Abstract
Organisms constantly undergo various stresses within their life span, which can damage their DNA. In order to maintain genomic stability and counteract the development of unwanted genomic mutations, organisms have evolved a DNA-damage response (DDR) to protect their genome. Due to the critical roles played by DDR in genomic stability, its defects can lead to cellular transformation and potentially tumorigenesis. Consequently, this also provides the opportunity to specifically target tumor cells due to a weakened ability to tolerate genotoxic stresses. In this lies a treatment strategy in which the inhibition of remaining DDR pathways can hyper-sensitize tumors to chemotherapeutic agents while minimizing deleterious effects to healthy cells. Therefore it is important to understand the genotypic background of specific tumors to determine which DDR pathways remain and can be targeted for inhibition. Tumor therapies based on the DDR are ideal not only as a means of increasing the effectiveness of current chemotherapies but also as a means to selectively target tumor cells while leaving healthy cells unharmed. Thus, targeting DDR components as a means of increasing effectiveness and discrimination of current chemotherapeutic tumor treatments is currently the focus of many studies and clinical trials.
Overview of DNA-Damage Responses and Signaling
The genome of living organisms is constantly under the attack of exogenous and endogenous agents that damage their DNA potentially leading to harmful events such as single- or double-strand DNA breaks (SSBs or DSBs).Citation1 It is imperative for the organisms to resolve such damage and protect their DNA to maintain genomic stability, allowing for proper replication and proliferation. To counteract the deleterious effects of DNA-damaging agents, organisms have developed DNA-damage responses (DDR) in order to maintain the stability of their genome and ensure proper replication and survival. This is achieved through the sensing of DNA-damaging events and transducing effecter kinases that regulate genes involved in various pathways that enable cells to tolerate their current state of stress.
The Ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) sensor kinases are recruited to sites of DNA damage. Upon recruitment they phosphorylate and activate a variety of target proteins that execute the DDR. The ATM sensor kinase has been implicated mainly in the activation of the DDR in response to DSBs in the genome induced by agents such as ionizing radiation.Citation2 ATM is recruited to sites of DSB through its interaction with Nbs1 of the Mre11-Rad50-Nbs1 (MRN) complex involved in DSB repair.Citation3–Citation5 The Ku70/Ku80 DNA-end-binding heterodimer has been implicated in the recruitment of the MRN complex allowing for the full activation of ATM in the presence of DSB.Citation6
The ATR sensor kinase is mainly involved in the activation of the DDR in response to single-strand DNA (ssDNA), which occurs upon stalling of a replication fork.Citation7 A stall may result from an adduct within the DNA caused by agents such as UV radiation.Citation7 The recruitment of ATR is mediated through its interaction with ATR-interacting protein (ATRIP) which in turn interacts with ssDNA-binding replication protein A (RPA).Citation8,Citation9 Complete ATR activation is achieved through interaction with the sensor Rad9Rad1-Hus1 (9-1-1) complex that also localizes to sites of stalled replication.Citation8,Citation9 This is mediated through independent recruitment of both the 9-1-1 complex and the ATRIP-ATR complex by RPA, as well as through interaction of the mediator protein TopBP1 with both the Rad9 subunit of the 9-1-1 complex and ATR.Citation8,Citation10–Citation14
The main substrates of the ATM and ATR sensor kinases are the effecter kinases Chk1 and Chk2 (). Chk1 can be activated by both ATM and ATR through phosphorylation in the presence of a variety of sources of DNA damage such as ionizing radiations, UV light, hydroxyurea and topoisomerase I inhibitors.Citation15,Citation16 Chk1 activation plays an important role in checkpoint regulation and can arrest the cell cycle at S and G2/M phases through its interaction with and phosphorylation of Cdc25 phosphatase, which is required for proper cell cycle progression.Citation17,Citation18 The phosphorylation signals degradation of Cdc25A during the G1/S phase transition and Cdc25C during the G2/M phase transition, effectively arresting the cell cycle though the increase in cyclin-dependent kinase phosphorylation.Citation19,Citation20 Furthermore, the Wee1 kinase, through Chk1 activation, aids in the G2 checkpoint through the phosphorylation and inhibition of Cdc2, which leads to cell cycle arrest.Citation21 Chk2 is involved in the activation of p53, an important tumor suppressor gene product, and multiple other substrates involved in cell cycle arrest and the induction of apoptosis.Citation22,Citation23 Chk2 may also activate the transcription factor E2F1, which can in turn lead to apoptosis in both a p53-dependent and -independent manner via positive feedback activation of ATM.Citation24–Citation26
Chk1 and Chk2 also share many redundant roles and substrates involved in the activation of the DDR and effector targets, such as the BRCA homologous recombination repair (HRR) pathway.Citation27 The Chk1/Chk2-dependent role in BRCA2-mediated HRR is achieved through its interaction with Rad51, a key component of the strand-invasion step of HRR.Citation28–Citation30 Brit1, another direct substrate of both ATM and ATR, is also a key regulator of the BRCA HRR pathway, both indirectly through Chk1 activation and through direct activation of BRCA1.Citation31
Due to important roles of DDR in maintaining genome stability and ensuring proper replication and proliferation, defects in this pathway are often associated with inherited diseases, particularly tumorigenesis. In order for tumors to develop, cell cycle checkpoint must be disarmed, which is often accompanied by defects in other DNA-repair mechanisms.Citation32 Thus many genes in the DDR pathway mediated by ATM and ATR are found to be defective to allow for tumor formation and progression.
The tight connection between DDR defects and tumors has been the focus of many anti-cancer therapies. Defects that have allowed the formation of the tumor have also left the deficient cells more vulnerable to genotoxic agents. Thus many therapies involve the selective killing of tumor cells due to their increased chemo- and radio-sensitivities in comparison to normal proliferating cells. By inhibiting the remaining repair pathway(s) tumor cells have, they would become extremely sensitive to the specific damaging agents, whereas normal cells contain multiple pathways to deal with genotoxic stresses and can still rely on their redundant repair pathways to tolerate the damage. Furthermore, the increased resistance normal cells display over tumor cells in the presence of specific inhibitors inducing a synthetic sensitivity may eventually lead to tumor therapies that leave normal cells entirely unharmed. These therapies based on DDR defects illustrate the importance of understanding the genotype of a specific tumor in order to best treat and remove cancer from infected organisms.
ATM and ATR Sensor Kinases as a Target for Inhibition and Cancer Therapy
The sensor kinases ATM and ATR have been well-characterized in protecting cells from exogenous sources of DNA damage, through activation of both checkpoint and other DNA-damage responses. As a result of the protective nature of the ATM and ATR pathways in genomic stability and inducing apoptosis, ATM and ATR themselves as well as other members of their respective signaling pathways have been chosen as targets for cancer-related therapies. The positive effect of the inhibition of ATM and ATR was first visualized through the radio- and chemo-sensitizing effects of caffeine.Citation33 It has also been shown that caffeine has the ability to selectively kill p53 mutant cells.Citation34 These studies laid the basis for the studies centered on the inhibition of the ATM and ATR signaling pathways as a means of cancer therapy.
As a result of caffeine's ability to inhibit phosphoinositide-3-kinase (PI3K)-related kinases such as ATM and ATR, and thus enhance DNA-damage sensitivity, a search for more potent inhibitors of the PI3K kinases (PIKK) began. The compounds LY294002 and wortmannin were also found to inhibit PIKK to varying degrees; however, like caffeine, they lacked specificity to ATM and/or ATR.Citation35,Citation36 Using these drugs as a template for more specific inhibitors resulted in the identification of 2-morpholin-4-yl-6-thianthren-1-pyran-4-one (KU-55933) as a highly specific inhibitor for ATM.Citation37 The KU-55933 compound was found to induce sensitivity to IR and other DSB-inducing chemicals such as topoisomerase II inhibitors.Citation37 Recently, KU-55933 inhibition of ATM has been shown to prevent the complete activation of Akt,Citation38 a signal transducer overexpressed in some cancers, which promotes increased cell proliferation and survival.Citation39 It was found that tumor cells treated in combination with KU-55933 and rapamycin, which targets mTOR, a downstream target of Akt, not only induced apoptosis, but also decreased cell proliferation more than either drug alone.Citation38
The effects of PIKK inhibitors were analyzed to ensure that their use would only affect cells being challenged under DNA-damaging conditions. This led to the characterization of ATM inhibition synthetic lethal phenotype, via KU-55933, with genes involved in the Fanconi anemia pathway.Citation40 The ATM and Fanconi anemia pathways appear to play parallel and compensatory roles in maintaining genomic stability and cell viability.Citation40 DNA replication-fork stalling can be resolved through activation of the Fanconi anemia pathway, which can stabilize and coordinate repair at the fork through the promotion of HRR and translesion synthesis.Citation41 Currently, the role of ATM in the tolerance of DNA replication-fork stalling is unclear. ATM may provide time for the resolution of the stalled fork through activation of DDR checkpoints or potentially play a direct role in repair through signaling non-homologous end joiningCitation42 or HRR.Citation43 The potential for targeting ATM, as a means of inducing synthetic lethality, is very promising as many Fanconi anemia complementation (FANC) group genes are often mutated in a variety of cancers, such as head, neck, lung, ovarian and cervical cancers.Citation40 The reverse synthetic lethality was demonstrated utilizing ATM-deficient tumor cells treated with EF24, a potent inhibitor for FANCD2.Citation44 This along with other synthetic lethal phenotypes has potential to be an effective means of combating specific tumors without harming normal cells.
Due to their radio- and chemo-sensitization phenotypes, the inhibition of both ATM and ATR has been pursued. The compound schisandrin B (SchB) was identified during a screen for potent inhibitors of ATR.Citation45 SchB, a dibenzocyclooctadiene, is derived from Fructus schisandrae, and has been utilized as a traditional Chinese remedy for the treatment of hepatitis and myocardial disorders.Citation46,Citation47 It has been shown to act as a specific inhibitor for ATR, causing decreased phosphorylation of downstream targets BRCA1 and Chk1, leading to the abrogation of the G2/M checkpoint following UV treatment.Citation45 The specific nature of ATR inhibition allows for a novel targeting of cancer cells to undergo apoptosis that have lost p53-dependent apoptotic function due to p53 mutations. This scenario is supported by the ability of SchB to enhance doxorubicin-induced apoptosis of tumor cells, while leaving normal cells unaffected.Citation48 Hence, SchB becomes an excellent candidate for cancer therapy, although further studies are required to determine its clinical feasibility.
Through studies showing the tendency of Atr+/− Mlh1−/− (a component of mismatch repair, MMR) mice to develop tumors,Citation49 it has been suggested that the ATR pathway may act in concert with MMR to maintain genome stability. This has been attributed to the accumulation of chromosome instability (CIN), as suppression of both ATR and MMR by siRNA resulted in an increased susceptibility to CIN.Citation50 The increase in CIN was also observed when either ATR or Chk1 was depleted in MMR-defective cell lines.Citation50 The clinical significance of this phenotype was addressed by showing that tumor cell lines deficient in both ATR and MMR pathways displayed an increased sensitivity over cells deficient in either of the individual pathways alone to the chemotherapeutic agent 5-fluorouracil (5-FU).Citation50 The mechanism of this synthetic relationship in increasing susceptibility to chemotherapeutic agents is not fully understood, but may be attributed to an increased incidence of DSB when both pathways are compromised.Citation50 The connection between ATR and MMR defects has therapeutic potential in treating nonpolyposis colon carcinomas that are characteristically resistant to 5-FU,Citation51 as well as the wide variety of other common MMR-deficient tumors such as colorectal, gastric, endometrial and bladder cancers.Citation52 Thus, the harnessing of this synthetic phenotype may lead to the ability to lower doses of chemotherapeutic drugs reducing damage to healthy cells.
Recently identified compounds, NU6027 and VE-821, have been shown to exhibit potent and specific inhibition of ATR while not affecting other PIKK. NU6027 has been shown to enhance hydroxyurea and cisplatin cytotoxicity in an ATR-dependent manner as well as display a synthetic lethal phenotype with defects in SSB repair, such as PARP-1 inhibition.Citation53 Although, NU6027 studies reinforce targeting ATR inhibition as a potential cancer therapy it shows insufficient aqueous solubility for in vivo evaluation and is thus unlikely to be clinically pursued.Citation53 The inhibition of ATR by VE-821 has been shown to be lethal to HCT116 cancer cells alone and increase sensitivity to various genotoxic agents and antimetabolites.Citation54 VE-821 also induces a strong conditional synthetic lethal phenotype between the ATR and ATM-p53 tumor suppressor pathway in combination with cisplatin.Citation54 These studies support the pursuit of ATR inhibition and involve synthetic lethal phenotypes as a novel means of improving current tumor therapies.
BEZ-235, a well-characterized inhibitor of PI3K and mTOR, has recently been shown to inhibit additional PIKK, ATM and ATR.Citation55 The sensitizing effects of BEZ-235 on tumor cells displayed in multiple pre-clinical, phase I and II trails may in fact be a result of the inhibition of the DDR kinases, ATM and ATR as their effect cannot be explained through PI3K and mTOR inhibition alone.Citation55–Citation57 Additional studies and clinical trials should be designed accounting for the inhibition of ATM and ATR by BEZ-235 to harness the full potential this compound has as a therapeutic agent.
Chk1 and Chk2 Effecter Kinases as a Target for Inhibition and Cancer Therapy
The effecter kinases Chk1 and Chk2 are important targets for cancer therapy, as they act as direct substrates for the sensor kinases ATM and ATR and mediate the transduction of damage signals to many effecter proteins. The manipulation of this signal transduction is not only a therapeutic target, but also a source of tumorigenesis. Thus the dual nature of manipulating the DNA-damage signal transduction pathways creates difficulties in determining whether a specific effecter is an adequate therapeutic target.
Chk1 inhibition has been widely studied due to its ability to induce p53-independent apoptosis in response to cytotoxic drugs or radiation.Citation58,Citation59 Chk1 inhibitors, such as UCN-01, have the ability to induce apoptosis in p53−/− cells following IR, thus showing the promise of Chk1 inhibitors as therapeutic agents, since they can selectively target p53−/− tumors leaving cells with a functional checkpoint unharmed.Citation60 Lidamycin (LDM), an inducer of DSBs currently undergoing phase II clinical trials, can induce p53-independent apoptosis which selectively targets p53−/− cells due to their lack of a functional Chk1-mediated G2/M checkpoint.Citation61,Citation62 Another Chk1 inhibitor, AZD7762, has been shown to sensitize multiple tumor xenographs by interfering with HRR and the abrogation of the G2/M checkpoint.Citation63
UCN-01, the first Chk1 inhibitor tested in human trials has experienced problems during clinical trials due to non-selective inhibition and poor in vivo activity, which raises questions regarding its ability to be utilized as an effective anti-cancer agent.Citation64,Citation65 Additional Chk1 inhibitors that show more promise, AZD7762, SCH900776, LY2603618 and LY2606368, are currently undergoing clinical trials. AZD7762 and SCH900776 are in phase I clinical trials in combination with gemcitabine, a nucleoside analog, and irinotecan, a topoisomerase I inhibitor.Citation66–Citation68 Further studies involving AZD7762 have supported its use in combination with gemcitabine and radiation as a therapy for advanced pancreatic cancers.Citation69 LY2606368 is undergoing a sole phase I trial, unaccompanied by any genotoxic agents, and LY2603618 is also being assessed in multiple phase I and II clinical trails in combination with a variety of chemotherapeutic agents. These clinical studies further support the use of Chk1 inhibition as a means to sensitize tumors to current therapies.
Chk2 inhibition has shown significantly less potential and more controversy as a target for cancer therapy in comparison to Chk1. Many inhibitors of Chk2 show very low specificity and display inhibition of Chk1 as well.Citation70 In addition, inhibition of Chk2 via complete knockdown in HCT116 cells expressing various levels of p53 showed no inhibition of G2/M checkpoint or LDM-induced apoptosis as well as no increase in sensitivity when combined with a Chk1 knockdown.Citation62 Multiple lines of evidence show that Chk2 inhibition by interference RNA offers no further therapeutic value over the inhibition of Chk1 alone,Citation71 or when combined with antimetabolites.Citation63
However, there are also studies that support the use of Chk2 inhibition as a tumor therapy as it leads to mitotic catastropheCitation72 and interference RNA or expression of a dominant negative CHK2 leads to enhanced apoptosis following IR or doxorubicin treatment.Citation73 Chk2 has recently been implicated in mitosis as well as genomic stability, as the loss of Chk2 is sufficient to induce CIN,Citation74 providing new phenotypes to potentially lead to novel therapeutic targets. The potential role(s) of Chk2 in genomic stability is further supported in murine studies showing inhibition of both p53 and Chk2 leads to an additive genomic instability following irradiation.Citation75
Chk2 inhibition shows potential in cancer therapies as a means of not only reducing tumor cells but also protecting normal proliferating cells. Chk2-deficient mice exhibit radio-resistance indicating a radio-protective role of Chk2 inhibitors for somatic cells.Citation76 Chk2 inhibition-induced radioprotection has been further validated through analysis of radio-resistance of normal proliferating cells upon treatment with Chk2 inhibitors, 2-arylbenzimidazoleCitation77 and VRX0466617.Citation78 However the radio-protective aspect of Chk2 inhibition must be extensively studied before being clinically considered due to the potential of increasing resistance to radiation therapy of tumor cells as well as healthy cells.
Downstream Effecters as Targets of Synthetic Lethality-Based Therapies
In order for tumor cells to develop, DDR defects must accumulate to allow for uncontrolled proliferation. These characteristics may be exploited in a number of ways such as radio- or chemosensitization or through synthetic lethality by simultaneous loss of two separate but nevertheless redundant DNA-repair pathways. Tumor treatments which harness synthetic lethal phenotypes are not only beneficial as they increase the effectiveness of the chemotherapeutic agent but also as a means of protecting healthy cells by selectively sensitizing the defective tumors. A well-characterized synthetic lethal phenotype that is currently utilized as a means of radio-sensitizing cancer cells consists of creating defects in both HRR and base excision repair (BER) pathways.
Poly (ADP-ribose) polymerase 1 (PARP-1) has been investigated as a target for cancer therapy and for inducing synthetic lethality due to its key regulatory role in the BER pathway through interaction with SSBs.Citation79,Citation80 PARP-1 inhibitors have exhibited the ability to sensitize tumor cells to cytotoxic agents such as temozolomide, a topoisomerase I inhibitor, platinum and radiations.Citation81–Citation83 PARP-1 inhibition has been evaluated in phase II clinical trials and tumors showed specific sensitization to temozolomide.Citation83–Citation86 Furthermore, PARP-1 inhibition caused selective killing of tumors deficient in BRCA1 or BRCA2 with minimal effect on normal cells,Citation87,Citation88 which is largely attributed to the defect in HRR.Citation89 However, PARP-1 inhibition is not limited to BRCA1 and BRCA2 mutations.Citation90–Citation93 In an HRR-deficient background, PARP-1 inhibition induces genomic instability and lethality probably through aberrant activation of the error-prone process of non-homologous end joining.Citation94 The requirement of non-homologous end joining and/or HRR for the synthetic lethal interactions with PARP-1 inhibition reinforces the need to determine the genetic background of tumor cell lines to determine optimal treatment. Additional phase I and II clinical trials have confirmed the ability of PARP-1 inhibition to sensitize BRCA1- and BRCA2-deficient tumors to cytotoxic agents as a result of the synthetic lethality due to the loss of both HRR and BER.Citation86,Citation95–Citation97 As a result of the success of PARP-1 inhibition in targeting HRR-deficient tumors, multiple phase I and II clinical trials are being conducted to examine other potential synthetic lethal phenotypes PARP-1 exhibits with DSB repair and ATM pathways.Citation93,Citation98 The PARP-1 inhibitor, BSI-201, also known as iniparib, is currently undergoing phase II clinical trials for treatment of BRCA-deficient tumors as well as phase III clinical trials administering iniparib in conjunction with chemotherapy drugs gemcitabine and carboplatin.Citation99 ABT-888, also known as Veliparib, is currently undergoing a wide variety of phase I and II clinical trials in combination with various genotoxic and radiation therapies.Citation100–Citation112
Cell cycle checkpoint inhibition is a common target for cancer therapy utilized to induce p53-independent apoptosis. Thus, Wee1, a tyrosine kinase involved in the G2/M checkpoint,Citation113 has been a focus of interest. Wee1 inhibition, just as Chk1 described previously, was able to preferentially sensitize p53−/− tumor cells in the presence of DNA damage due to the loss of both p53-dependent and independent cell cycle checkpoints.Citation114 The synthetic lethal phenotype was further confirmed through small molecule inhibition of Wee1 in p53-deficient tumor cells.Citation115,Citation116 Wee1 inhibition shows promising results as a therapeutic agent in preclinical and its first clinical trials utilizing MK-1775, a small molecule inhibitor of Wee1.Citation117 Recent studies have confirmed Wee1 inhibition by MK-1775 to selectively sensitize p53-deficient tumor-derived cells to the chemotherapeutic drug gemcitabine.Citation118 Other Wee1 inhibitors have been shown to increase the effect of selective radiation killing in specific tumors.Citation119
Synthetic lethal phenotypes are of great interest in the field of cancer therapy, as they allow for selective killing of tumor cells deficient in various DDR pathways. As a result of this potential benefit, many screens are being undertaken to find novel targets of inhibition to induce synthetic lethality in tumors. Brit1, a positive regulator of homologous recombination repair which is activated through both ATM and ATR, is found to be misregulated in many murine tumorsCitation31 and is thus the current subject of highthroughput screening to identify inhibitor compounds that may induce a novel synthetic lethal phenotype to be potentially used as a therapeutic agent.Citation120
Conclusion
Conditional synthetic lethality as a means of treating tumors is becoming an increasingly powerful tool in the fight against cancer. This strategy has two appealing aspects that make it superior to classical treatment of tumors that rely solely on chemo- and radiotherapies. The synthetic lethality induced by targeting the DDR sensitizes tumor cell lines to classical treatments while normal cells are affected to a far lesser extent. This permits selective killing of tumor cells and may potentially even allow for chemo- and radiotherapies that completely abolish negative effects to normal healthy cells.
The current state of knowledge with regard to DDR has allowed for the development of many genetically inspired therapies specifically targeting tumor cells based on their unique phenotypes. Some selected DDR inhibitors undergoing clinical trials are listed in . However, the understanding of DDR and other differentially regulated pathways within tumor cells is still incomplete. Furthermore, the known components of DDR have yet to be exhaustively tested as targets to allow for the selective killing of tumor cells. Thus, continued research is required to hopefully identify novel components of DDR that may serve as targets for tumor therapies. The search for chemicals and drugs acting on known targets within DDR must also be expanded in the hope of identifying novel selective therapies.
Figures and Tables
Figure 1 ATM/ATR DNA-damage response pathway. Open boxes indicate damage signals; gray boxes indicate the genetic components; filled boxes represent the downstream consequences of the response pathway. Arrows indicate the ability of one component of the pathway to lead to the activation of a subsequent component.
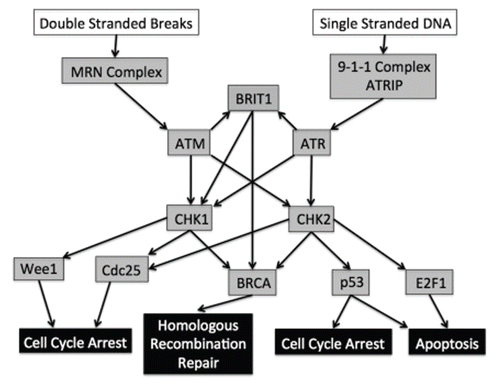
Table 1 Inhibitors of the DDR undergoing clinical trials
Acknowledgments
We wish to thank Michelle Hanna for proofreading the manuscript and other laboratory members for helpful discussion. This work was supported by Natural Sciences and Engineering Research Council of Canada Discovery Grant No. 138338-2009 to W.X., M.B. is a recipient of the Graduate Scholarship from the College of Medicine, University of Saskatchewan.
References
- Featherstone C, Jackson SP. DNA double-strand break repair. Curr Biol 1999; 9:759 - 761; PMID: 10531043; http://dx.doi.org/10.1016/S0960-9822(00)80005-6
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177 - 2196; PMID: 11544175; http://dx.doi.org/10.1101/gad.914401
- D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317 - 327; PMID: 11988766; http://dx.doi.org/10.1038/nrm805
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499 - 506; PMID: 12556884; http://dx.doi.org/10.1038/nature01368
- Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 2003; 13:458 - 462; PMID: 12946624; http://dx.doi.org/10.1016/S0962-8924(03)00170-3
- Ball LG, Xiao W. Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacol Sin 2005; 26:897 - 907; PMID: 16038621; http://dx.doi.org/10.1111/j.1745-7254.2005.00165.x
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA 2007; 104:19855 - 19860; PMID: 18077418; http://dx.doi.org/10.1073/pnas.0707579104
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542 - 1548; PMID: 12791985; http://dx.doi.org/10.1126/science.1083430
- Rouse J, Jackson SP. Interfaces between the detection, signaling and repair of DNA damage. Science 2002; 297:547 - 551; PMID: 12142523; http://dx.doi.org/10.1126/science.1074740
- Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 2001; 294:867 - 870; PMID: 11679674; http://dx.doi.org/10.1126/science.1063827
- Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem 2006; 281:27855 - 27861; PMID: 16864589; http://dx.doi.org/10.1074/jbc.M605176200
- Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 2007; 282:28036 - 28044; PMID: 17636252; http://dx.doi.org/10.1074/jbc.M704635200
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 2007; 21:1472 - 1477; PMID: 17575048; http://dx.doi.org/10.1101/gad.1547007
- Warmerdam DO, Kanaar R. Dealing with DNA damage: relationships between checkpoint and repair pathways. Mutat Res 2010; 704:2 - 11; PMID: 20006736; http://dx.doi.org/10.1016/j.mrrev.2009.12.001
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 1459; PMID: 10859164
- Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Bio Chem 2003; 278:14806 - 14811; PMID: 12588868; http://dx.doi.org/10.1074/jbc.M210862200
- Sørensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3:247 - 258; PMID: 12676583; http://dx.doi.org/10.1016/S1535-6108(03)00048-5
- Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 2002; 21:5911 - 5920; PMID: 12411508; http://dx.doi.org/10.1093/emboj/cdf567
- Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol 2006; 16:150 - 159; PMID: 16360315; http://dx.doi.org/10.1016/j.cub.2005.11.066
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842 - 847; PMID: 11298456; http://dx.doi.org/10.1038/35071124
- Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol 1996; 134:963 - 970; PMID: 8769420; http://dx.doi.org/10.1083/jcb.134.4.963
- Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer 2004; 4:216 - 225; PMID: 14993903; http://dx.doi.org/10.1038/nrc1296
- Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res 2007; 35:7475 - 7484; PMID: 17942417; http://dx.doi.org/10.1093/nar/gkm744
- Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol 2003; 5:401 - 409; PMID: 12717439; http://dx.doi.org/10.1038/ncb974
- Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, et al. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol 2004; 24:2968 - 2977; PMID: 15024084; http://dx.doi.org/10.1128/MCB.24.7.2968-77.2004
- Powers JT, Hong S, Mayhew CN, Rogers PM, Knudsen ES, Johnson DG. E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation and apoptosis. Mol Cancer Res 2004; 2:203 - 214; PMID: 15140942
- Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008; 27:3977 - 3985; PMID: 18317453; http://dx.doi.org/10.1038/onc.2008.17
- Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Bio Chem 1997; 272:31941 - 31944; PMID: 9405383; http://dx.doi.org/10.1074/jbc.272.51.31941
- Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA 1998; 95:5287 - 5292; PMID: 9560268; http://dx.doi.org/10.1073/pnas.95.9.5287
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4:435 - 445; PMID: 12778123; http://dx.doi.org/10.1038/nrm1127
- Rai R, Dai H, Multani AS, Li K, Chin K, Gray J, et al. BRIT1 regulates early DNA damage response, chromosomal integrity and cancer. Cancer Cell 2006; 10:145 - 157; PMID: 16872911; http://dx.doi.org/10.1016/j.ccr.2006.07.002
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26:7773 - 7779; PMID: 18066090; http://dx.doi.org/10.1038/sj.onc.1210881
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 1999; 59:4375 - 4382; PMID: 10485486
- Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D, et al. Differential sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res 1995; 55:1643 - 1648; PMID: 7712468
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide-3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 1998; 58:4375 - 4382; PMID: 9766667
- Izzard RA, Jackson SP, Smith GC. Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res 1999; 59:2581 - 2586; PMID: 10363977
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152 - 9159; PMID: 15604286; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727
- Li Y, Yang DQ. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther 2010; 9:113 - 125; PMID: 20053781; http://dx.doi.org/10.1158/1535-7163.MCT-08-1189
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14:381 - 395; PMID: 11882383; http://dx.doi.org/10.1016/S0898-6568(01)00271-6
- Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest 2007; 117:1440 - 1449; PMID: 17431503; http://dx.doi.org/10.1172/JCI31245
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 2004; 15:607 - 620; PMID: 15327776; http://dx.doi.org/10.1016/j.molcel.2004.08.009
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis and proteins locating to gamma-H2AX foci. Mol Cell 2004; 16:715 - 724; PMID: 15574327; http://dx.doi.org/10.1016/j.molcel.2004.10.029
- Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000; 19:463 - 471; PMID: 10654944; http://dx.doi.org/10.1093/emboj/19.3.463
- Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, Turker MS, et al. Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 2009; 8:133; PMID: 20043851; http://dx.doi.org/10.1186/1476-4598-8-133
- Nishida H, Tatewaki N, Nakajima Y, Magara T, Ko KM, Hamamori Y, et al. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res 2009; 37:5678 - 5689; PMID: 19625493; http://dx.doi.org/10.1093/nar/gkp593
- Liu GT. Pharmacological actions and clinical use of fructus schizandrae. Chin Med J (Engl) 1989; 102:740 - 749; PMID: 2517053
- Ko KM, Mak DH, Chiu PY, Poon MK. Pharmacological basis of ‘Yang-invigoration’ in Chinese medicine. Trends Pharmacol Sci 2004; 25:3 - 6; PMID: 14723971; http://dx.doi.org/10.1016/j.tips.2003.11.002
- Li L, Lu Q, Shen Y, Hu X. Schisandrin B enhances doxorubicin-induced apoptosis of cancer cells but not normal cells. Biochem Pharmacol 2006; 71:584 - 595; PMID: 16405922; http://dx.doi.org/10.1016/j.bcp.2005.11.026
- Fang Y, Tsao CC, Goodman BK, Furumai R, Tirado CA, Abraham RT, et al. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J 2004; 23:3164 - 3174; PMID: 15282542; http://dx.doi.org/10.1038/sj.emboj.7600315
- Jardim MJ, Wang Q, Furumai R, Wakeman T, Goodman BK, Wang XF. Reduced ATR or Chk1 expression leads to chromosome instability and chemosensitization of mismatch repair-deficient colorectal cancer cells. Mol Biol Cell 2009; 20:3801 - 3809; PMID: 19570909; http://dx.doi.org/10.1091/mbc.E09-04-0303
- Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer 2003; 106:66 - 73; PMID: 12794758; http://dx.doi.org/10.1002/ijc.11176
- Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat Rev Clin Oncol 2010; 7:197 - 208; PMID: 20177404; http://dx.doi.org/10.1038/nrclinonc.2010.18
- Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 2011; 105:372 - 381; PMID: 21730979; http://dx.doi.org/10.1038/bjc.2011.243
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428 - 430; PMID: 21490603; http://dx.doi.org/10.1038/nchembio.573
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 2011; 18:721 - 727; PMID: 21552262; http://dx.doi.org/10.1038/nsmb.2076
- Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, et al. Dual phosphoinositide-3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res 2009; 69:7644 - 7652; PMID: 19789349; http://dx.doi.org/10.1158/0008-5472.CAN-09-0823
- Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res 2010; 70:9693 - 9702; PMID: 21098704; http://dx.doi.org/10.1158/0008-5472.CAN-10-2286
- Janetka JW, Ashwell S, Zabludoff S, Lyne P. Inhibitors of checkpoint kinases: from discovery to the clinic. Curr Opin Drug Discov Devel 2007; 10:473 - 486; PMID: 17659489
- Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 2008; 7:2955 - 2966; PMID: 18790776; http://dx.doi.org/10.1158/1535-7163.MCT-08-0492
- Petersen L, Hasvold G, Lukas J, Bartek J, Syljuasen RG. p53-dependent G(1) arrest in 1st or 2nd cell cycle may protect human cancer cells from cell death after treatment with ionizing radiation and Chk1 inhibitors. Cell Prolif 2010; 43:365 - 371; PMID: 20590661; http://dx.doi.org/10.1111/j.1365-2184.2010.00685.x
- Shao RG, Zhen YS. Enediyne anticancer antibiotic lidamycin: chemistry, biology and pharmacology. Anticancer Agents Med Chem 2008; 8:123 - 131; PMID: 18288918; http://dx.doi.org/10.2174/187152008783497055
- Pan Y, Ren KH, He HW, Shao RG. Knockdown of Chk1 sensitizes human colon carcinoma HCT116 cells in a p53-dependent manner to lidamycin through abrogation of a G2/M checkpoint and induction of apoptosis. Cancer Biol Ther 2009; 8:1559 - 1566; PMID: 19502782; http://dx.doi.org/10.4161/cbt.8.16.8955
- Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle 2006; 5:1983 - 1988; PMID: 16931916; http://dx.doi.org/10.4161/cc.5.17.3184
- Kortmansky J, Shah MA, Kaubisch A, Weyerbacher A, Yi S, Tong W, et al. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J Clin Oncol 2005; 23:1875 - 1884; PMID: 15699481; http://dx.doi.org/10.1200/JCO.2005.03.116
- Fuse E, Kuwabara T, Sparreboom A, Sausville EA, Figg WD. Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol 2005; 45:394 - 403; PMID: 15778420; http://dx.doi.org/10.1177/0091270005274549
- Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the Replication Checkpoint Using SCH 900776, a Potent and Functionally Selective CHK1 Inhibitor Identified via High Content Screening. Mol Cancer Ther 2011; 10:591 - 602; PMID: 21321066; http://dx.doi.org/10.1158/1535-7163.MCT-10-0928
- Ashwell S, Zabludoff S. DNA damage detection and repair pathways—recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res 2008; 14:4032 - 4037; PMID: 18593978; http://dx.doi.org/10.1158/1078-0432.CCR-07-5138
- Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer 2008; 98:523 - 528; PMID: 18231106; http://dx.doi.org/10.1038/sj.bjc.6604208
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972 - 4981; PMID: 20501833; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004; 3:513 - 519; PMID: 15078995
- Xiao Z, Xue J, Sowin TJ, Zhang H. Differential roles of checkpoint kinase 1, checkpoint kinase 2 and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Mol Cancer Ther 2006; 5:1935 - 1943; PMID: 16928813; http://dx.doi.org/10.1158/1535-7163.MCT-06-0077
- Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R, et al. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 2004; 23:4353 - 4361; PMID: 15048074; http://dx.doi.org/10.1038/sj.onc.1207573
- Ghosh JC, Dohi T, Raskett CM, Kowalik TF, Altieri DC. Activated checkpoint kinase 2 provides a survival signal for tumor cells. Cancer Res 2006; 66:11576 - 11579; PMID: 17178848; http://dx.doi.org/10.1158/0008-5472.CAN-06-3095
- Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 2010; 12:492 - 499; PMID: 20364141; http://dx.doi.org/10.1038/ncb2051
- MacLaren A, Slavin D, McGowan CH. Chk2 protects against radiation-induced genomic instability. Radiat Res 2009; 172:463 - 472; PMID: 19772467; http://dx.doi.org/10.1667/RR1603.1
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, et al. Chk2-deficient mice exhibit radio-resistance and defective p53-mediated transcription. EMBO J 2002; 21:5195 - 5205; PMID: 12356735; http://dx.doi.org/10.1093/emboj/cdf506
- Arienti KL, Brunmark A, Axe FU, McClure K, Lee A, Blevitt J, et al. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J Med Chem 2005; 48:1873 - 1885; PMID: 15771432; http://dx.doi.org/10.1021/jm0495935
- Carlessi L, Buscemi G, Larson G, Hong Z, Wu JZ, Delia D. Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther 2007; 6:935 - 944; PMID: 17363488; http://dx.doi.org/10.1158/1535-7163.MCT-06-0567
- Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709 - 715; PMID: 8469282; http://dx.doi.org/10.1038/362709a0
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res 2007; 13:1383 - 1388; PMID: 17332279; http://dx.doi.org/10.1158/1078-0432.CCR-06-2260
- Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst 2004; 96:56 - 67; PMID: 14709739; http://dx.doi.org/10.1093/jnci/djh005
- Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, et al. Chemopotentiation of temozolomide, irinotecan and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther 2003; 2:371 - 382; PMID: 12700281
- Miknyoczki S, Chang H, Grobelny J, Pritchard S, Worrell C, McGann N, et al. The selective poly(ADP-ribose) polymerase-1(2) inhibitor, CEP-8983, increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol Cancer Ther 2007; 6:2290 - 2302; PMID: 17699724; http://dx.doi.org/10.1158/15357163.MCT-07-0062
- Plummer R, Lorigan P, Evans J, Steven N, Middleton M, Wilson R, et al. First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM). J Clin Oncol 2006; 24:8013
- Plummer R, Middleton M, Wilson R, Jones C, Evans J, Robson L, et al. First in human phase I trial of the PARP inhibitor AG-014699 with temozolomide (TMZ) in patients (pts) with advanced solid tumors. J Clin Oncol 2005; 23:3065
- Penning TD. Small-molecule PARP modulators—current status and future therapeutic potential. Curr Opin Drug Discov Devel 2010; 13:577 - 586; PMID: 20812149
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913 - 917; PMID: 15829966; http://dx.doi.org/10.1038/nature03443
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 921; PMID: 15829967; http://dx.doi.org/10.1038/nature03445
- Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev 2008; 18:80 - 86; PMID: 18343102; http://dx.doi.org/10.1016/j.gde.2008.01.016
- Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009; 1:315 - 322; PMID: 20049735; http://dx.doi.org/10.1002/emmm.200900041
- Sourisseau T, Maniotis D, McCarthy A, Tang C, Lord CJ, Ashworth A, et al. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol Med 2010; 2:130 - 142; PMID: 20373286; http://dx.doi.org/10.1002/emmm.201000068
- Williamson CT, Muzik H, Turhan AG, Zamo A, O'Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther 2010; 9:347 - 357; PMID: 20124459; http://dx.doi.org/10.1158/1535-7163.MCT-09-0872
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010; 116:4578 - 4587; PMID: 20739657; http://dx.doi.org/10.1182/blood-2010-01-265769
- Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA 2011; 108:3406 - 3411; PMID: 21300883; http://dx.doi.org/10.1073/pnas.1013715108
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 2010; 376:245 - 251; PMID: 20609468; http://dx.doi.org/10.1016/S0140-6736(10)60893-8
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010; 376:235 - 244; PMID: 20609467; http://dx.doi.org/10.1016/S0140-6736(10)60892-6
- Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem 2009; 52:7170 - 7185; PMID: 19873981; http://dx.doi.org/10.1021/jm901188v
- Löser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther 2010; 9:1775 - 1787; PMID: 20530711; http://dx.doi.org/10.1158/1535-7163.MCT-09-1027
- Maxmen A. Beyond PARP inhibitors: agents in pipelines target DNA repair mechanisms. J Natl Cancer Inst 2010; 102:1110 - 1111; PMID: 20668266; http://dx.doi.org/10.1093/jnci/djq294
- Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, et al. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS ONE 2011; 6:24148; PMID: 21912620; http://dx.doi.org/10.1371/journal.pone.0024148
- Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, et al. Phase I Study of PARP Inhibitor ABT-888 in Combination with Topotecan in Adults with Refractory Solid Tumors and Lymphomas. Cancer Res 2011; 71:5626 - 5634; PMID: 21795476; http://dx.doi.org/10.1158/0008-5472.CAN-11-1227
- Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol 2011; 99:331 - 338; PMID: 21719137; http://dx.doi.org/10.1016/j.radonc.2011.05.084
- Shibata H, Miuma S, Saldivar JC, Huebner K. Response of subtype-specific human breast cancer-derived cells to poly(ADP-ribose) polymerase and checkpoint kinase 1 inhibition. Cancer Sci 2011; 102:1882 - 1888; PMID: 21707865; http://dx.doi.org/10.1111/j.1349-7006.2011.02016.x
- Huehls AM, Wagner JM, Huntoon CJ, Geng L, Erlichman C, Patel AG, et al. Poly(ADP-Ribose) polymerase inhibition synergizes with 5-fluorodeoxyuridine but not 5-fluorouracil in ovarian cancer cells. Cancer Res 2011; 71:4944 - 4954; PMID: 21613406; http://dx.doi.org/10.1158/0008-5472.CAN-11-0814
- Barreto-Andrade JC, Efimova EV, Mauceri HJ, Beckett MA, Sutton HG, Darga TE, et al. Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol Cancer Ther 2011; 10:1185 - 1193; PMID: 21571912; http://dx.doi.org/10.1158/1535-7163.MCT-11-0061
- Kruse V, Rottey S, De Backer O, Van Belle S, Cocquyt V, Denys H. PARP inhibitors in oncology: a new synthetic lethal approach to cancer therapy. Acta Clin Belg 2011; 66:2 - 9; PMID: 21485757
- Li X, Delzer J, Voorman R, de Morais SM, Lao Y. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos 2011; 39:1161 - 1169; PMID: 21436403; http://dx.doi.org/10.1124/dmd.110.037820
- Strimpakos AS, Syrigos KN, Saif MW. Translational research in pancreatic cancer. Highlights from the “2011 ASCO Gastrointestinal Cancers Symposium”. San Francisco, CA USA. January 20–22, 2011. JOP 2011; 12:120 - 122; PMID: 21386635
- Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, et al. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neurooncol 2011; 13:471 - 486; PMID: 21377995; http://dx.doi.org/10.1093/neuonc/nor011
- Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res 2011; 71:2632 - 2642; PMID: 21300766; http://dx.doi.org/10.1158/0008-5472.CAN-10-1120
- Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res 2011; 39:3607 - 3620; PMID: 21227924; http://dx.doi.org/10.1093/nar/gkq1304
- Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat 2011; 125:627 - 636; PMID: 21161370; http://dx.doi.org/10.1007/s10549-010-1293-1
- Rowley R, Hudson J, Young PG. The wee1 protein kinase is required for radiation-induced mitotic delay. Nature 1992; 356:353 - 355; PMID: 1549179; http://dx.doi.org/10.1038/356353a0
- Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther 2004; 3:305 - 313; PMID: 14726685; http://dx.doi.org/10.4161/cbt.3.3.697
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009; 8:2992 - 3000; PMID: 19887545; http://dx.doi.org/10.1158/1535-7163.MCT-09-0463
- Mizuarai S, Yamanaka K, Itadani H, Arai T, Nishibata T, Hirai H, et al. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer 2009; 8:34; PMID: 19500427; http://dx.doi.org/10.1186/1476-4598-8-34
- Stathis A, Oza A. Targeting Wee1-like protein kinase to treat cancer. Drug News Perspect 2010; 23:425 - 429; PMID: 20862394
- Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a Potent Wee1 Inhibitor, Synergizes with Gemcitabine to Achieve Tumor Regressions, Selectively in p53-Deficient Pancreatic Cancer Xenografts. Clin Cancer Res 2011; 17:2799 - 2806; PMID: 21389100; http://dx.doi.org/10.1158/1078-0432.CCR-10-2580
- PosthumaDeBoer J, Wurdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, et al. WEE1 inhibition sensitizes Osteosarcoma to Radiotherapy. BMC Cancer 2011; 11:156; PMID: 21529352; http://dx.doi.org/10.1186/1471-2407-11-156
- Liang Y, Lin SY, Brunicardi FC, Goss J, Li K. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg 2009; 33:661 - 666; PMID: 19034564; http://dx.doi.org/10.1007/s00268-008-9840-1