDNA reaction and gene-specific oligonucleotides under conditions of linear amplification. PCR was performed in duplicate and PCR products were run on 3% agarose gel and visualized by ethidium bromide staining using UV light. The 28S mRNA, used as internal standard, was amplified from the same cDNA reaction mixture. Densitometric analysis was applied to quantify mRNA levels normalized with values obtained for 28S amplification. The primer sequences used were as follow: human vimentin forward, 5′-GACAATGCGTCTCTGGCACGTCTT-3′ and reverse, 5′-TCCTCCGCCTCCTGCAGGT TCTT-3′;Citation24 28S forward, 5′-GTTCACCCACTAATAG GGAACGTGA-3′ and reverse, 5′-GGATTCTGACTTAGAGGC GTTCAGT-3′.Citation24 HIPK2 forward, 5′-AGGAAGAGTAA GCAGCACCAG-3′ and reverse, 5′-TGCTGATGGTGATGA CACTGA-3′.Citation45
Transfection, plasmids and transactivation assay
H1299 cells were transiently transfected by using the N,N-bis-(2- hydroxyethyl)-2-amino-ethanesulphonic acid-buffered saline (BBS) version of the calcium phosphate procedure,Citation7 while MCF7 and the MDA-MB-231 were transfected using the cationic polymer LipofectaminePlus method (Invitrogen), according to manufacturers’ instructions.
The expression plasmids used were: wild-type Flag-HIPK2, kinase-deficient Flag-K221R, GFP-HIPK2, empty-Flag and empty-GFP vectors.Citation3 For HIPK2 interference, the pSUPER vectors carrying HIPK2 or aspecific RNA-interfering sequencesCitation45 were used.
The plasmid reporter used in co-transfection with HIPK2 or K221R vectors, was the vimentin promoter luciferase vector (kindly provided by C. Gilles, University of Liege).Citation24 The amount of plasmid DNA in each sample was equalized by supplementing with empty vector. Transfection efficiency was normalized with the use of a co-transfected β-galactosidase (β-gal) plasmid. Luciferase activity was assayed on whole cell extract and the luciferase values were normalized to β-gal activity and protein content and expressed as relative luciferase unit (RLU) ± SD.
Cell proliferation and viability
Cell were transfected with GFP-HIPK2, GFP-K221R expression vectors with the LipofectaminePlus method (Invitrogen). Soon after transfection, cells were trypsinized, counted and equal cell number re-plated in triplicate in 60 mm Petri dishes. For hypoxia treatment, 12 h after transfection CoCl2 (200 μM) was added to the cell medium for 24 h. Both floating and adherent cells were collected and cell viability was determined by Trypan blue exclusion, by direct counting with a hemocytometer, as reported.Citation7
Protein gel immunoblotting
Total cell extracts were prepared by incubating at 4°C for 30 min in lysis buffer [50 mmol/L TRIS-HCl (pH 7.5), 150 mmol/L NaCl, 5 mmol/L EDTA, 150 mmol/L KCl, 1 mmol/L DTT, 1% NP40] and a mix of protease inhibitors (Sigma) and resolved by 10% SDS-PAGE. Immunoblotting was performed with mouse monoclonal anti-vimentin (V9) (Santa Cruz Biotechnology) and mouse monoclonal anti-Hsp70 (Stressgene). Immunoreactivity was detected by enhanced chemiluminescence kit (ECL; Amersham).
Chromatin Immunoprecipitation (ChIP) assays and RT-PCR
Chromatin Immunoprecipitation (ChIP) analysis was performed essentially as described.Citation8 Protein complexes were cross-linked to DNA in living cells by adding formaldehyde directly to the cell culture medium at 1% final concentration. Chromatin extracts containing DNA fragments with an average size of 500 bp were incubated overnight at 4°C using rabbit polyclonal anti-HIPK2 (Santa Cruz Biotechnology) and anti-Histone deacetylase 1 (HDAC1, Sigma) antibodies. Before use, protein G (Pierce) was blocked with 1 μg/μL sheared herring sperm DNA and 1 μg/μL BSA for 3 h at 4°C and then incubated with chromatin and antibody for 2 h at 4°C. PCR was performed with HOT-MASTER Taq (Eppendorf) using 2 μL of immuniprecipitated DNA and promoter-specific primers for human vimentin promoter: forward 5′-GCTAGGTCCCGATTGGCT-3′ and reverse 5′-CGAGGGCGCTGTTTTTAT-3′, resulting in a PCR product of 92 bp in length.Citation25 Promoter-specific primers for human GAPDH promoter were used as control. Immunoprecipitation with non-specific immunoglobulins (No Ab) (IgG; Santa Cruz Biotechnology) was performed as negative controls. The amount of precipitated chromatin measured in each PCR was normalized with the amount of chromatin present in the input of each immunoprecipitation. PCR products were run on a 3% agarose gel and visualized by ethidium bromide staining using UV light.
Chemoinvasion assay
Chemoinvasion was assessed using a 48-well modified Boyden chamber (NeuroProbe) and 8-μm pore polyvinyl pyrrolidone-free polycarbonate Nucleopore filters (Costar), as reported.Citation46 The filters were coated with an even layer of 3 mg/mL Cultrex Basement Membrane Extract (a soluble form of basement membrane purified from Engelbreth-Holm-Swarm-EHS-tumor) (Trevigen). The lower compartment of chamber was filled with conditioned serum-free medium produced by NIH3T3 fibroblasts or with 0.1% BSA as negative control. MDA-MB-231 and MCF7 cells were transiently transfected with GFP-HIPK2, GFP-K221R expression vector or empty vector. Serum-starved (2% fetal bovine serum for 24 h) transfected cells (2 × 106 cells/sample) untreated or treated for 24 h with chemical hypoxia condition were harvested and placed in the upper compartment (45 μl/well) of the Boyden chamber. After 8 and 24 h of incubation at 37°C for, respectively, MDA-MB-231 and MCF7 cells, the filters were removed and then stained with Diff-Quick (Biomap), and the migrated cells in 12 high-power fields were counted. Each assay was performed in quadruplicate and repeated at least three times. The ability of the cells to adhere to the filters was verified by staining the upper side of the filter.
Statistical analyses
All experiment unless indicated were performed at least three times. All experimental results were expressed as the arithmetic mean and standard deviation (S.D.) of measurements was shown. Student’s t-test was used for statistical significance of the differences between treatment groups. Statistical analysis was performed using analysis of variance at 5% (p < 0.05) or 1% (p < 0.01).
| Abbreviations: | ||
| BSA | = | bovine serum albumin |
| β-gal | = | β-galactosidase |
| CoCl2 | = | cobalt chloride |
| ChIP | = | chromatin immunoprecipitation |
| EMT | = | epithelial mesenchymal transition |
| FBS | = | fetal bovine serum |
| GFP | = | green fluorescent protein |
| HDAC1 | = | histone deacetylase 1 |
| HIPK2 | = | homeodomain-interacting protein kinase-2 |
| RLU | = | relative luciferase units |
| RT-PCR | = | reverse-transcription polymerase chain reaction |
Additional material
Download Zip (145 KB)Acknowledgments
This work was supported by Research Grants from the Italian Association for Cancer Research (AIRC, to G. D’Orazi and R. Falcioni) and by Flight Attendants Medical Research Institute (FAMRI) (to G. Rechavi). V.F. is a recipient of a FIRC Fellowship. A.A. is supported by funding from Susan G. Komen for the Cure Foundation. We thank C. Gilles for the vimentin luciferase promoter and G. Bossi and A. Farsetti for helpful discussion.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Calzado MA, Renner F, Roscic A, Schmitz ML. HIPK2: a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 2007; 6:139 - 43; http://dx.doi.org/10.4161/cc.6.2.3788; PMID: 17245128
- Rinaldo C, Prodosmo A, Siepi F, Soddu S. HIPK2: a multitalented partner for transcription factors in DNA damage response and development. Biochem Cell Biol 2007; 85:411 - 8; http://dx.doi.org/10.1139/O07-071; PMID: 17713576
- D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser46 and mediates apoptosis. Nat Cell Biol 2002; 4:11 - 9; http://dx.doi.org/10.1038/ncb714; PMID: 11780126
- Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 2003; 115:177 - 86; http://dx.doi.org/10.1016/S0092-8674(03)00802-X; PMID: 14567915
- Hofmann TG, Stollberg N, Schmitz ML, Will H. HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res 2003; 63:8271 - 7; PMID: 14678985
- Doxakis E, Huang EJ, Davies AM. Homeodomain-interacting protein-2 regulates apoptosis in developing sensory and sympathetic neurons. Curr Biol 2004; 14:1761 - 5; http://dx.doi.org/10.1016/j.cub.2004.09.050; PMID: 15458648
- Nardinocchi L, Puca R, Sacchi A, D’Orazi G. Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol Cancer 2009; 8:1 - 9; http://dx.doi.org/10.1186/1476-4598-8-1; PMID: 19128456
- Nardinocchi L, Puca R, Sacchi A, Rechavi G, Givol D, D’Orazi G. Targeting hypoxia in cancer cells by restoring homeodomain interacting protein kinase 2 and p53 activity and suppressing HIF-1alpha. PLoS ONE 2009; 4:e6819; http://dx.doi.org/10.1371/journal.pone.0006819; PMID: 19714248
- Steinert PM, Roop DR. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem 1988; 57:593 - 625; http://dx.doi.org/10.1146/annurev.bi.57.070188.003113; PMID: 3052284
- Ivaska J. Vimentin: Central hub in EMT induction?. Small GTPases 2011; 2:51 - 3; http://dx.doi.org/10.4161/sgtp.2.1.15114; PMID: 21686283
- Thiery JP. Epithelial-mesenchimal transition in tumor progression. Nat Rev Cancer 2002; 2:442 - 54; http://dx.doi.org/10.1038/nrc822; PMID: 12189386
- Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res 2004; 10:4125 - 33; http://dx.doi.org/10.1158/1078-0432.CCR-0578-03; PMID: 15217949
- Zhao Y, Yan Q, Long X, Chen X, Wang Y. Vimentin affects the mobility and invasiveness of prostate cancer cells. Cell Biochem Funct 2008; 26:571 - 7; http://dx.doi.org/10.1002/cbf.1478; PMID: 18464297
- Sommers CL, Walker-Jones D, Heckford SE, Worland P, Valverius E, Clark R, et al. Vimentin rather than keratin expression in some hormone-independent breast cancer cell lines and in oncogenes-transformed mammary epithelial cells. Cancer Res 1989; 49:4258 - 63; PMID: 2472876
- Sommers CL, Heckford SE, Skerker JM, Worland P, Torri JA, Thompson EW, et al. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res 1992; 52:5190 - 7; PMID: 1382837
- Gilles C, Polette M, Piette J, Delvigne AC, Thompson EW, Foidart JM, et al. Vimentin expression in cervical carcinomas: association with invasive and migratory potential. J Pathol 1996; 180:175 - 80; http://dx.doi.org/10.1002/(SICI)1096-9896(199610)180:2<175::AID-PATH630>3.0.CO;2-G; PMID: 8976877
- Domagala W, Lasota J, Bartkowiak J, Weber K, Osborn M. Vimentin is preferentially expressed in human breast carcinomas with low estrogen receptor and high Ki-67 growth fraction. Am J Pathol 1990; 136:219 - 27; PMID: 2153347
- Thomas PA, Kirschmann DA, Cerhan JR, Folberg R, Seftor EA, Sellers TA, et al. Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clin Cancer Res 1999; 5:2698 - 703; PMID: 10537332
- Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi J-P, Nevo J, Gjerdrum C, et al. Vimentin regulates EMT induction by Slug and oncogenes H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011; 30:1436 - 8; http://dx.doi.org/10.1038/onc.2010.509; PMID: 21057535
- Satelli A, Li S. Vimentin in cancer and its potential as molecular target for cancer therapy. Cell Mol Life Sci 2011; 68:3033 - 46; http://dx.doi.org/10.1007/s00018-011-0735-1; PMID: 21637948
- Sommers CL, Byers SW, Thompson EW, Torri JA, Gelmann EP. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res Treat 1994; 31:325 - 35; http://dx.doi.org/10.1007/BF00666165; PMID: 7881109
- Janda E, Nevolo M, Lehmann K, Downward J, Beung H, Grieco M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006; 25:7117 - 30; http://dx.doi.org/10.1038/sj.onc.1209701; PMID: 16751808
- Fabre-Guillevin E, Malo M, Cartier-Michaud A, Peinado H, Moreno-Bueno G, Vallee B, et al. PAI-1 and functional blockade of SNAI1 in breast cancer cell migration. Breast Cancer Res 2008; 10:R100; http://dx.doi.org/10.1186/bcr2203; PMID: 19055748
- Gilles C, Polette Mestdagt M, Nawrocki-Raby B, Ruggeri P, Birembaut P, Foidart JM. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res 2003; 63:2658 - 64; PMID: 12750294
- Wu Y, Zhang X, Salmon M, Zehner ZE. The zinc finger repressor, ZBP-89, recruits histone deacetylase 1 to repress vimentin gene expression. Genes Cells 2007; 12:905 - 18; http://dx.doi.org/10.1111/j.1365-2443.2007.01104.x; PMID: 17663720
- Puca R, Nardinocchi L, Givol D, D’Orazi G. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in cancer cells. Oncogene 2010; 29:4378 - 87; http://dx.doi.org/10.1038/onc.2010.183; PMID: 20514025
- Smith L, Watson MB, O’Kane SL, Drew PL, Lind MJ, Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol Cancer Ther 2006; 5:2115 - 20; http://dx.doi.org/10.1158/1535-7163.MCT-06-0190; PMID: 16928833
- Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 2008; 10:295 - 305; http://dx.doi.org/10.1038/ncb1691; PMID: 18297062
- Nardinocchi L, Puca R, Guidolin D, Belloni AS, Bossi G, Michiels C, et al. Transcriptional regulation of hypoxia-inducible factor 1alpha by HIPK2 suggests a novel mechanism to restrain tumor growth. Biochim Biophys Acta 2009; 1793:368 - 77; http://dx.doi.org/10.1016/j.bbamcr.2008.10.013; PMID: 19046997
- Wang GL, Jiang H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510 - 4; http://dx.doi.org/10.1073/pnas.92.12.5510; PMID: 7539918
- Puca R, Nardinocchi L, D’Orazi G. Regulation of vascular endothelial growth factor expression by homeodomain-interacting protein kinase-2. J Exp Clin Cancer Res 2008; 27:22; http://dx.doi.org/10.1186/1756-9966-27-22; PMID: 18644116
- Choi CY, Kim YH, Kwon HJ, Kim Y. The Homeodomain protein NK-3 recruits Groucho and histone deacetylase complex to repress transcription. J Biol Chem 1999; 274:33194 - 7; http://dx.doi.org/10.1074/jbc.274.47.33194; PMID: 10559189
- Hendrix MJ, Seftor EA, Seftor RE, Trevor KT. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behaviour. Am J Pathol 1997; 150:483 - 95; PMID: 9033265
- Gilles C, Polette M, Zahn JM, Tournier JM, Volders L, Foidart JM, et al. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci 1999; 112:4615 - 25; PMID: 10574710
- Singh S, Sadacharan S, Su S, Belldegrun A, Persad S, Singh G. Overexpression of vimentin: role in the invasive phenotype in an androgen-independent model of prostate cancer. Cancer Res 2003; 63:2306 - 11; PMID: 12727854
- McInroy L, Maatta A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun 2007; 360:109 - 14; http://dx.doi.org/10.1016/j.bbrc.2007.06.036; PMID: 17585878
- Calzado MA, de la Vega L, Moller A, Bowtell DD, Schmitz ML. An inducible autoregulatory loop between HIPK2 and Siah2 at the apex of the hypoxic response. Nat Cell Biol 2009; 11:85 - 91; http://dx.doi.org/10.1038/ncb1816; PMID: 19043406
- Michieli P. Hypoxia, angiogenesis and cancer therapy. Cell Cycle 2009; 8:3291; http://dx.doi.org/10.4161/cc.8.20.9741; PMID: 19770588
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010; 29:625 - 34; http://dx.doi.org/10.1038/onc.2009.441; PMID: 19946328
- Nardinocchi L, Puca R, Givol D, D’Orazi G. HIPK2: A therapeutical target to be (re)activated for tumor suppression. Role in p53 activation and HIF-1α inhibition. Cell Cycle 2010; 9:1 - 2; http://dx.doi.org/10.4161/cc.9.7.11125; PMID: 20016263
- Puca R, Nardinocchi L, Pistritto G, D’Orazi G. Overexpression of HIPK2 circumvents the blockade of apoptosis in chemoresistant ovarian cancer cells. Gynecol Oncol 2008; 109:403 - 10; http://dx.doi.org/10.1016/j.ygyno.2008.02.018; PMID: 18395248
- Bon G, Di Carlo S, Folgiero V, Avetrani P, Lazzari C, D’Orazi G, et al. Negative regulation of b4 integrin transcription by homeodomain-interacting protein kinase 2 and p53 inpmairs tumor progression. Cancer Res 2009; 69:5978 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-09-0244; PMID: 19567674
- Ballman KV, Grill DE, Oberg AL, Therneau TM. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics 2004; 20:2778 - 86; http://dx.doi.org/10.1093/bioinformatics/bth327; PMID: 15166021
- Zeisel A, Amir A, Kostler WJ, Domany E. Intensity dependent estimation of noise in microarrays improves detection of differentially expressed genes. BMC Bioinformatics 2010; 11:400; http://dx.doi.org/10.1186/1471-2105-11-400; PMID: 20663218
- Di Stefano V, Rinaldo C, Sacchi A, Soddu S, D’Orazi G. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Exp Cell Res 2004; 293:311 - 20; http://dx.doi.org/10.1016/j.yexcr.2003.09.032; PMID: 14729469
- Falk W, Goodwin RH Jr., Leonard EJ. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte results. J Immunol Methods 1980; 33:201 - 12; PMID: 6966301