Abstract
There has been considerable interest in targeting cell cycle checkpoints particularly in emerging and alternative anticancer strategies. Here, we show that checkpoint abrogation by AZD7762, a potent and selective CHK1/2 kinase inhibitor enhances genotoxic treatment efficacy in immature KG1a leukemic cell line and in AML patient samples, particularly those with a complex karyotype, which display major genomic instability and chemoresistance. Furthermore, these data suggest that constitutive DNA-damage level might be useful markers to select AML patients susceptible to receive checkpoint inhibitor in combination with conventional chemotherapy. Moreover, this study demonstrates for the first time that AZD7762 inhibitor targets the CD34+CD38-CD123+ primitive leukemic progenitors, which are responsible for the majority of AML patients relapse. Finally, CHK1 inhibition does not seem to affect clonogenic potential of normal hematopoietic progenitors.
Key words:
Introduction
Cell cycle checkpoints represent central orchestrators of the DNA damage response (DDR) pathway, playing a critical role in the maintenance of genetic stability by sensing DNA damage or aberrant replication structures and slowing down cell cycle progression to facilitate repair. Constitutive activation of the DDR pathway has been identified as a biological barrier against activated oncogenes and tumor progression in early human lesions.Citation1–Citation3 DDR pathway is organized as sensors, mediators, transducers and effectors.Citation4 The DNA damage sensing kinases ATM and ATR kinase are required for initiation of the checkpoint response by phosphorylating down-stream transducer kinases CHK1 and CHK2. The activated CHK1 and CHK2 kinases phosphorylate and promote degradation or sequestration of effectors CDC25 phosphatases, which are consequently unable to activate cyclin-dependent kinases thus leading to cell cycle arrest.
There has been considerable interest in targeting DDR pathway considering its central role in regulating cell cycle arrest, DNA repair, transcriptional programs and apoptosis.Citation5 Specific inhibition of key components of the DDR can thus increase the efficacy of conventional chemo- or radiotherapy. The strategy relies on the knowledge that high rate of the tumors are p53-deficients thus placing CHK1, which contributes to all cell cycle checkpoints and is essential for the maintenance of genomic integrity (whereas CHK2 is conditional), as a core mediator of the cellular response to DNA damage. This has prompted the development of CHK1 inhibitors that are able to enhance the efficacy of genotoxic cancer therapies by allowing premature entry into the cell cycle following DNA damage and insufficient DNA repair. The first CHK1 inhibitor to be tested extensively in human cells was UCN-01 (7-hydroxystaurosporine) but because of its lack of specificity and its poor drug-like properties, new agents have been synthesized and several of them are currently tested in clinical trials in combination with clastogenic agents. Recently, a novel CHK1/2 inhibitor, AZD7762 was shown to enhance the cytoxoxicity of DNA-damaging agentsCitation6 as well as radiation in p53-compromised cells both in vitro and in vivo.Citation7 Moreover, Morgan et al. demonstrated that radiosensitization by AZD7762 was associated with G2 checkpoint abrogation and homologous recombination repair inhibition.Citation8
Only few works have documented the use of CHK1 inhibitor in hematologic cancers, in particular Acute Myeloid Leukemia (AML). It was first shown that targeting CHK1 sensitizes AML progenitors to 1-β-D-arabinofuranosylcytosine (Ara-C).Citation9 More recently, we reported a beneficial effect of CHK1 inhibitionCitation10 by UCN-01 in high-DNA damage level AML samples, particularly those with complex karyotypes and treated by Ara-C.Citation11 These data prompted us to evaluate more specific inhibitor to determine whether the CHK1/2 inhibitor AZD7762 sensitizes AML cells with complex karyotypes to ara-C with the same extent.
Collectively, the data presented in this study support the evaluation of AZD7762 in hematologic malignancies as chemo-sensitizer as well as the interest of such novel therapeutic strategies.
Results
Inhibition of checkpoint kinases by AZD7762 allows illegitimate G2/M checkpoint exit.
To determine whether AZD7762 inhibitor would modulate an activated G2/M cell cycle checkpoint, we analyzed the capability of U2OS osteosarcoma cell line and KG1a leukemic cells to illegitimately enter into mitosis after activation of the G2/M checkpoint following etoposide (VP-16) treatment. To monitor entry into mitosis, we analyzed cell cycle progression using the previously described mitotic trap assay.Citation10,Citation13 Cells were treated with etoposide for 1 h and then released in drug-free media with or without AZD7762 inhibitor for 23 h. Nocodazole was added to the media, thus trapping the cells that could overcome or exit the G2/M checkpoint in a mitosis-like state (). The percentage of mitotic cells was evaluated by mitotic phosphorylation labeling (3.12.I.22 antibody) Citation24 and DNA content was monitored using flow cytometry. As presented in , U2OS (B) and KG1a (C) cells mostly proceeded into mitosis in absence of DNA damage and cell cycle checkpoint activation. Following etoposide treatment, U2OS as well as KG1a cells arrested their cell cycle at G2/M. In contrast, when treated with etoposide and AZD7762 (300 nM) cells did not arrest at G2/M, proceeded into mitosis and were trapped by nocodazole. We found substantial differences in the percentage of mitotic cells accumulated over time in presence of AZD7762 treatment for both cell line. These results are in agreement with previous reports showing that checkpoint kinase inhibitor potentiates the effects of the DNA-damaging agents.Citation11,Citation14
AZD7762 enhances VP-16-induced apoptosis on human leukemia cells.
To determine the influence of AZD7762 on VP-16 treated leukemic KG1a cells, apoptotic processes were evaluated by monitoring mitochondrial membrane potential changes, cytochrome c release from mitochondria and cell membrane permeability (, respectively). KG1a cells were treated with 70 µM VP16 for 1 h, and then released in fresh media in presence or not of 300 nM AZD7762. After 24 h, cells were subjected to immunofluorescence assay using Cellomics multiparameter cytotoxicity 3 kit. Quantification was performed with a Cellomics Array Scan device (ThermoScientific). The combination of AZD7762 and VP-16 significantly increased apoptosis 24 h post-treatment on KG1a cells. These data clearly show a correlation between the abrogation of the G2 arrest and AZD7762-mediated DNA damage sensitization.
Checkpoint kinases inhibition potentiated ara-C treatment on AML blast cells but not on normal granulomonocyte progenitors (CFU-GM).
We then examined the effect of CHK1 inhibition on the sensitization of 11 complex karyotypes AML patient samples (see and S1) to ara-C treatment in clonogenic assays. In light of our previous reports, we monitored constitutive DNA damage level assessed by phospho-H2AX labeling (as well as CHK1/CHK1-P and ATM-ATR substrates data not shown). We evaluated the IC50 for ara-C alone and in combination with AZD7762, from the proliferation dose-response curves after clonogenic assays. These results are presented in , together with the constitutive (untreated cells) phospho-H2AX levels and the sensitization factor (SF), which is a ratio between the IC50 for ara-C alone and in combination with AZD7762. These results show that AZD7762 mediated enhancement of chemotherapy treatment, ara-C, with a marked decrease of IC50 in all tested AML samples. As seen in , the highest sensitization was observed in the samples harbouring the highest constitutive DNA damage level. Indeed, reveals a strong correlation between the SF and the DNA damage level for the 11 AML samples screened (***p = 0.001). These findings suggest that AZD7762-mediated checkpoint inhibition and may offer considerable selective leukemic genotoxic sensitization. Considering that AZD7762 did not exert appreciable cytotoxicity alone or in combination with DNA damage agent on normal granulomonocyte progenitors (CD34+ CFU-GM). As seen in (right sample), no significative change in the number of colony-forming CFU-GM was observed after ara-C treatment in combination with AZD7762 inhibitor.
Checkpoint kinases inhibitor enhances ara-C-induced apoptosis including in CD34+ CD38− CD123+ primitive leukemia progenitors compartment.
To assess apoptosis in immature hematopoietic subpopulations, AML samples were subjected to 2 µM ara-C treatment alone or in combination with 100 nM AZD7762 for 48 h. After this period, AML samples were processed using four colors immunostaining (CD34-PE-Cy7, CD38-APC, CD123-PE and CD45-V450H). Induction of apoptosis in AML samples was assessed using annexin-V-FITC and 7-AAD after gating on CD34+CD38−CD123+ cells. AZD7762 alone did not induce significant apoptotic response. Interestingly, the combination of ara-C and AZD7762 induced significant cell death (20 to 30%) in CD34+CD38−CD123+ primitive leukemic progenitors from four AML primary samples (). Apoptotic response was also detected in the bulk (data not shown).
Discussion
In the present study, we have shown that CHK1/2 inhibition by AZD7762 enhances genotoxic treatment efficacy in immature KG1a leukemic cell line and conventional chemotherapy in AML patient samples, particularly those with a complex karyotype, which display major genomic instability and chemoresistance.Citation15 These data support the clinical investigation of CHK1 inhibitors, specifically with cytogenetic high-risk AML patients group, in combination with standard therapy. Furthermore, these data suggest that γH2AX and with less extent S317 CHK1 and phosphorylated ATM/ATR substrates ( and ), might be useful markers to select AML patients susceptible to receive this type of combination therapy. In addition, this study demonstrates for the first time that AZD7762 inhibitor targets the CD34+CD38−CD123+ primitive leukemic progenitors compartment, which is responsible for the majority of AML patients' relapse.Citation16 Finally, CHK1 inhibition with this compound does not seem to affect clonogenic potential of normal hematopoietic progenitors.
In AML, recent studies have shown that patients with complex aberrant karyotype display frequent p53 mutations or alterations (78%), compared with those without complex aberrant karyotype (14%).Citation17 These complex aberrant karyotype group's characteristics may offer a therapeutic window for selective sensitization of complex karyotype leukemic cells to conventional chemotherapy by CHK1 inhibitor. Our work strongly supports this attractive pharmacologic strategy. We reported that complex karyotype samples are highly sensitive to CHK1 inhibition by UCN-01 or by RNA interference in combination with ara-C treatment.Citation11 Similar results were obtained when UCN-01 and VP-16 were used together on primary leukemic cells from AML sample belonging to the M1 and M2 classes of the FrenchAmerican-British classification.Citation10 Moreover, UCN-01 has been evaluated in a pilot clinical trial in combination with ara-C in eight patients with relapsed AML. Thus, use of this pharmacological combination decreases CHK1 phosphorylation, JNK activation and inhibits the Akt survival pathway demonstrating cytotoxic effect.Citation9 However, despite interesting effects, UCN-01 presents poor selectivity, unfavorable pharmacokinetics and toxicity. In the present report, we demonstrate that AZD7762 activity enhances ara-c treatment on complex aberrant karyotype samples.
A major function of CHK1 is to monitor the integrity of the replication fork during the replication fork during cell division cycle even in the absence of exogenously introduced DNA damage.Citation18 Further studies specifically dedicated to the investigation of long-term effects of CHK1 inhibition in normal hematopoietic progenitors will be necessary to evaluate precisely its impact. Here we have demonstrated that combination of AZD7762 and ara-C has no effect on the growth and clonogenic capacity of normal hematopoietic cells. Nevertheless, a recent report has demonstrated a role for CHK1 in erythropoiesis by maintaining the balance between erythroid progenitors and enucleated erythroid cells. These data indicate that disruptions in CHK1 levels can lead to anemia.Citation19 Elucidation of exact CHK1's role may lead to a better understanding of its possible use in clinical trial (see Fig. S1).
A critical next step will be to determine the molecular profile of individual tumoral population (including stem cells populations) in order to select appropriate patient population that would benefit of this combinative therapy. This approach will be the key to the successful development and application of these new generation potent and selective CHK1 inhibitors.Citation20
Materials and Methods
Cells.
Fresh and thawed samples from AML patients have been obtained after informed consent and stored at the HIMIP collection. According to the French law, HIMIP collection has been declared to the Ministry of Higher Education and Research (DC 2008-307 collection 1) and obtained a transfer agreement (AC 2008-129) after approbation by the “Comité de Protection des Personnes Sud-Ouest et Outremer II” (ethical committee). Clinical and biological annotations of the samples have been declared to the CNIL (Comité National Informatique et Libertés i.e., Data processing and Liberties National Committee). Frozen cells were thawed in IMDM medium and immediately processed for clonogenic assays.
Normal bone marrow CD34+ hematopoietic progenitor cells (HPCs) were obtained after informed consent from discarded fragments from hematologically healthy patients who had undergone hip surgery. Mononuclear cells from bone marrow were obtained by Ficoll-Hypaque density-gradient centrifugation, after which isolation of HPCs was performed by positive selection of CD34-expressing cells using magnetic separation and EasySep procedure (StemCell Technologies).
The human KG1a, U937 and Jurkat leukemic cell lines were cultured in Iscove modified Dulbecco medium containing 20% fetal calf serum (FCS; KG1a), RPMI/10% FCS (U937 and Jurkat), Dulbeccos modified Eagles medium/10% FCS (UT7-Epo1) at 37°C with 5% CO2.Citation10
Human U2OS osteosarcoma cells obtained from the American Type Culture Collections were maintained at 37°C/5% CO2 in DMEM containing 10% FCS.
Pharmacologic inhibitor and antibodies.
AZD7762 was a kind gift from AstraZeneca.Citation6 Stock solutions of AZD7762 is at 10 mM in DMSO.
Antibodies were monoclonal and polyclonal anti-phosphoS139-H2AX (1:100 and 1:25 respectively) (Upstate Biotechnology and Cell Signaling), anti-phospho-S317-CHK1 (1:75) (Cell signaling), anti-CHK1 (1:75) (Santa Cruz Biotechnology).
Flow cytometry.
Immunostaining was performed according to the previously described protocol.Citation11 Samples were analyzed in an FC500 flow cytometer (Beckman Coulter) and subsequently processed using the CXP software. All flow cytometry results are expressed as the ratio between mean fluorescence intensity of the stained sample and that of its control isotypic antibody.
Clonogenic assays.
The percentage of leukemic cells present in patients' bone marrow is reported in . Clonogenic assays on AML cells and on fresh human bone marrow CD34+ HPC cells were made according to a method previously described in reference Citation12.
Apoptosis assays.
Apoptosis was assessed using cellomics multiparameter cytotoxicity 3 kit (Thermoscientific) in leukemic cell lines. The acquisition of images was obtained on an ArrayScan HCS reader with a 20x objective lens, and effective image analysis were done by Cellomics Technologies (Compartmental Bioapplication-Thermo Scientific).
For AML patient samples and particularly leukemic subpopulation, apoptosis was determined as follow. Leukemic blasts were incubated with or without 2 or 5 µM ara-C for 48 h in the presence or absence of 100 nM AZD7762. After incubation, 300,000 cells were washed in PBS-1% BSA and then incubated for 15 min at room temperature with annexin-V-FITC (Biolegend)/7-AAD (Sigma). Apoptosis was quantified by LSRII flow cytometer (Beckman Coulter). To identify the CD34+CD38−CD123+ primitive leukemia progenitors, AML samples were stained with CD34-PE-Cy7, CD38-APC, CD123-PE and CD45-V450H antibodies (BD Biosciences).
Statistics.
Results are expressed as means ± SEM. Statistical analyses were performed by the unpaired Student's t-test and Pearson correlation test with Prism4 software (Graph Pad Software). Differences were considered as significant for p values less than or equal to 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Inhibition of CHK1 by AZD7762 allows G2/M checkpoint exit. (A) Experimental procedure adapted from references Citation10 and Citation13. (B) U2Os cells were exposed to an etoposide pulse (40 µM for 1 h) as previously described in reference Citation23, and released into nocodazole (200 ng/ml) with or without 200 nM AZD7762 for 23 h, after which the percentage of mitotic cells was analyzed by 3-12-I-22 monoclonal antiboby stainingCitation24 together with cell cycle distribution determination (propidium iodide). The results are the average of three independent experiments. (C) KG1a cells were treated with different etoposide concentrations for 1 h then, released in 200 ng/ml nocodazole alone or in combinaison with 300 nM AZD7762 for 23 h. The proportion of mitosis cells is evaluated as in (B). The results are the average of four independent experiments.
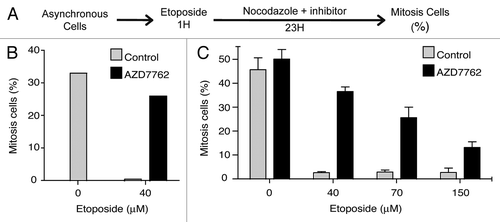
Figure 2 Inhibition of CHK1 by AZD7762 allows apoptosis. KG1a cells were treated with VP-16 (70 µM) for 1 h then released into fresh medium in the absence or presence of 300 nM AZD7762 for 20 h. Cells were then fixed and stained using cytotoxicity 3 HCS Reagent Kit, and the following indicators of cytotoxicity were measured by Cellomics Arrayscan instrument (Cellomics Inc.): mitochondrial membrane potential (A), mitochondrial cytochrome c release (B), membrane permeability (C). The arrayscan captures fluorescent images and performs automated image analysis using Compartmental analysis BioApplication. Statistical analyses were performed using a non-parametrical unpaired t-test (***p < 0.0001).
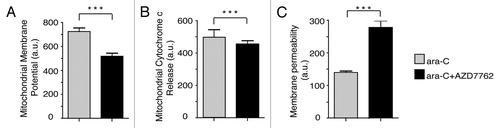
Figure 3 AZD7762 potentiated ara-C treatment on AML blast cells but not on normal granulomonocyte progenitors (CFU-GM). (A) AML blast cells (no. 1–11) and normal CD34+ HPC were grown in clonogenic assays in the presence of ara-C, alone or together with AZD7762 (10 nM). The clonogenic survival was assessed after 7 d and the IC50 for ara-C alone and in combination with AZD7762 was then calculated. The SF (sensitization factor) is the ratio between the IC50 for ara-C alone and in combination with AZD7762. (B) Correlation between SF and phospho-H2AX level (rMFI) (r = 0.85; ***p = 0.001). Statistical analyses were performed using a Pearson test.
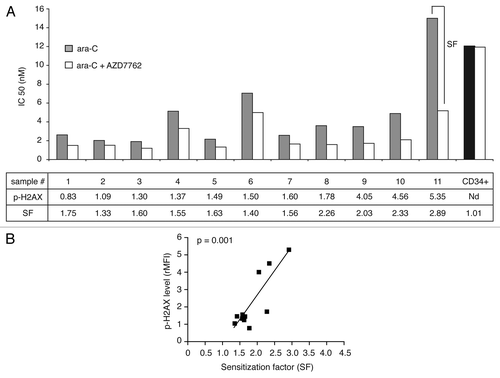
Figure 4 AZD7762 potentiated ara-C treatment on CD34+CD38−CD123+ stem cells compartments. (A) Primary leukemic cells from four patients were incubated for 48 h with or without 100 nM AZD7762 and/or 2 µM and then processed for apoptosis studies using annexin-V/7AAD staining after gating on CD34+CD38−CD123+. A representative AML sample is shown (B). Results are presented as percentage of survival cells.
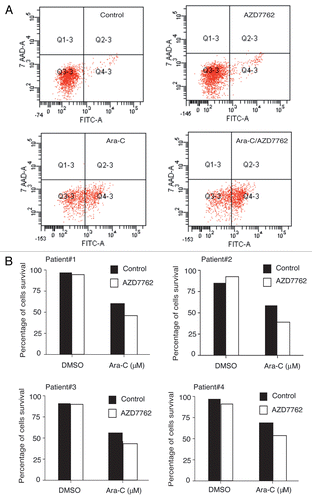
Table 1 Characteristics of AML patients
Additional material
Download Zip (169.5 KB)Acknowledgments
The authors acknowledge Dr. J. Brown (AstraZeneca) for generously providing the AZD7762 compound. We gratefully acknowledge N. Dastugue for karyotype analyze and F. Verges for FACS analysis advice. This work was supported by the C.N.R.S., the Université Paul Sabatier-Toulouse, the Institut National du Cancer (Programme libre 2005 et 2008) and la région Midi-Pyrénées to B.D.
References
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907 - 913; http://dx.doi.org/10.1038/nature03485; PMID: 15829965
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864 - 870; http://dx.doi.org/10.1038/nature03482; PMID: 15829956
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352 - 1355; http://dx.doi.org/10.1126/science.1140735; PMID: 18323444
- Zhou BB, Anderson HJ, Roberge M. Targeting DNA checkpoint kinases in cancer therapy. Cancer Biol Ther 2003; 2:16 - 22; PMID: 14508077
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19:238 - 245; http://dx.doi.org/10.1016/j.ceb.2007.02.009; PMID: 17303408
- Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 2008; 7:2955 - 2966; http://dx.doi.org/10.1158/1535-7163.MCT-08-0492; PMID: 18790776
- Mitchell JB, Choudhuri R, Fabre K, Sowers AL, Citrin D, Zabludoff SD, et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res 2010; 16:2076 - 2084; http://dx.doi.org/10.1158/1078-0432.CCR-09-3277; PMID: 20233881
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972 - 4981; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573; PMID: 20501833
- Sampath D, Cortes J, Estrov Z, Du M, Shi Z, Andreeff M, et al. Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood 2006; 107:2517 - 2524; http://dx.doi.org/10.1182/blood-2005-08-3351; PMID: 16293603
- Didier C, Cavelier C, Quaranta M, Galcera MO, Demur C, Laurent G, et al. G2/M checkpoint stringency is a key parameter in the sensitivity of AML cells to genotoxic stress. Oncogene 2008; 27:3811 - 3820; http://dx.doi.org/10.1038/sj.onc.1211041; PMID: 18212737
- Cavelier C, Didier C, Prade N, Mansat-De Mas V, Manenti S, Recher C, et al. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res 2009; 69:8652 - 8661; http://dx.doi.org/10.1158/0008-5472.CAN-09-0939; PMID: 19843865
- Récher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, et al. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005; 105:2527 - 2534; http://dx.doi.org/10.1182/blood-2004-06-2494; PMID: 15550488
- Roberge M, Berlinck RG, Xu L, Anderson HJ, Lim LY, Curman D, et al. High-throughput assay for G2 checkpoint inhibitors and identification of the structurally novel compound isogranulatimide. Cancer Res 1998; 58:5701 - 5706; PMID: 9865726
- Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol 2009; 76:208 - 214; http://dx.doi.org/10.1124/mol.109.055178; PMID: 19403702
- Chen CC, Yang CF, Lee KD, You JY, Yu YB, Ho CH, et al. Complex karyotypes confer a poor survival in adult acute myeloid leukemia with unfavorable cytogenetic abnormalities. Cancer Genet Cytogenet 2007; 174:138 - 146; http://dx.doi.org/10.1016/j.cancergencyto.2006.12.002; PMID: 17452256
- Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene 2004; 23:7178 - 7187; http://dx.doi.org/10.1038/sj.onc.1207935; PMID: 15378078
- Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the Tp53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008; 22:1539 - 1541; http://dx.doi.org/10.1038/leu.2008.143; PMID: 18528419
- Scorah J, McGowan CH. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle 2009; 8:1036 - 1043; http://dx.doi.org/10.4161/cc.8.7.8040; PMID: 19270516
- Boles NC, Peddibhotla S, Chen AJ, Goodell MA, Rosen JM. Chk1 haploinsufficiency results in anemia and defective erythropoiesis. PLoS ONE 2010; 5:8581; http://dx.doi.org/10.1371/journal.pone.0008581; PMID: 20052416
- Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011; 17:88 - 96; http://dx.doi.org/10.1016/j.molmed.2010.10.009; PMID: 21087899
- Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 1998; 92:2322 - 2333; PMID: 9746770
- Schlenk RF, Benner A, Hartmann F, del Valle F, Weber C, Pralle H, et al. Risk-adapted postremission therapy in acute myeloid leukemia: results of the German multicenter AML HD93 treatment trial. Leukemia 2003; 17:1521 - 1528; http://dx.doi.org/10.1038/sj.leu.2403009; PMID: 12886238
- Jullien D, Bugler B, Dozier C, Cazales M, Ducommun B. Identification of N-terminally truncated stable nuclear isoforms of CDC25B that are specifically involved in G2/M checkpoint recovery. Cancer Res 2011; 71:1968 - 1977; http://dx.doi.org/10.1158/0008-5472.CAN-10-2453; PMID: 21363925
- Cazales M, Quaranta M, Lobjois V, Jullien D, Al Saati T, Delsol G, et al. A new mitotic-cell specific monoclonal antibody. Cell Cycle 2008; 7:267 - 268; http://dx.doi.org/10.4161/cc.7.2.5306; PMID: 18256524