Abstract
The carcinogenic activity of bisphenol A (BPA) is responsible for stimulating growth in estrogen-dependent breast cancer tissues, cell lines and rodent studies. However, it is not fully understood how this compound promotes mammary carcinogenesis. In our study, we examined the effect of BPA on cellular proliferation and senescence in human mammary epithelial cells (HMEC). Exposure to BPA for 1 week at the early stage at passage 8 increased the proliferation and sphere size of HMEC at the later stage up to passage 16, suggesting that BPA has the capability to modulate cell growth in breast epithelial cells. Interestingly, the number of human heterochromatin protein-1γ positive cells, which is a marker of senescence, was also increased among BPA-treated cells. Consistent with these findings, the protein levels of both p16 and cyclin E, which are known to induce cellular senescence and promote proliferation, respectively, were increased in BPA-exposed HMEC. Furthermore, DNA methylation levels of genes related to development of most or all tumor types, such as BRCA1, CCNA1, CDKN2A (p16), THBS1, TNFRSF10C and TNFRSF10D, were increased in BPA-exposed HMEC. Our findings in the HMEC model suggested that the genetic and epigenetic alterations by BPA might damage HMEC function and result in complex activities related to cell proliferation and senescence, playing a role in mammary carcinogenesis.
Introduction
Among the known estrogen-like compounds, bisphenol A (BPA) has received a great deal of attention, because it is commonly found in the environment, as well as in human tissues and fluids.Citation1,Citation2 BPA has been detected in 92% of urine samples (0.4–149 μg/L) in a US reference population, suggesting people may be continuously exposed to this compound in their daily lives.Citation3,Citation4 Epidemiology studies have highlighted the correlation between BPA exposure and human cancers.Citation5 Animal studies have also shown that these low levels of BPA exposure may alter developmental programs of sensitive end organs during critical stages of early development, and increase mammary cancer risk in mouse models of breast cancer.Citation6,Citation7
The underlying mechanism involved in the carcinogenic activity of BPA has been studied in several animal and cell models. Betancourt et al.Citation8 demonstrated that changes in mammary gland protein expression of signaling pathways such as in the cell cycle, apoptosis, differentiation and migration are consistent with increased susceptibility for cancer development in rats prenatally exposed to BPA. Ptak et al.Citation9 also reported that exposure to environmental relevant concentrations of BPA can affect the cellular proliferation and expression of genes involved in the cell cycle and apoptosis in human ovarian cancer cells. However, studies with human breast cancer cells have yielded conflicting data,Citation10-Citation12 and the molecular mechanisms by which exposure to BPA at the early passage can affect breast cells at later passages are still unknown.Citation13 Furthermore, to our knowledge, there are a lack of data concerning the action of BPA on gene expression involved in cell proliferation and apoptosis in normal human mammary endothelial cells (HMEC).
There is general consensus that accumulation of cellular damage is the initiating event of both cancer and aging.Citation14 Tumorigenesis is fuelled by the accumulation of genetic and epigenetic damage. Similarly, aging occurs, at least in part, because of an accumulation of macromolecular damage, which initially affects cellular proteins, lipids and DNA, but eventually impairs tissue regeneration. Accordingly, those mechanisms that protect cells from damage could, in principle, protect against cancer and aging simultaneously.Citation15 In this regard, one potential mechanism by which estrogenic agents such as BPA may promote carcinogenesis might include increased DNA damage.Citation16 DNA damage induced by 17β-estradiol (E2) is more than 1000 times greater than that of BPA.Citation16 However, when BPA is compared with E2, a difference in biological effects is observed. For example, Bouskine et al.Citation17 reported that the human testicular seminoma cell-promoting effect of BPA is mediated through two signaling pathways of estrogen receptors and G-protein-coupled receptors (GPCRs). The GPCR pathway is not activated by stimulation of E2. Therefore, it is likely that the effects of BPA are based on estrogenic activities, but are not identical to those of E2. To further elucidate the biological effects of BPA on the mammary gland, gene expression profiling has also been performed using in vivo animal models.Citation18 The resulting data indicated that a high dose of BPA induces changes in genes related to differentiation, suggesting that this compound may have an adverse effect on developmental processes in the mammary gland.Citation18 However, although gene expression profiles are very informative in terms of detecting expression changes in target organs, it is difficult to precisely determine the corresponding molecular mechanisms in various differentiated cells such as ductal epithelial, stromal, and acinar cells in the in vivo system. To avoid issues of complexity when using in vivo systems, gene expression profiling has been performed using cultured cells treated with several environmental carcinogens.Citation19 In a similar manner, gene expression profiling following BPA exposure was performed in an earlier study using MCF-7 human breast cancer cells.Citation20 However, since MCF-7 cells are immortalized, they already harbor chromosomal abnormalities that are frequently observed in human malignant lesions.
In the current study, we evaluated the potential carcinogenic activity of BPA in HMEC, which are derived from normal human mammary epithelium, and therefore contain a normal karyotype.Citation21-Citation23 The long-term effects of BPA exposure at “low doses” were focused on in this study, with a low dose currently considered as < 2.19 × 10−7 M for in vitro cell or organ culture studies.Citation24,Citation25 We examined the effect of BPA exposure at early passage on proliferation, senescence, gene expression, and DNA methylation in HMEC at later passages.
Results
Effects of BPA exposure on the proliferation of HMEC
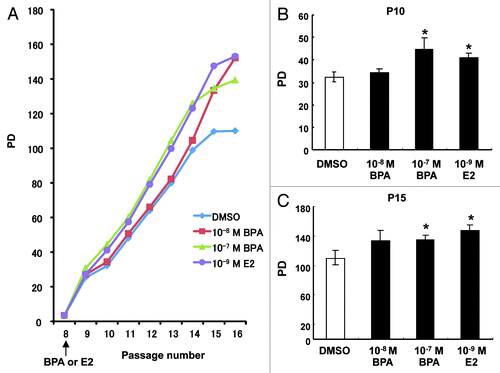
An important aspect of our experimental design was to expose HMEC to BPA for 7 d at passage 8 and then examine its effects at later passages. Exposure to BPA and E2 enhanced cell proliferation of HMEC and increasing effects were still evident until passage 16, even after removing BPA and E2 from the cell culture medium from passage 9 (). We statistically evaluated the differences in the speed of cell proliferation at the time points of passages 10 and 15. As shown in , proliferation of HMEC was significantly enhanced through treatment with 10−7 M BPA and 10−9 M E2 at passage 10 and passage 15, respectively. These observations indicated that increased proliferation induced by exposure to BPA or E2 at an early passage persists during later cell divisions.
Figure 1. Effects of BPA exposure on the proliferation of HMEC. (A) Cumulative PDs of cells treated with DMSO, 10−7 M or 10−8 M BPA, or 10−9 M E2 at passage 8 (7 d period). Representative results from one series of experiments are shown. (B) Calculation of the PDs of HMEC at passages 10 and 15. *p < 0.05 vs. the DMSO control.
Effects of BPA exposure on the colony formation of HMEC
We found that BPA and E2 treatment enhanced cell proliferation, possibly also resulting in enhanced cellular senescence. To elucidate whether the enhanced cell proliferation by BPA and E2 had any effect on the carcinogenic status of HMEC, we employed a 3D “on-top” assay that offers the advantages of both 2D and 3D analysis.Citation27
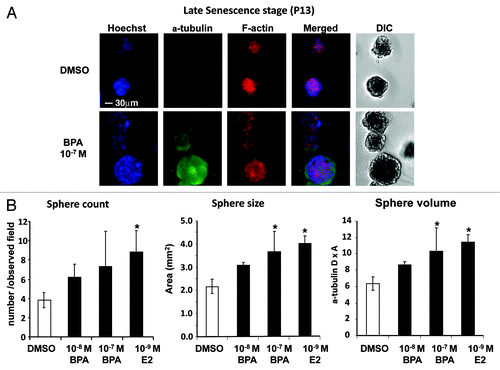
Cells covered with Matrigel produced rounded ductal colonies similar to spheres comprising a monolayer of epithelial cells, indicating that ductal formation had successfully occurred through coating with the Matrigel. The morphological status of HMEC at passage 13, which had been treated with BPA at passage 8, was assessed via the 3D “on-top” assay (). Significant increases in sphere size of HMEC were found in the 10−7 M BPA and 10−9 M E2 treatment groups (). The 3D “on-top” assay demonstrated that exposure to BPA and E2 at passage 8 can affect sphere formation in HMEC at later passages.
Figure 2. Effects of BPA exposure on colony formation of HMEC. Cells were treated with BPA or E2 at passage 8 (7 d period). (A) Morphology of colonies derived from HMEC at passage 13. Cells were stained with α-tubulin and F-actin antibodies. Cells were also counterstained with Hoechst, and the merged images and differential interference contrast (DIC) images are also shown (30 μm magnification). (B) Statistical analysis of sphere count, size, and volume of colonies derived from HMEC at passage 13. *p < 0.05 vs. the DMSO control.
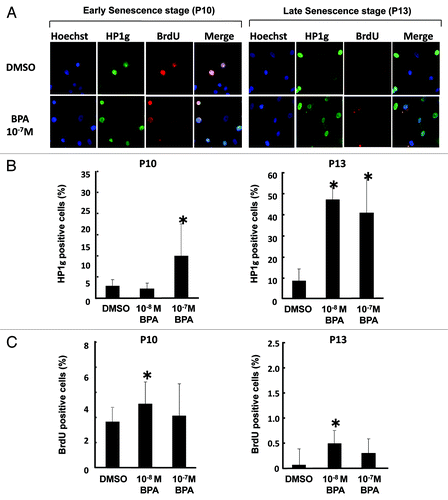
Recent studies have described that the balance between proliferation and senescence is important to develop cancer when normal cells are damaged by exogenous stimuli.Citation14,Citation15 We then wished to determine whether BPA exposure could alter the balance between proliferation and senescence in HMEC. Therefore, we simultaneously examined the expression of cell cycle, proliferation and senescence markers in HMEC at passages 10 and 13, 2 to 5 weeks post-chemical treatment. Three-color fluorescence imaging analyses characterized the distribution of cells of various stages with Hoechst, HP1γ, and BrdU (). Hoechst staining indicates the number of nuclei in all cells. BrdU incorporation into nuclei represents identification of cells in the early S phase. HP1γ-positive cells at the early senescence stage (passage 10) were significantly increased by exposure to 10−7 M BPA (, left panel). At the late senescence stage (passage 13), a significant increase in HP1γ-positive cells was observed by exposure to both 10−8 M and 10−7 M BPA (, right panel). A significant increase in BrdU-positive cells was demonstrated by exposure to 10−8 M BPA at passages 10 and 13 ().
Figure 3. Effects of BPA exposure on cellular senescence of HMEC. Cells were treated with BPA at passage 8 (7 d period). (A) Staining of HMEC at passages 10 and 13 with HP1 γ or BrdU antibodies. Cells were also counterstained with Hoechst, and the merged images are also shown (200 × magnification). (B) Number of HP1γ-positive cells. (C) Number of BrdU positive cells. *p < 0.05 vs. the DMSO control.
Gene and protein expression analysis
To determine the effects of BPA on the cellular growth of HMEC at the transcriptional level, gene expression analyses using a PCR array for mammary cancer-related genes were performed in HMEC at passage 11. Genes showing differential expression with BPA exposure are summarized in . It is noteworthy that the downregulated genes are associated with cell cycle control in many cases. A knowledge-based gene interaction network was then analyzed to determine how BPA at a dose of 10−8 M plays a role in signaling associated with cell cycle control (). CCNE1, CCNA2 and CDKN2A, which are among the key molecules underlying G1-S control during the cell cycle, were downregulated or unchanged in BPA-treated HMEC. Other factors related to cell growth such as EGFR, ERBB2, PTGS2, and IGFBP2 were increased by BPA treatment at 10−8 M or 10−7 M (). These observations indicate that the increased proliferation we observed in the BPA-treated cells may have been due to enhanced G1-S progression resulting from the decreased expression of negative cell cycle regulators.
Figure 4. Gene expression and network analysis. HEMC were treated with BPA or E2 at passage 8 (7 d period) and the relative mRNA expression of selected genes was measured at passage 11. (A) Effects of BPA exposure on cancer signaling gene expression. The results are expressed as the average of two independent experiments. Relative mRNA expression normalized to β-actin is shown as the log2 ratio, with the fold-change referring to the DMSO control cells. (B) Gene networks representing key genes for BPA exposure at 10−8 M were identified using GNCPro (SA Biosciences).
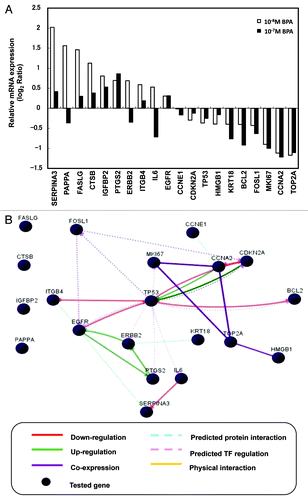
Knowledge-based network analysis was then performed to further clarify these findings. Network analysis for gene expression profiling at 10−8 M of BPA found that TP53 upregulated CDKN2A and CCNA2, and indirectly interacted with CCNE1, and that the upregulation of FASLG, CTSB, IGFBP2 and PAPPA were not associated with TP53.
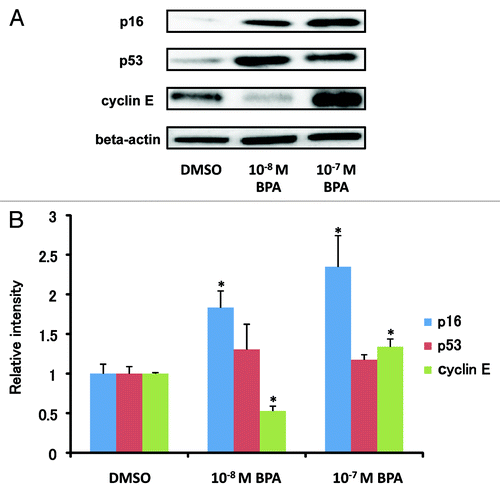
We also investigated the effects of BPA exposure on protein expression of p16 (CDKN2A), p53 (TP53) and Cyclin E (CCNE1) in HMEC using western blot analysis. As shown in , BPA exposure did not have a significant effect on p53 protein expression, while a dose-dependent increase in p16 protein expression was observed. Downregulation at 10−8 M BPA and upregulation at 10−7 M BPA of Cyclin E protein expression were also observed.
Figure 5. Modulation of the protein expression of p16, p53 and cyclin E in HEMC by BPA exposure. (A) Cells were treated with 10−8 M or 10−7 M BPA at passage 8 (7 d period) and protein expression of p16, p53 and cyclin E was measured at passage 11. (B) The cellular protein levels were calculated using ImageJ densitometry software and are expressed as the mean ± SD relative to DMSO control after normalizing the bands to β-actin. *p < 0.05 vs. the DMSO control.
DNA methylation patterns
Alterations in DNA methylation patterns are associated with the development of a variety of human cancers, including breast cancer.Citation28,Citation29 Previous studies have found promoter hypermethylation of in situ lesions and identified aberrant methylation at the promoters of candidate genes, which include GSTP1, CCND2, RARB2, TWIST1, RASSF1A, HIC1, CDKN2A, SFN (TP53), BRCA1, CCNA1, THBS1, TNFRSF and APC1.Citation30-Citation33 Therefore, BPA-induced changes in the methylation status of 24 gene promoters were investigated via quantitative real-time PCR arrays in this study. shows that 7 gene promoters exhibited changes of more than 10% in hypermethylation status between DMSO control and BPA-exposed cells (). These genes exhibited an increased percentage of promoter hypermethylation by BPA exposure including BRCA1, CCNA1, CDKN2A, THBS1, TNFRSF10C and TNFRSF10D, while HIC1 had decreased promoter hypermethylation by BPA exposure.
Table 1. Promotor methylation status of genes in HMEC at passage 12
Discussion
To investigate how BPA affects the carcinogenesis process in normal breast cells, we exposed HMEC to this agent at an early passage for a duration of 1 week and examined subsequent effects on cell proliferation, gene expression, and DNA methylation at the stage of later passages. HMEC are a model system for studying early events in mammary tumorigenesis, and they have a normal karyotype and enter senescence after a lengthy culture, while neoplastic cells are allowed to continuously grow, thus overcoming the barrier of cellular senescence.Citation34 HMEC morphology and growth status in in vitro culture assay have been shown to be closely connected with malignancy in a comparison involving 25 mammary-gland-derived cell lines.Citation27
In the current study, we focused on the effects of early-passage exposure to BPA in later-passage HMEC. In non-treated HMEC, the rate of cell growth slowed at approximately passage 15 (), indicating the onset of cellular senescence, as HMEC are not immortalized. In contrast, HMEC treated with E2 or BPA did not show a reduced cell proliferation rate at approximately passage 15 (). Our previous study showed that the telomerase activity of BeWo cells was enhanced by their exposure to E2 or 2,3,7,8-tetrachlorodibenzo-p-dioxin, a known endocrine disrupting agent.Citation35 This suggests that BPA might also upregulate telomerase activity in HMEC, resulting in an extended lifespan until a crisis point is eventually reached and cell division ceases.
To clarify the effects of BPA and E2 treatment on cell growth, a 3D “on-top” assay was performed using Matrigel, which is the optimal coating material for HMEC in this assay. Our optimized 3D “on-top” assay showed that BPA and E2 enhanced the nuclear count for each HMEC colony and increased the area of these colonies at passage 13. Kenny et al.Citation27 reported morphological differences among colonies under 3D “on-top” analysis of 25 mammary cancer-derived cell lines, and they could be classified into four groups of “round,” “mass,” “grape-like” and “satellite.” Relatively fewer malignant cells show round-shaped colonies in the 3D “on-top” assay, and most malignant grades result in grape-like or satellite shapes.Citation27 In our study, HMEC showed a “mass” shape (), and treatment with BPA or E2 did not change the shape of the colonies, but increased the size and cell numbers for each colony. These results indicated that BPA exposure at passage 8 had a subsequent effect on the cell growth of HMEC. The increased nuclear count and body area of spheres indicated overgrowth of the differentiated colony, possibly indicating a hyperplastic state. This may provide a key insight into the potential adverse effects of BPA upon the status of the mammary gland, which may result in carcinogenesis.
BPA exposure was found to increase the number of HP1γ-positive cells in HMEC at passages 10 and 13 in our study (). At the late senescence stage (passage 13), both 10−8 M and 10−7 M BPA exposure increased HP1γ-positive cells but not BrdU-positive cells. A previous study reported that the onset of senescence induces an increase in the number of positive nuclear bodies that contain HP1γ.Citation36,Citation37 HP1γ protein is also known to be positive for the entire nuclear area in the early S stageCitation38 and the G2/S stage of the cell cycle.Citation39,Citation40 When non-immortalized cells are close to their cell division limit, they often show positive SA-βgal activity in the cytoplasm and senescence-associated DNA foci in nuclei containing HP1γ.Citation37 Therefore, there are several possible explanations for the increased nuclear HP1γ-positive nuclei by BPA exposure in this study, such as enhanced cellular senescence or a delayed cell cycle progression during the G1/S stage.
To elucidate the effects of BPA on enhanced cellular senescence or a delayed cell cycle progression during the G1/S stage in HMEC, gene and protein expressions of mammary cancer-related genes were investigated in this study. Cyclin E protein expression was increased by exposure to BPA at the high dose (10−7 M), but not at the low dose (10−8 M) (). This might partly explain our findings that BPA exposure at the high dose but not the low dose at passage 8 significantly increased HEMC growth at passages 10 and 13 (). Cyclin E is cyclically expressed during the cell cycle, and it binds and activates the cyclin-dependent kinase Cdk2 and catalyzes the transition from the G1 phase to the S phase.Citation41,Citation42 Our findings suggest that Cyclin E might play an important role in BPA-induced cell growth of HMEC. However, a discrepancy between Cyclin E gene and protein expression following BPA treatment was observed in our study ( and ). The regulation of Cyclin E is through transcriptional regulatory mechanisms or through protein degradation by the proteasome pathway.Citation43 It is known that SCF (Skp1-Cullin-F-box) ubiquitin ligases regulate the degradation of many proteins involved in the control of cell division and growth.Citation44,Citation45 Indeed, it has been reported that the amount of Cyclin E protein present in the cell is tightly controlled by ubiquitin-mediated proteolysis and one ubiquitin ligase responsible for Cyclin E ubiquitination is known as SCFFbw7.Citation46,Citation47 Furthermore, it was found that phosphorylation within N- and C-terminal legions of Cyclin E plays a critical role in the binding of Cyclin E to SCFFbw7 and thus its ubiquitination and proteasomal degradation.Citation48 Therefore, one possible explanation of our results is that BPA exposure might affect the phosphorylation of Cyclin E. Interestingly, a recent study that analyzed the effects of a low dose of BPA in a testicular cell line revealed that this compound induces the activation of cAMP response-element-binding protein and strongly induces the phosphorylation of retinoblastoma protein (Rb).Citation17 These findings support our hypothesis that the effect of BPA exposure on mammary cell proliferation is related with the dysregulation of cell cycle regulatory genes, since the activation of Rb is also known to enhance the cell cycle, particularly at the G1-S transition. An animal study recently found that BPA exposure can significantly accelerate mammary tumorigenesis and metastasis in MMTV-erbB2 mice, and one of the underlying mechanisms includes the regulation of phosphorylation of proteins involved in the Akt pathway.Citation49 Because the overexpression of Cyclin E has been related to progression of a variety of cancers and constitutive expression of Cyclin E leads to genomic instability, further study on the mechanism by which long-term BPA exposure could mediate Cyclin E expression might provide an insight into the potential carcinogenic activity of BPA in the mammary gland.
Another interesting finding of our study is that BPA exposure appeared to promote cellular senescence and proliferation of HEMC simultaneously (, and ). This was supported by our findings that BPA exposure increased p16 protein expression in a dose-dependent manner; p16 has recently been found to promote aging in murine cells.Citation50 Indeed, there has been increased attention on the balance between convergent and divergent mechanisms of cancer and aging.Citation14,Citation15 Both cancer and aging are fuelled by the accumulation of cellular damage. Additionally, one concern regarding BPA is that differing doses of BPA may function in distinctly different, and sometimes opposing, manners at both the tissue and molecular level.Citation49 Our study suggests that effects of BPA exposure at differing doses on the balance of cancer and aging in the mammary gland might partly contribute to this issue.
Eckhardt et al.Citation51 reported that approximately one-third of the differentially methylated 50-UTRs were inversely correlated with transcription in normal tissues. Radpour et al. and other researchers also reported that 10 hypermethylated genes (APC, BIN1, BMP6, BRCA1, CST6, ESRb, GSTP1, CDKN2A, CDKN1A and TIMP3) were identified for distinguishing between cancerous and normal tissues.Citation52-Citation54 Therefore, hypermethylation of tumor suppressor genes causes the inactivation of genes that are important in suppressing the development of most or all tumor types. In our study, we observed increases in DNA hypermethylation of BRCA1, CCNA1, CDKN2A, THBS1, and TNFRSF in BPA-exposed HMEC (). We were interested in CDKN2A hypermethylation status by exposure to BPA because CDKN2A and GSTP1 showed significantly (p < 0.002) higher mean methylation levels in increasing grades (I, II, III) of invasive ductal breast cancer in a study by Moelan et al.Citation55 Interestingly, both of the two CDKN2A probes increased levels of DNA hypermethylation by exposure to BPA, although DNA hypermethylation levels of GSTP1 were not altered in our study. CDKN2A (p16), an inhibitor of the cyclin D-dependent protein kinases, is a tumor suppressor gene, and is altered in several tumor types.Citation56 Correlation of CDKN2A hypermethylation with CDKN2A protein loss has been previously reported and its loss of function has been associated with the development of a variety of cancers.Citation56-Citation58 Our finding is inconsistent with these previous studies, which found that exposure to BPA in HMEC at passage 8 induced CDKN2A protein expression at passage 11 and promoter hypermethylation at passage 12. However, a study using tumor tissues derived from patients diagnosed with endometrial carcinoma found that loss of nuclear p16 protein expression is not associated with promoter methylation.Citation59 Our study indicates that the mechanism by which BPA upregulates CDKN2A protein expression appears to be complicated and further study is required for clarification.
In summary, our present study is the first to address the long-lasting effects of BPA exposure over multiple cellular passages of HMEC. The underlying mechanism might include genetic and epigenetic dysregulation of cell cycle regulatory genes or tumor suppressor genes.
Materials and Methods
Chemicals
Dimethyl sulfoxide (DMSO) and E2 were obtained from Sigma Chemical Co. BPA was obtained from Wako Industries. DMSO was used as the primary solvent for all chemicals, and DMSO solutions were further diluted in cell culture media for treatment. The final concentrations of DMSO in the media did not exceed 0.1% (vol/vol).
Cell culture and chemical treatment
HMEC were obtained from Cambrex Bio Science and maintained in accordance with the supplier's instructions. Briefly, the cells were cultured in plastic dishes with MEGM SingleQuots medium in an incubator at 37°C in 5% CO2. HMEC were supplied at passage 7, and were grown to passage 8 prior to use in the experiments. BPA at final concentrations of 10−7 M and 10−8 M or E2 at 10−9 M was added to the culture media of passage 8 in HMECs and maintained for a period of 1 week. During this week, the culture media were changed twice with media containing the original BPA concentrations. The culture was then further grown until it was harvested for phenotype and gene expression analysis in media without chemicals. The cumulative population doublings (PDs) as a determinant of cellular lifespan were measured as previously described.Citation26 Briefly, the total number of cells harvested from each subculture was calculated and the number of accumulated PDs per passage was determined by the equation PD = (A/B)/log2, where A is the number of harvested cells and B is the number of plated cells.Citation26 Data were obtained from 6 duplicate cultures at each passage. The cumulative PDs from passage 8 to 16 were measured twice to validate the reproducibility of the results. The 3D “on-top” in vitro culture assay was performed essentially as described previously with different biocoated platesCitation27 (BD BioCoat™ Cellware Matrigel™ or Collagen Type I Coated Cellware plate, BD Biosciences).
Immunofluorescence cytochemistry
HMEC were transferred to 3D culture systems at passage 11 and maintained until passage 13. HMEC were then fixed in 4% neutralized paraformaldehyde solution for 60 min and blocked with 3% normal goat serum (NGS)/0.5% Triton X-100 in phosphate-buffered saline (PBS). The primary antibodies used were mouse monoclonal antibodies for human heterochromatin protein-1γ (HP1γ, S-19; Santa Cruz Biotechnology, sc-101004), and a rabbit polyclonal antiserum to α-tubulin (Abcam, ab15246). The secondary antibodies were anti-mouse Alexa 546 and Alexa Fluor 488 goat anti-rabbit IgG antibodies (Invitrogen). For the measurement of 5-bromo-2'-deoxyuridine (BrdU) incorporation during DNA synthesis, a cell proliferation fluorescence kit (GE Healthcare, 25–9001–89) was used according to the instruction manual. DNA was visualized by Hoechst staining (Wako Industries) and F-actin was visualized using Alexa Fluor 568 phalloidin (Invitrogen). Immunofluorescence staining signals were detected with an IN Cell Analyzer 1000 (GE Healthcare), a multiple-imaging analyzer, and morphological analysis was performed using IN Cell Investigator image analysis software (GE Healthcare).
Gene expression analysis
Total RNA was extracted using an RNeasy kit (Qiagen) when cells at passage 11 were approximately 70% confluent. These preparations were then used to detect the expression of 83 genes (Table S1), which have been reported to be frequently expressed in mammary cancers using a real-time RT-PCR method, Superarray-qPCR (Estrogen Receptor Signaling PCR Array, SA Biosciences). Gene expression was normalized by β-actin expression and set to 1 for the control DMSO-treated cells.
Western blot analysis
HMEC treated with BPA for a period of 1 week at passage 8 were lysed at passage 11 using RIPA buffer (Santa Sruz Biotechnology). After boiling at 99°C for 5 min, the protein samples were resolved by sodium dodecyl sulfate (SDS) PAGE on a 4–20% gel and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories). After blotting in tris-buffered saline (TBS) with 5% nonfat dry milk–Tris buffered saline and 0.1% Tween, the membrane was probed with p16 (1:10000 dilution, Abcam, ab51243), p53 (1:200 dilution, Santa Cruz Biotechnology, sc-126), cyclin E (1:200 dilution, Santa Cruz Biotechnology, sc-198), and β-actin (1:200 dilution, Santa Cruz Biotechnology, sc-7210) primary antibodies. Blots were then incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (ECL plus western blotting reagent pack, 1:10000 dilution, GE Healthcare, RPN2124). The immune complex was detected with the Amersham ECL PlusTM western blotting Detection System (GE Healthcare, RPN2132). The blots were exposed to Hyperfilm (Amersham Pharmacia Biotech), and bands were quantified with ImageJ densitometry software (National Institutes of Health).
DNA methylation pattern assay
Genomic DNA from HMEC at passage 12 was isolated using the Qiagen DNeasy kit according to the manufacturer’s instructions. Differentially methylated fractions of DNA were then prepared using a Methyl-Profiler DNA Methylation Enzyme Kit (SA Biosciences). After DNA had been digested, digested DNA samples were prepared with real-time PCR using a Methyl-Profiler DNA Methylation PCR Array (SA Biosciences, MEAH-011A). The MEAH-011A was loaded with 24 gene promoters.
Bioinformatics and statistical analysis
Gene networks representing key genes for BPA exposure were identified using GNCPro (SA Biosciences), which is a free online software and an in silico research tool for collating gene and pathway interactions with integrating collective biological knowledge through text mining, data mining, data acquisition and computational prediction. The interactions among a group of genes are represented graphically and are interactive. All experiments in this study were performed independently two or more times to test the reproducibility of the results. Quantitative data are expressed as the means ± SD, except those for mRNA expression levels, which are expressed as the mean of two independent experiments. A non-parametric test, the Mann-Whitney U test, was applied to test for statistical significance. Values of p < 0.05 were considered to indicate statistical significance.
Additional material
Download Zip (116.8 KB)Acknowledgments
This study was supported in part by a Grant in Aid for Scientific Research from the Ministry of the Health and Labour, Japan. The authors gratefully acknowledge the critical advice of Dr. Tohru Inoue (National Institute of Health Sciences, Japan), and the technical support of Ms. Noriko Oshima (GE Healthcare Japan Corporation) in the analysis using the IN Cell Analyzer 1000. The authors also thank Ms. Yumi Matsumoto for her technical assistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA). Reprod Toxicol 2007; 24:139 - 77; http://dx.doi.org/10.1016/j.reprotox.2007.07.010; PMID: 17825522
- Brotons JA, Olea-Serrano MF, Villalobos M, Pedraza V, Olea N. Xenoestrogens released from lacquer coatings in food cans. Environ Health Perspect 1995; 103:608 - 12; http://dx.doi.org/10.1289/ehp.95103608; PMID: 7556016
- Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect 2005; 113:391 - 5; http://dx.doi.org/10.1289/ehp.7534; PMID: 15811827
- Krishnan AV, Stathis P, Permuth SF, Tokes L, Feldman D. Bisphenol-A: an estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology 1993; 132:2279 - 86; http://dx.doi.org/10.1210/en.132.6.2279; PMID: 8504731
- Keri RA, Ho SM, Hunt PA, Knudsen KE, Soto AM, Prins GS. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod Toxicol 2007; 24:240 - 52; http://dx.doi.org/10.1016/j.reprotox.2007.06.008; PMID: 17706921
- Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod 2001; 65:1215 - 23; PMID: 11566746
- Weber Lozada K, Keri RA. Bisphenol A increases mammary cancer risk in two distinct mouse models of breast cancer. Biol Reprod 2011; 85:490 - 7; http://dx.doi.org/10.1095/biolreprod.110.090431; PMID: 21636739
- Lamartiniere CA, Jenkins S, Betancourt AM, Wang J, Russo J. Exposure to the endocrine disruptor bisphenol a alters susceptibility for mammary cancer. Horm Mol Biol Clin Investig 2011; 5:45 - 52; http://dx.doi.org/10.1515/HMBCI.2010.075; PMID: 21687816
- Ptak A, Wrobel A, Gregoraszczuk EL. Effect of bisphenol-A on the expression of selected genes involved in cell cycle and apoptosis in the OVCAR-3 cell line. Toxicol Lett 2011; 202:30 - 5; http://dx.doi.org/10.1016/j.toxlet.2011.01.015; PMID: 21277958
- Dairkee SH, Seok J, Champion S, Sayeed A, Mindrinos M, Xiao W, et al. Bisphenol A induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res 2008; 68:2076 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-07-6526; PMID: 18381411
- Singleton DW, Feng Y, Yang J, Puga A, Lee AV, Khan SA. Gene expression profiling reveals novel regulation by bisphenol-A in estrogen receptor-alpha-positive human cells. Environ Res 2006; 100:86 - 92; http://dx.doi.org/10.1016/j.envres.2005.05.004; PMID: 16029874
- Diel P, Olff S, Schmidt S, Michna H. Effects of the environmental estrogens bisphenol A, o,p'-DDT, p-tert-octylphenol and coumestrol on apoptosis induction, cell proliferation and the expression of estrogen sensitive molecular parameters in the human breast cancer cell line MCF-7. J Steroid Biochem Mol Biol 2002; 80:61 - 70; http://dx.doi.org/10.1016/S0960-0760(01)00173-X; PMID: 11867264
- Weng YI, Hsu PY, Liyanarachchi S, Liu J, Deatherage DE, Huang YW, et al. Epigenetic influences of low-dose bisphenol A in primary human breast epithelial cells. Toxicol Appl Pharmacol 2010; 248:111 - 21; http://dx.doi.org/10.1016/j.taap.2010.07.014; PMID: 20678512
- Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol 2007; 8:715 - 22; http://dx.doi.org/10.1038/nrm2242; PMID: 17717516
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223 - 33; http://dx.doi.org/10.1016/j.cell.2007.07.003; PMID: 17662938
- Iso T, Futami K, Iwamoto T, Furuichi Y. Modulation of the expression of bloom helicase by estrogenic agents. Biol Pharm Bull 2007; 30:266 - 71; http://dx.doi.org/10.1248/bpb.30.266; PMID: 17268063
- Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect 2009; 117:1053 - 8; PMID: 19654912
- Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J Endocrinol 2008; 196:101 - 12; http://dx.doi.org/10.1677/JOE-07-0056; PMID: 18180321
- de Waard WJ, Aarts JM, Peijnenburg AA, Baykus H, Talsma E, Punt A, et al. Gene expression profiling in Caco-2 human colon cells exposed to TCDD, benzo[a]pyrene, and natural Ah receptor agonists from cruciferous vegetables and citrus fruits. Toxicol In Vitro 2008; 22:396 - 410; http://dx.doi.org/10.1016/j.tiv.2007.10.007; PMID: 18061397
- Buterin T, Koch C, Naegeli H. Convergent transcriptional profiles induced by endogenous estrogen and distinct xenoestrogens in breast cancer cells. Carcinogenesis 2006; 27:1567 - 78; http://dx.doi.org/10.1093/carcin/bgi339; PMID: 16474171
- Garbe JC, Holst CR, Bassett E, Tlsty T, Stampfer MR. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle 2007; 6:1927 - 36; http://dx.doi.org/10.4161/cc.6.15.4519; PMID: 17671422
- Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc Natl Acad Sci USA 1985; 82:2394 - 8; http://dx.doi.org/10.1073/pnas.82.8.2394; PMID: 3857588
- Sudo H, Garbe J, Stampfer MR, Barcellos-Hoff MH, Kronenberg A. Karyotypic instability and centrosome aberrations in the progeny of finite life-span human mammary epithelial cells exposed to sparsely or densely ionizing radiation. Radiat Res 2008; 170:23 - 32; http://dx.doi.org/10.1667/RR1317.1; PMID: 18582160
- Qin XY, Zaha H, Nagano R, Yoshinaga J, Yonemoto J, Sone H. Xenoestrogens down-regulate aryl-hydrocarbon receptor nuclear translocator 2 mRNA expression in human breast cancer cells via an estrogen receptor alpha-dependent mechanism. Toxicol Lett 2011; 206:152 - 7; http://dx.doi.org/10.1016/j.toxlet.2011.07.007; PMID: 21771643
- Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, et al. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 2007; 24:178 - 98; http://dx.doi.org/10.1016/j.reprotox.2007.05.010; PMID: 17628395
- Nijjar T, Bassett E, Garbe J, Takenaka Y, Stampfer MR, Gilley D, et al. Accumulation and altered localization of telomere-associated protein TRF2 in immortally transformed and tumor-derived human breast cells. Oncogene 2005; 24:3369 - 76; http://dx.doi.org/10.1038/sj.onc.1208482; PMID: 15735711
- Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol 2007; 1:84 - 96; http://dx.doi.org/10.1016/j.molonc.2007.02.004; PMID: 18516279
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 28; PMID: 12042769
- Fleming JM, Miller TC, Meyer MJ, Ginsburg E, Vonderhaar BK. Local regulation of human breast xenograft models. J Cell Physiol 2010; 224:795 - 806; http://dx.doi.org/10.1002/jcp.22190; PMID: 20578247
- Bruder ED, Lee JJ, Widmaier EP, Raff H. Microarray and real-time PCR analysis of adrenal gland gene expression in the 7-day-old rat: effects of hypoxia from birth. Physiol Genomics 2007; 29:193 - 200; http://dx.doi.org/10.1152/physiolgenomics.00245.2006; PMID: 17213367
- Evron E, Dooley WC, Umbricht CB, Rosenthal D, Sacchi N, Gabrielson E, et al. Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR. Lancet 2001; 357:1335 - 6; http://dx.doi.org/10.1016/S0140-6736(00)04501-3; PMID: 11343741
- Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, et al. DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int J Cancer 2003; 107:970 - 5; http://dx.doi.org/10.1002/ijc.11508; PMID: 14601057
- Lee JS, Fackler MJ, Teo WW, Lee JH, Choi C, Park MH, et al. Quantitative promoter hypermethylation profiles of ductal carcinoma in situ in North American and Korean women: Potential applications for diagnosis. Cancer Biol Ther 2008; 7:1398 - 406; http://dx.doi.org/10.4161/cbt.7.9.6425; PMID: 18769130
- Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001; 409:633 - 7; http://dx.doi.org/10.1038/35054579; PMID: 11214324
- Sarkar P, Shiizaki K, Yonemoto J, Sone H. Activation of telomerase in BeWo cells by estrogen and 2,3,7,8-tetrachlorodibenzo-p-dioxin in co-operation with c-Myc. Int J Oncol 2006; 28:43 - 51; PMID: 16327978
- Bandyopadhyay D, Curry JL, Lin Q, Richards HW, Chen D, Hornsby PJ, et al. Dynamic assembly of chromatin complexes during cellular senescence: implications for the growth arrest of human melanocytic nevi. Aging Cell 2007; 6:577 - 91; http://dx.doi.org/10.1111/j.1474-9726.2007.00308.x; PMID: 17578512
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703 - 16; http://dx.doi.org/10.1016/S0092-8674(03)00401-X; PMID: 12809602
- Malyavantham KS, Bhattacharya S, Barbeitos M, Mukherjee L, Xu J, Fackelmayer FO, et al. Identifying functional neighborhoods within the cell nucleus: proximity analysis of early S-phase replicating chromatin domains to sites of transcription, RNA polymerase II, HP1gamma, matrin 3 and SAF-A. J Cell Biochem 2008; 105:391 - 403; http://dx.doi.org/10.1002/jcb.21834; PMID: 18618731
- Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y. Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci 2003; 116:3327 - 38; http://dx.doi.org/10.1242/jcs.00635; PMID: 12840071
- Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 1999; 108:220 - 34; http://dx.doi.org/10.1007/s004120050372; PMID: 10460410
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003; 36:131 - 49; http://dx.doi.org/10.1046/j.1365-2184.2003.00266.x; PMID: 12814430
- Sherr CJ. Cancer cell cycles. Science 1996; 274:1672 - 7; http://dx.doi.org/10.1126/science.274.5293.1672; PMID: 8939849
- Roussel-Gervais A, Bilodeau S, Vallette S, Berthelet F, Lacroix A, Figarella-Branger D, et al. Cooperation between cyclin E and p27(Kip1) in pituitary tumorigenesis. Mol Endocrinol 2010; 24:1835 - 45; http://dx.doi.org/10.1210/me.2010-0091; PMID: 20660298
- Minella AC, Clurman BE. Mechanisms of tumor suppression by the SCF(Fbw7). Cell Cycle 2005; 4:1356 - 9; http://dx.doi.org/10.4161/cc.4.10.2058; PMID: 16131838
- Kitagawa K, Kotake Y, Kitagawa M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci 2009; 100:1374 - 81; http://dx.doi.org/10.1111/j.1349-7006.2009.01196.x; PMID: 19459846
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 2007; 26:131 - 43; http://dx.doi.org/10.1016/j.molcel.2007.02.022; PMID: 17434132
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294:173 - 7; http://dx.doi.org/10.1126/science.1065203; PMID: 11533444
- Minella AC, Loeb KR, Knecht A, Welcker M, Varnum-Finney BJ, Bernstein ID, et al. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev 2008; 22:1677 - 89; http://dx.doi.org/10.1101/gad.1650208; PMID: 18559482
- Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. Chronic oral exposure to bisphenol a results in a non-monotonic dose response in mammary carcinogenesis and metastasis in mmtv-erbb2 mice. Environ Health Perspect 2011; 119:1604 - 9; http://dx.doi.org/10.1289/ehp.1103850; PMID: 21988766
- Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009; 460:1136 - 9; http://dx.doi.org/10.1038/nature08290; PMID: 19668188
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378 - 85; http://dx.doi.org/10.1038/ng1909; PMID: 17072317
- Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene 2009; 28:2969 - 78; http://dx.doi.org/10.1038/onc.2009.149; PMID: 19503099
- Dejeux E, Ronneberg JA, Solvang H, Bukholm I, Geisler S, Aas T, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer 2010; 9:68; http://dx.doi.org/10.1186/1476-4598-9-68; PMID: 20338046
- Pal R, Srivastava N, Chopra R, Gochhait S, Gupta P, Prakash N, et al. Investigation of DNA damage response and apoptotic gene methylation pattern in sporadic breast tumors using high throughput quantitative DNA methylation analysis technology. Mol Cancer 2010; 9:303; http://dx.doi.org/10.1186/1476-4598-9-303; PMID: 21092294
- Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, BRCA1, MSH6, PAX5, PAX6, and WT1 in ductal carcinoma in situ and invasive breast cancer. J Pathol 2011; 225:222 - 31; http://dx.doi.org/10.1002/path.2930; PMID: 21710692
- Shim YH, Kang GH, Ro JY. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab Invest 2000; 80:689 - 95; http://dx.doi.org/10.1038/labinvest.3780072; PMID: 10830779
- Taghavi N, Biramijamal F, Sotoudeh M, Khademi H, Malekzadeh R, Moaven O, et al. p16INK4a hypermethylation and p53, p16 and MDM2 protein expression in esophageal squamous cell carcinoma. BMC Cancer 2010; 10:138; http://dx.doi.org/10.1186/1471-2407-10-138; PMID: 20388212
- Zang JJ, Xie F, Xu JF, Qin YY, Shen RX, Yang JM, et al. P16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol 2011; 17:3043 - 8; http://dx.doi.org/10.3748/wjg.v17.i25.3043; PMID: 21799651
- Salvesen HB, Das S, Akslen LA. Loss of nuclear p16 protein expression is not associated with promoter methylation but defines a subgroup of aggressive endometrial carcinomas with poor prognosis. Clin Cancer Res 2000; 6:153 - 9; PMID: 10656444