Abstract
Epidermal growth factor receptor (EGFR) mutation is the best marker of sensitivity to the EGFR tyrosine kinase inhibitor gefitinib, but a marker for the anti-EGFR antibody cetuximab has not been identified in lung cancer. The present study investigated markers for sensitivity to cetuximab. Sensitivity to cetuximab and gefitinib was compared with EGFR expression, EGFR and KRAS mutation, and EGFR gene copy numbers in lung cancer cell lines. We also studied the effect of these agents on the activation of EGFR, ERK, AKT, and STAT3 in cetuximab-sensitive and -resistant cell lines. We found one cetuximab-sensitive cell line with EGFR mutation among 19 lung cancer cell lines. Analysis of molecules downstream from EGFR revealed that AKT phosphorylation was suppressed in this cell line. Augmentation of AKT phosphorylation by transfection of a plasmid induced resistance to cetuximab. Acquisition of cetuximab resistance was associated with AKT activation in this cell line, while pharmacological inhibition of AKT markedly enhanced the growth inhibitory effect of cetuximab. Dephosphorylation of AKT in association with EGFR mutation is a candidate marker for sensitivity to cetuximab, and combined use of an AKT pathway inhibitor with cetuximab could be a novel therapeutic strategy for lung cancer.
Introduction
Lung cancer is the leading cause of cancer death in the world. Few patients are diagnosed at an early stage when curative resection is possible and the objective response rate of advanced disease to systemic chemotherapy is very low.Citation1 Therefore, more effective and less toxic therapeutic agents are being sought. Recently, along with accumulation of more knowledge about the molecular pathogenesis of lung cancer, the concept of molecular targeting therapy has been developed. Epidermal growth factor receptor (EGFR) has emerged as the most attractive target for the treatment of lung cancer, because it is a key receptor in the processes of cell growth and proliferation.Citation2 In addition, EGFR is frequently overexpressed in non-small cell lung cancer (NSCLC) and its overexpression is correlated with a poor prognosis.Citation3 Therefore, EGFR-targeting drugs have been developed and have come into clinical use recently.
There are two possible strategies for EGFR-targeting therapy. One is the use of low molecular weight tyrosine kinase inhibitors (TKIs) that inhibit the tyrosine kinase activity of the cytoplasmic catalytic domain of EGFR, and the other is to employ a monoclonal antibody (mAb) directed against the ligand-binding site of the extracellular domain of EGFR. For the clinical treatment of lung cancer, TKIs were developed first and gefitinib was approved for the treatment of NSCLC in Japan in January 2002. Though some patients showed a dramatic response, two large-scale clinical studies revealed that addition of gefitinib to conventional chemotherapy provided no survival benefit for lung cancer patients.Citation4,Citation5 Therefore, an anti-EGFR mAb, cetuximab, has been developed as a new alternative for anti-EGFR therapy.
Cetuximab is a chimeric human-mouse mAb that has a human IgG1 backbone combined with the variable region of a mouse mAb obtained from mice immunized with A431 cells by Mendelson in 1983.Citation6 Cetuximab has a high affinity for the extracellular ligand-binding domain of EGFR. Its binding to this domain inhibits the phosphorylation and activation of EGFR, downregulates of EGFR by receptor internalization, and induces immunological antitumor activity such as antibody-dependent cellular cytotoxicity (ADCC).Citation7,Citation8 Cetuximab has been approved by the US. Food and Drug Administration (FDA) for metastatic EGFR-positive colorectal cancer and locally advanced head and neck cancer. In the lung cancer field, a recent randomized, multicenter, phase III study (the FLEX study) revealed that addition of cetuximab to chemotherapy improved the survival of patients with advanced NSCLC irrespective of the histological subtype,Citation9 which is the first time that an EGFR inhibitor has demonstrated survival benefit as a first-line treatment for NSCLC. However, the actual increase of survival among patients who received cetuximab was only five weeks and the cost/benefit ratio of this therapy was not very good.Citation10 Therefore, identification of the subset of patients with NSCLC who will show a clinically meaningful response to cetuximab is now needed.
Selection of patients based on the molecular characteristics of their cancer is an attractive possibility for molecular-targeting therapy, but remains a challenge. This was first successfully accomplished for TKIs in the case of anti-EGFR therapy. In 2004, two groups from the USA reported that somatic mutations affecting the tyrosine kinase domain of EGFR are promising predictors of the response to gefitinib.Citation11,Citation12 In 2004, an increase of the EGFR copy numberCitation13 and KRAS mutationCitation14 were reported as positive and negative predictors of the response to gefitinib, respectively. Based on these reports, recent clinical studies have selected eligible patients by using such molecular markers, especially EGFR mutations, and have demonstrated a significant survival benefit of TKIs in NSCLC patients.Citation15,Citation16 However, a molecular marker for responsiveness to cetuximab has not yet been identified in NSCLC patients. Though the benefit of cetuximab treatment was largely confined to patients with wild-type KRAS in the case of colorectal cancer,Citation17 the KRAS mutation status was recently reported to not be useful as a marker in NSCLC patients.Citation18
In order to identify a molecular marker for cetuximab responsiveness of lung cancer, we analyzed the EGFR signaling system in 19 NSCLC cell lines and determined the association of various potential markers with sensitivity to cetuximab or gefitinib. Our results indicate that a combination of EGFR signaling pathway status (as typified by EGFR mutation) and the status of AKT activation can be a molecular marker for the efficacy of cetuximab.
Results
EGFR expression, mutation status, and gene copy number, as well as KRAS mutation status, and their relationship with sensitivity of a panel of lung cancer cell lines to EGFR-targeting drugs
We first examined the molecular status of EGFR and KRAS, as well as the effect of EGFR-targeting drugs on 19 lung cancer cell lines and A431 epidermoid carcinoma (EC) as the positive control (). Cell surface expression of EGFR was measured by flow cytometric analysis,Citation19 revealing abundant EGFR expression on the NSCLC cell lines, whereas there were few EGFR molecules on small cell lung cancer (SCLC) cell lines. Among NSCLC cell lines, expression of EGFR was highest for large cell carcinoma cell lines (LA), followed by adenocarcinoma cell lines (AD) and squamous cell carcinoma cell lines (SQ). We next analyzed activating mutations of EGFR and KRAS, as well as the EGFR copy number. PCR and direct sequencing revealed that 11–18 cells had a point mutation (L858R) in exon 21 of EGFR, while PC9, PC14, and Ma1 cells had a deletion mutation (delE746-A750) in exon 19. Mutation of codon 12 of KRAS was only found in cells with wild-type EGFR, A549, LK87, and Lu99, corresponding to the previous report that these mutations are mutually exclusive.Citation20 EGFR copy numbers were analyzed by fluorescence in situ hybridization (FISH), with FISH positivity being defined according to the standard of Hirsch and Cappuzzo et al.Citation13 An increase of EGFR copy numbers was observed in eight of 12 AD cell lines (67%), one of three SQ cell lines (33%), and EC cells, but not in LA or SCLC cell lines. Interestingly, all cell lines with EGFR mutation had amplification of the EGFR gene.
Table 1. Comparison of EGFR expression, EGFR and KRAS mutation, and EGFR FISH and gefitinib- and cetuximab-induced growth inhibition in various lung cancer cell lines
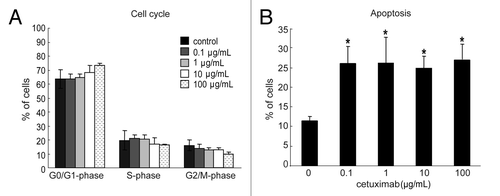
The sensitivity of these cell lines to EGFR-targeting drugs was measured by the WST-8 assay. Three of four cell lines with EGFR mutation and FISH positivity (11–18, PC9 and PC14) were highly sensitive to gefitinib (IC50 = 0.1 μmol/L) and the other cell line (Ma1) was moderately sensitive (IC50 = 7.1 μmol/L). Among the six cell lines with wild-type EGFR and FISH positivity, four (67%) lines (LCSC#2, LK-1, LK79, and A431) were moderately sensitive to gefitinib, with IC50 values of 1.9, 2.9, 2.8, and 1.8 μmol/L, respectively. The other 12 cell lines with wild-type EGFR and FISH negativity were resistant to gefitinib and had IC50 values of 9.3–44.7 μmol/L. For cetuximab, only 11–18 cells with EGFR mutation and an increased EGFR copy number were highly sensitive (IC50 = 0.1 μg/mL), whereas the other 19 cell lines were completely resistant and had high IC50 values (> 500 μg/mL). To explore the mechanism of the growth inhibitory effect of cetuximab in 11–18 cells, we analyzed cell cycle progression and apoptosis under the treatment of cetuximab. In the cell cycle analysis, we observed a tendency of decreased percentage of cells in G2-M/S phases and an increased number of cells in G0/G1 population. However, these changes were trivial and not significant (). On the other hand, we detected significantly increased levels of apoptosis at an IC50 value (0.1 μg/mL) of cetuximab (). Therefore, cetuximab inhibits cell proliferation mainly through the induction of apoptosis rather than through cell cycle arrest. These data suggested that EGFR mutation accompanied by an increase of the copy number is the most important marker for gefitinib sensitivity, followed by an increased gene copy number alone. However, EGFR mutation together with an increased copy number was not a good marker for cetuximab sensitivity, and there seems to be another mechanism that sensitizes lung cancer cells to cetuximab. Expression of EGFR was not correlated with sensitivity to either cetuximab or gefitinib, corresponding to previous reports.Citation21
Figure 1. Effect of cetuximab on the cell cycle and apoptosis of sensitive cell line. (A) 11–18 cells were treated with indicated concentrations of cetuximab for 72 h, fixed, and stained with propidium iodide. The percentage of cells were analyzed in each phase of cell cycle by FACSCalibur. Experiments were done in triplicate. Bars indicate SD (B) 11–18 cells were treated with indicated concentrations of cetuximab for 72 h, and stained with PE Annexin V and 7-AAD. The percentage of apoptotic cells were analyzed using FACSCalibur. Data are the summary of percentages of early and late apoptotic cells (Annexin V positive), and expressed as mean ± SD of three independent experiments. *p < 0.05 vs. control by Mann-Whitney U test.
Activation of EGFR and its downstream molecules in cetuximab-sensitive and -resistant lung cancer cell lines
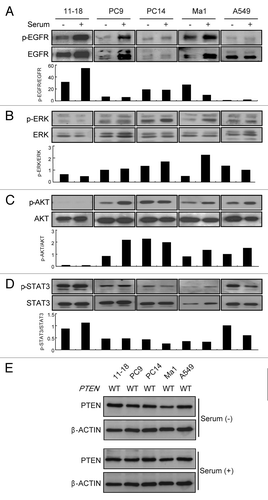
To explore additional markers for cetuximab, we assessed the activation of EGFR and its downstream molecules in lung cancer cell lines. For this assay, we used five representative cell lines, including four (11–18, PC9, PC14 and Ma1) with EGFR mutation that were sensitive to gefitinib and one (A549) with wild-type EGFR that was resistant to gefitinib. Among them, 11–18 was the only cell line showing sensitivity to cetuximab. Assessment of activation was done by western blotting with specific antibodies for EGFR, ERK, AKT and STAT3 after incubation in the presence and absence of serum. In the cells with EGFR mutation (11–18, PC9, PC14 and Ma1), EGFR was phosphorylated in the basal state and its phosphorylation increased after stimulation with serum. In the wild-type EGFR cell line (A549), EGFR showed little phosphorylation under both basal and serum-stimulated conditions (), suggesting that mutant EGFR was markedly activated in the cell lines even without serum stimulation. Regarding the downstream molecules, ERK and AKT showed varying levels of phosphorylation under basal and serum-stimulated conditions () irrespective of the presence or absence of EGFR mutation, expect in 11–18 cells. This suggests that these downstream molecules might also be controlled by various upstream signaling pathways other than EGFR in the EGFR mutant cell lines. In 11–18 cells (which were highly sensitive to cetuximab), AKT was expressed, but showed little phosphorylation irrespective of serum stimulation (), suggesting that the AKT pathway was completely inactivated in this cetuximab-sensitive cell line. Phosphorylation of STAT3 was observed in all cell lines, but was not altered by serum stimulation, suggesting limited involvement of this molecule in determining cetuximab sensitivity (). To determine the contribution of PTEN on the inactivation status of AKT in cetuximab sensitive cell line, we further determined the expression and mutation status of PTEN in cetuximab-sensitive and cetuximab-resistant cell lines. As shown in , significant alternation of PTEN expression was not observed between these cell lines, and any mutation of coding sequences of PTEN was not detected in any cell line. Therefore, AKT inactivation seen in 11–18 cells might be caused other than by PTEN hyperactivity, such as by direct AKT dephosphorylation, by protein phosphatase A2 (PP2A) or by PH domain leucine-rich repeat protein phosphatase (PHLPP).Citation22 In any case, these results suggested the possible involvement of activation of AKT in sensitivity to cetuximab among cell lines with mutant and highly activated EGFR.
Figure 2. Phosphorylation of EGFR, ERK, AKT and STAT3, and expression and mutation status of PTEN in EGFR mutant and wild-type NSCLC cell lines. (A–D) Cells were incubated with/without serum for 24 h, and immunoblotting was done with antibodies targeting phosphorylated (p) or total EGFR, ERK, and AKT. Densitometric analysis was performed with TotaLab software (Nonlinear Dynamics), and the relative pEGFR/EGFR, pERK/ERK, pAKT/AKT, pSTAT3/STAT3 ratios are indicated as bar graphs under each blot. Experiments were repeated three times and representative results are shown. (E) PTEN protein status in each cell line was determined by immunoblotting in the presence or absence of serum. The β-actin was used as loading control. Experiments were repeated three times and representative results are shown. The mutation status of PTEN in each cell line is also indicated. WT, wild type.
Influence of gefitinib and cetuximab on activation of EGFR signaling molecules in cetuximab-sensitive and -resistant lung cancer cell lines
To further explore the involvement of molecules downstream from EGFR in cetuximab sensitivity, we next examined the influence of gefitinib and cetuximab on activation of these molecules in cells with EGFR mutation in the presence of EGF. In this analysis, we used 11–18 and PC9 cells as representative cetuximab-sensitive and -resistant cell lines, respectively. First, when we treated the cells with gefitinib, phosphorylation of EGFR, ERK, and AKT induced by EGF was decreased in both cell lines (). In particular, gefitinib could inhibit basal phosphorylation of EGFR, ERK, and AKT, and inhibition was nearly complete at a concentration of 1 μmol/L.
Figure 3. Influence of gefitinib and cetuximab on phosphorylation status of EGFR, ERK, AKT, in cetuximab-sensitive and -resistant NSCLC cell lines. Cetuximab-sensitive (11–18) and -resistant (PC9) cells with EGFR mutation were incubated with gefitinib (A) or cetuximab (B) at the indicated concentrations in serum-free medium for 2 h, after which cells were stimulated with EGF (100 ng/mL) for 10 min. Then the cells were lyzed, and subjected to immunoblot analysis with antibodies to phosphorylated (p) or total EGFR, ERK, and AKT, as well as an antibody targeting β-actin (loading control). Experiments were repeated three times and representative results are shown.
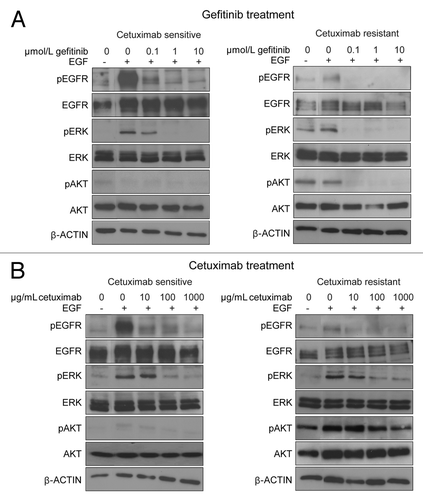
On the other hand, when we treated the cells with cetuximab, it reduced the elevated phosphorylation of EGFR or ERK due to EGF stimulation to the basal level, but could not further decrease phosphorylation (). This suggests that cetuximab only inhibits ligand-dependent activation of EGFR and ERK. Regarding the AKT pathway, inhibition of the phosphorylation of AKT by cetuximab was very weak in both cetuximab-sensitive and -resistant cell lines, and an extremely high concentration of cetuximab (1,000 μg/mL) was needed to achieve apparent inhibition. Thus, there was an obvious difference phosphorylation status of AKT. In the cetuximab-sensitive cell line, because basal phosphorylation of AKT was constitutively suppressed, phosphorylations of both ERK and AKT after cetuximab treatment was effectively suppressed and was similar to that seen after gefitinib treatment. In the resistant cell line, however, AKT was still phosphorylated even after cetuximab treatment. Similar incomplete suppression of the AKT pathway by cetuximab was observed in the other cetuximab-resistant cell lines (PC14 and Ma1) (data not shown). Namely, in cells with endogenously inactivated AKT, inhibition of the ERK pathway by cetuximab is able to suppress both the AKT and ERK pathways, and this may be the mechanism of cetuximab sensitivity.
Role of AKT in sensitivity to cetuximab of EGFR mutant cell lines
To confirm our hypothesis that endogenously inactivated AKT is a marker for cetuximab in cells with EGFR mutation, we first examined the effect of overexpression of AKT in the sensitive cell line. As shown in transfection of the AKT expression vector enhanced the level of AKT expression and accordingly phosphorylated AKT was also increased in the cetuximab-sensitive cell line, whereas other signaling molecules from the EGFR axis were fundamentally unaffected. The cell viability assay revealed that the cetuximab sensitivity of AKT-transfected cells was markedly increased and IC50 was 100-fold larger compared with that of mock-transfected pCEFL cells (). We next examined the effect of downregulation of AKT in cetuximab-resistant cell line on cetuximab sensitivity. AKT silencing by siRNA reduced the expression of AKT in cetuximab-resistant Ma1 cells (, left panel), and accordingly significantly increased the sensitivity of this cell line for cetuximab (, right panel). To further explore the contribution of AKT to cetuximab sensitivity, we next established a cetuximab-resistant cell line (11–18R) by culturing sensitive cells (11–18) in the presence of increasing concentrations of cetuximab over a period of 6 months (, left panel). Western blotting analysis revealed that phosphorylation of AKT was markedly increased in this resistant clone although the level of AKT expression was unchanged (, right panel). Finally, we tested the effect of pharmacological inhibition of the AKT pathway in the resistant clone on cetuximab sensitivity. Treatment of parental sensitive cells with the PI3-K inhibitor LY294002 enhanced the growth-inhibiting effect of cetuximab (), but significant and more profound enhancement of this effect of cetuximab was seen with resistant cells (). Data were analyzed by the method of Chou and TalalayCitation23 using CalcuSyn software. The resulting CI values were less than 1 for all combinations of doses, showing that addition of LY294002 to cetuximab had a synergistic inhibitory effect on both cetuximab-sensitive and -resistant cells (). These data suggest that the state of AKT activation is the critical factor determining cetuximab sensitivity for lung cancer cell lines with EGFR mutation.
Figure 4. Effect of the modulation of AKT expression on cetuximab sensitivity of NSCLC cell lines. (A) A cetuximab-sensitive cell line (11–18) was transfected with the AKT expression vector (pCEFL-HA-AKT) or a mock control plasmid (pCEFL). After 24 h, the cells were lyzed and subjected to immunoblot analysis with the indicated antibodies. (B) Cells were treated with various concentrations of cetuximab for 72 h and then cell growth was determined by the WST-8 assay. Then, IC50 values of cetuximab were calculated for each cell. Experiments in triplicate were done three times separately and IC50 values from each experiment are combined. Bars indicate SD *p < 0.05 vs. control by Mann-Whitney U test. (C) A cetuximab-resistant cell line (Ma1) was transfected with indicated concentrations of the AKT siRNA or negative control siRNA. Cell lysates were subjected to immunoblotting with the indicated antibodies 48 h after transfection (left panel). For cell growth inhibition assay, 24 h after transfection with 50 μM of AKT siRNA or negative control siRNA, the cells were started incubating with various concentration of cetuximab for 72 h, and then cell viability was determined by the WST-8 assay and plotted as a percentage of that for untreated control cells. Bars are the SD of triplicate cultures. Data are representative of three independent experiments (right panel).
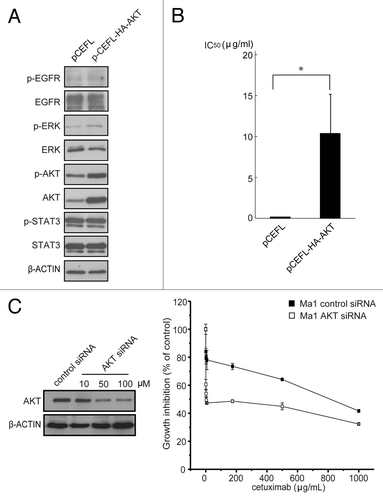
Figure 5. Effect of the combination of cetuximab and PI3K/AKT inhibitor to cetuximab resistance. Cetuximab-sensitive cells (11–18) were exposed to increasing concentrations (0.1–1,000 μg/mL) of cetuximab for six months and resistant cells (11–18R) were established. (A) Parental cells and established resistant cells were treated with the indicated concentrations of cetuximab for 72 h. Then cell viability was measured by WST-8 assay and plotted as a percentage of that for untreated control cells (left panel). Cells were grown for 24 h in the presence of serum, lysed, and subjected to immunoblotting with the indicated antibodies (right panel). (B) Parental cetuximab-sensitive cells (11–18) were treated with various concentrations of cetuximab in combination with 0, 1, 10 or 20 μM of LY294002 for 72 h. Cell viability was measured by the WST-8 assay and plotted as a percentage of that for untreated control cells. (C) The same experiment was repeated using the established cetuximab-resistant cells (11–18R). Bars are the SD of triplicate cultures. Data are representative of four independent experiments.
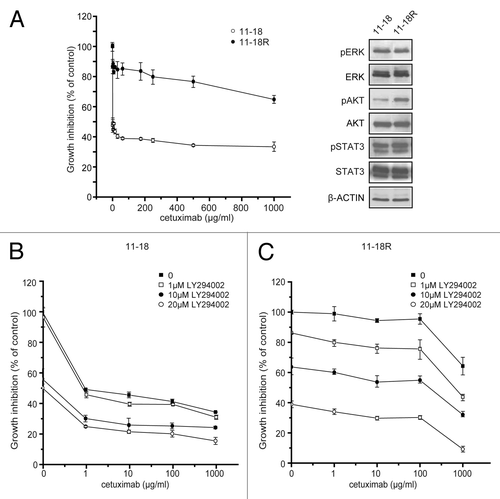
Table 2. Synergistic effect of LY294002 with cetuximab in cetuximab sensitive and resistant cell lines
Discussion
In this study, we explored the factors associated with sensitivity to cetuximab in lung cancer cell lines, and compared the findings with those for gefitinib. We found that inactivation of AKT in cells with EGFR mutation could be a marker of sensitivity to cetuximab. We also showed that cetuximab can inhibit activation of ERK induced by EGFR stimulation, but not that of AKT. Therefore, cells with inactivation of AKT are susceptible to inhibition of growth by cetuximab treatment, while the cells with activation of AKT are resistant to cetuximab. These observations suggest that the AKT status of lung cancer might be a novel biological marker for predicting the effectiveness of cetuximab. This finding could be particularly useful, because a recent report indicated that potential markers based on experience with gefitinib in NSCLC patients or cetuximab in CRC patients (such as KRAS or EGFR mutation, EGFR protein expression, or EGFR copy number) were not associated with the clinical benefit of cetuximab for NSCLC in a large-scale phase III clinical study.Citation18
The cetuximab-sensitive cell line that we used in this study had EGFR mutation in addition to inactivation of AKT. We believe that EGFR mutation also contributed to cetuximab sensitivity to some extent, although it is not a definitive marker. In general molecular-targeting drugs show effectiveness against cancer cells with activation of the relevant signaling pathway for each molecular agent. This concept is known as “oncogene addiction,” and it was advocated by Weinstein in 1997.Citation24 For example, chronic myeloid leukemia, pancreatic cancer, and breast cancer have been shown to depend on abnormal activation of tyrosine kinase Bcr-Abl,Citation25 KRAS mutation,Citation26 and Her-2 amplification,Citation27 respectively. Experience with gefitinib also supports this concept. Because EGFR mutation leads to abnormal activation of EGFR and gefitinib effectively inhibits this mutant EGFR, cells with EGFR mutation are highly sensitive to growth inhibition by gefitinib.Citation11 Therefore, the same may be true for the effectiveness of cetuximab. “Addiction” of cancer cells to the EGFR pathway by mutation might be a basal requirement for the effectiveness of cetuximab, because cetuximab can inhibit mutant EGFR, as shown in this study and by others.Citation28
Moreover, our data indicated that loss of AKT activity in cells with EGFR mutation is necessary and sufficient for responsiveness to cetuximab. Though very few cell lines have been shown as cetuximab-sensitive by the viability assay, some evidence reported previously supports our findings. A well-characterized cetuximab-sensitive cell line, HCC827, possesses EGFR mutation, and the phosphorylation of AKT in these cells is reported to be nearly completely inhibited by cetuximab treatment.Citation29 This inactivation is similar to the inhibition of AKT observed in 11–18 cells. Another sensitive cell line, H292, has very little constitutive phosphorylation of AKT, which is analogous to 11–18 cells, and its phosphorylation is also completely inhibited by treatment with cetuximab.Citation28,Citation30 Though no data on the relation between AKT activation and clinical responsiveness of NSCLC to cetuximab have been reported yet, observations have been described for patients with metastatic colorectal cancer (mCRC). Loss of PTEN expression by mCRC was reported to be associated with nonresponsiveness to cetuximab,Citation31 though activation of AKT was not examined in that study. For another mAb targeting EGFR (EMD72000), a phase I trial in patients with mCRC showed that only tumors with low baseline phosphorylation of AKT were responsive to EMD72000.Citation32 These observations suggest that the level of AKT activation might play a central role in determining the antitumor effect of anti-EGFR antibodies. Because available clinical data are limited to mCRC, in which EGFR rarely shows mutation, clinical studies are needed to elucidate the role of the combination of AKT activation and EGFR mutation as marker for cetuximab sensitivity of NSCLC.
Identification of AKT inactivation plus EGFR mutation as a marker of cetuximab responsiveness for NSCLC cells may assist the selection of patients for treatment with this agent and lead to a novel therapeutic approach to NSCLC. Aiming to achieve simultaneous multiple inhibition of EGFR, use of cetuximab in combination with erlotinib or gefitinib has been suggested,Citation33,Citation34 and the feasibility of combination with gefitinib was shown by a phase I study.Citation35 Because cetuximab is reported to inhibit the activity of cells with gefitinib- and erlotinib-resistant EGFR mutation (T790M) both in vitroCitation28 and in vivo,Citation36 it is expected to be useful as an alternative therapy when tumors become refractory to gefitinib or erlotinib by acquisition of T790M mutation. In such clinical settings, selection of patients with inactivated, dephosphorylated, AKT would be particularly beneficial, because most of the eligible patients would have EGFR activating mutations. A recent study has suggested that the frequency of the tumor with lack of AKT activation in EGFR-mutated lung adenocarcinoma is 40%.Citation37 Therefore, a considerable portion of patients treated with TKIs might be suitable target of cetuximab treatment. As a possible screening modality for the selection of these eligible patients, we might utilize enzyme immunoassay technology, such as two-site chemiluminescence-linked immunosorbent assay (CLISA) for measuring phosphorylated AKT,Citation38 besides conventional immunohistochemistry (IHC). CLISA would more advantageous than IHC, because it allows us to obtain more precise and quantitative results. Moreover, because this procedure is based on homogenized samples, it is possible to simultaneously obtain DNAs for mutation analysis and proteins for phosphorylated AKT quantification from a single tumor tissue. In addition, our results suggest that patients with phosphorylation of AKT could be treated by the combination of cetuximab and an AKT pathway inhibitor. Moreover, the combination of cetuximab with PI3K or mTOR inhibitors may be promising in patients with EGFR mutations.
In summary, our results indicate that dephosphorylation of AKT in EGFR-addicted NSCLC cells is a possible candidate marker for cetuximab. Our study also showed that the combination of an AKT pathway inhibitor with cetuximab would be promising to overcome the limited effect of TKIs against tumors with EGFR mutations. These findings still need to be validated in clinical trials, but suggest a new strategy for selecting NSCLC patients to receive treatment with cetuximab.
Materials and Methods
Cell lines and cell culture
The 11–18, LCSC#1, LCSC#2, LC-2/ad, EBC-1, LK-2, Lu99 lung cancer cell lines and A431 epidermoid carcinoma cell line were provided by the Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University). The PC9 cell line was provided by Dr. N. Saijyo from the National Cancer Center Hospital, while PC14 cells were obtained from the RIKEN Cell Bank. LK87, LK-1, LK79, and 86–2 cells were provided by Dr. S. Kobayashi from the Miyagi Prefectural Semine Hospital through the Cell Resource Center. A549 was from the American Type Culture Collection (ATCC). N417 was provided by Dr. A.F. Gazdar and Dr. H. Oie (National Cancer Institute-Navy Medical Oncology Branch, NIH). Ma1, Ma10, Ma29, and Ms1 cells were provided by Dr. T. Hirashima (Osaka Prefectural Habikino Hospital). Ma1, Ma10, Ma29, EBC-1, 86–2, N417 and Ms1 cells were maintained in DMEM (Wako) supplemented with 10% fetal bovine serum (FBS) (Life technologies), 50 units/mL penicillin, and 50 units/mL streptomycin. The other cell lines were cultured in RPMI 1640 medium (Wako) supplemented with 10% FBS, 50 units/mL penicillin, and 50 units/mL streptomycin. All cells were cultured at 37°C in humidified air with 5% CO2.
Reagents
Gefitinib was provided by AstraZeneca, and was dissolved in dimethyl sulfoxide (DMSO) and stored at -20°C until use. Cetuximab (2 mg/mL) was provided by Bristol-Myers Squibb. Epidermal growth factor (21-leu EGF) and LY294002 were purchased from Progen and Calbiochem, respectively. A plasmid construct expressing epitope-tagged AKT (pCEFL-HA-AKT) was kindly provided by Dr. Silvio Gutkind (NIH).
Growth inhibition assay
Cell viability was assessed by using the 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)2H-tetrazolium monosodium salt (WST-8) assay (Dojindo). Cells were plated at 3⋅104/well in triplicate in 96-well plates overnight in complete medium. After 24 h, DMEM or RPMI-1640 supplemented with various concentrations of gefitinib and cetuximab was added. After 72 h, WST-8 solution was added to each well and incubation was done for 4 h at 37°C, after which the absorbance was measured at 450 nm. Cell viability was calculated as the mean absorbance of the wells containing treated cells divided by the mean for the untreated control wells. The concentration of gefitinib or cetuximab resulting in 50% growth inhibition (IC50) was calculated. Multiple drug effect analysis was done with CalcuSyn Software (Biosoft), which calculated the Chou and Talaly combination index (CI) for various drug combinations (Fa = fraction affected by dose).Citation23 CI values of < 1, 1, or > 1 indicate synergistic, additive, and antagonistic effects, respectively. All experiments were done at least in triplicate and were repeated three times or more.
Sequence analysis
DNA was extracted by using a Wizard genomic DNA purification kit according to the manufacturer’s instructions (Promega). Exons 18 to 21 of EGFR and exon 2 of KRAS were amplified by PCR using the primers and conditions described previously.Citation11,Citation39 Exons 1 to 9 of PTEN were amplified by PCR using a number of primer sets (Table S1). The PCR products were purified with a multiscreen PCR filter plate (Millipore) and sequenced by using a BigDye Terminator v3.1 Cycle Sequencing kit and an ABI PRISM 3130 × 1 Genetic Analyzer (Applied Biosystems).
Fluorescence in situ hybridization
Cells were fixed with methanol and acetic acid, and EGFR gene copy number was determined by FISH using the EGFR/CEN7 FISH probe (Dako) according to the manufacturer’s instructions. Fluorescence signals (green for CEP 7 and red for EGFR) were counted under an Olympus IX70 fluorescence microscope with appropriate filters (Olympus). For each hybridization, 100 nonoverlapping nuclei were assessed. The results were classified into six groups: disomy, low trisomy, high trisomy, low polysomy, high polysomy, and gene amplification, and high polysomy and gene amplification were classified as FISH positive according to Hirsh’s scoring guidelines.Citation13
Flow cytometric analysis
To determine the absolute number of EGFR molecules per cell, quantitative flow cytometric analysis was performed with a DAKO QIFIKIT (Dako) using FACSCalibur flow cytometer (BD Biosciences), as described previously.Citation8 For cell cycle analysis, cells treated with indicated concentrations of cetuximab for 72 h were harvested and resuspended in 70% ethanol at 4°C for 2 h. After washing twice with cold PBS, cells were treated with 0.25 mg/mL of RNase (Wako) at 37°C for 1 h and stained with 50 μg/mL propidium iodide (Wako) at 4°C for 30 min. DNA content was analyzed by FACSCalibur. For apoptosis analysis, cetuximab treated cells were harvested and stained with PE Annexin V and 7-Amino-Actinomycin D (7-AAD) using PE Annexin V Apoptosis Detection Kit I (BD Bioscience) according to the manufacturer’s protocol. The percentage of apoptotic cells were analyzed by FACSCalibur. Data were analyzed using FlowJo Software (Tree Star Inc.).
Western blotting
Cells lysates were fractioned by SDS-PAGE, after which the proteins were transferred to Immobilon-P (Millipore) membranes. Antigen-antibody complexes were detected with an ECL plus western blotting detection system (GE Healthcare). The following primary antibodies were used: pEGFR (Tyr 1068), pAKT (Ser 473), AKT, pSTAT3 (Tyr 705), STAT3, PTEN (Cell Signaling, MA), EGFR (1005) (Santa Cruz), β-ACTIN (Sigma), pERK, and ERK (Promega).
Transfection
Transfection was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, 3 × 105 cells were added to each well of 6-well plates at 24 h before transfection. Then the cells were transfected with the plasmid DNA (pCEFL-HA-AKT), harvested after 24 h, and analyzed by western blotting and the WST-8 assay. Cells transfected with the empty vector (pCEFL) were used as the mock control. Small interfering RNA (siRNA)-mediated gene silencing was done by transfection of Ma1 cells with the AKT siRNA (Cell Signaling) using Lipofectamine 2000. Allstars negative control siRNA (QIAGEN) was used in controlled experiments.
Establishment of acquired resistance to cetuximab
Over a period of six months, cultures of 11–18 cells were continuously exposed to increasing concentrations of cetuximab. Starting at the IC50 (0.1 μg/mL) of cetuximab, the concentration was progressively doubled every 10–14 d until it reached 1,000 μg/mL. Simultaneously, parental cells were cultured with PBS as a control. The established resistant cells (11–18R) were then maintained in continuous culture with the maximal concentration of cetuximab.
Statistical analysis
Comparisons between the two groups were made using the Mann-Whitney U test. The statistical analysis was performed using IBM SPSS Statistics 19 software (IBM) and Ps of < 0.05 were considered statistically significant.
| Abbreviations: | ||
| EGFR | = | epidermal growth factor receptor |
| NSCLC | = | non-small cell lung cancer |
| TKIs | = | tyrosine kinase inhibitors |
| mAb | = | monoclonal antibody |
| ADCC | = | antibody-dependent cellular cytotoxicity |
| FDA | = | U.S. Food and Drug Administration |
| EC | = | epidermal carcinoma |
| flow cytometric analysis | = | FACS |
| SCLC | = | small cell lung cancer |
| LA | = | large cell carcinoma |
| AD | = | adenocarcinoma |
| SQ | = | squamous cell carcinoma |
| FISH | = | in situ hybridization |
| FBS | = | fetal bovine serum |
| DMSO | = | dimethyl sulfoxide |
| IC50 | = | 50% growth inhibition |
| CI | = | combination index |
Additional material
Download Zip (63.1 KB)Acknowledgments
The authors thank Dr. Silvio Gutkind (National Institutes of Health/NIDCR) for providing the pCEFL plasmids and for his advice and discussion. This study was supported by grants-in-aid for Scientific Research (C) 21590994 (to H. Chikumi and E. Shimizu) and 22590863 (to E. Shimizu and H. Chikumi) from the Ministry of Education, Science, and Culture, Sports, Science and Technology, Japan.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
References
- Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, et al, Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002; 346:92 - 8; http://dx.doi.org/10.1056/NEJMoa011954; PMID: 11784875
- Singh AB, Harris RC. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 2005; 17:1183 - 93; http://dx.doi.org/10.1016/j.cellsig.2005.03.026; PMID: 15982853
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37:Suppl 4 S9 - 15; http://dx.doi.org/10.1016/S0959-8049(01)00231-3; PMID: 11597399
- Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol 2004; 22:777 - 84; http://dx.doi.org/10.1200/JCO.2004.08.001; PMID: 14990632
- Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol 2004; 22:785 - 94; http://dx.doi.org/10.1200/JCO.2004.07.215; PMID: 14990633
- Kawamoto T, Sato JD, Le A, Polikoff J, Sato GH, Mendelsohn J. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci U S A 1983; 80:1337 - 41; http://dx.doi.org/10.1073/pnas.80.5.1337; PMID: 6298788
- Mendelsohn J. Blockade of receptors for growth factors: an anticancer therapy--the fourth annual Joseph H Burchenal American Association of Cancer Research Clinical Research Award Lecture. Clin Cancer Res 2000; 6:747 - 53; PMID: 10741693
- Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 2007; 13:1552 - 61; http://dx.doi.org/10.1158/1078-0432.CCR-06-1726; PMID: 17332301
- Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al, FLEX Study Team. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009; 373:1525 - 31; http://dx.doi.org/10.1016/S0140-6736(09)60569-9; PMID: 19410716
- Joerger M, Matter-Walstra K, Früh M, Kühnel U, Szucs T, Pestalozzi B, et al. Addition of cetuximab to first-line chemotherapy in patients with advanced non-small-cell lung cancer: a cost-utility analysis. Ann Oncol 2011; 22:567 - 74; http://dx.doi.org/10.1093/annonc/mdq431; PMID: 20843984
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 39; http://dx.doi.org/10.1056/NEJMoa040938; PMID: 15118073
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 500; http://dx.doi.org/10.1126/science.1099314; PMID: 15118125
- Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005; 97:643 - 55; http://dx.doi.org/10.1093/jnci/dji112; PMID: 15870435
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2:e17; http://dx.doi.org/10.1371/journal.pmed.0020017; PMID: 15696205
- Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al, North-East Japan Study Group. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010; 362:2380 - 8; http://dx.doi.org/10.1056/NEJMoa0909530; PMID: 20573926
- Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al, West Japan Oncology Group. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11:121 - 8; http://dx.doi.org/10.1016/S1470-2045(09)70364-X; PMID: 20022809
- Russo A, Rizzo S, Bronte G, Silvestris N, Colucci G, Gebbia N, et al. The long and winding road to useful predictive factors for anti-EGFR therapy in metastatic colorectal carcinoma: the KRAS/BRAF pathway. Oncology 2009; 77:Suppl 1 57 - 68; http://dx.doi.org/10.1159/000258497; PMID: 20130433
- Khambata-Ford S, Harbison CT, Hart LL, Awad M, Xu LA, Horak CE, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol 2010; 28:918 - 27; http://dx.doi.org/10.1200/JCO.2009.25.2890; PMID: 20100958
- Brockhoff G, Hofstaedter F, Knuechel R. Flow cytometric detection and quantitation of the epidermal growth factor receptor in comparison to Scatchard analysis in human bladder carcinoma cell lines. Cytometry 1994; 17:75 - 83; http://dx.doi.org/10.1002/cyto.990170110; PMID: 8001460
- Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64:8919 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-04-2818; PMID: 15604253
- Hanna N, Lilenbaum R, Ansari R, Lynch T, Govindan R, Jänne PA, et al. Phase II trial of cetuximab in patients with previously treated non-small-cell lung cancer. J Clin Oncol 2006; 24:5253 - 8; http://dx.doi.org/10.1200/JCO.2006.08.2263; PMID: 17114658
- Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle 2010; 9:487 - 97; http://dx.doi.org/10.4161/cc.9.3.10508; PMID: 20081374
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27 - 55; http://dx.doi.org/10.1016/0065-2571(84)90007-4; PMID: 6382953
- Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006; 3:448 - 57; http://dx.doi.org/10.1038/ncponc0558; PMID: 16894390
- Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344:1084 - 6; http://dx.doi.org/10.1056/NEJM200104053441409; PMID: 11287980
- Aoki K, Yoshida T, Matsumoto N, Ide H, Sugimura T, Terada M. Suppression of Ki-ras p21 levels leading to growth inhibition of pancreatic cancer cell lines with Ki-ras mutation but not those without Ki-ras mutation. Mol Carcinog 1997; 20:251 - 8; http://dx.doi.org/10.1002/(SICI)1098-2744(199710)20:2<251::AID-MC12>3.0.CO;2-9; PMID: 9364215
- Colomer R, Lupu R, Bacus SS, Gelmann EP. erbB-2 antisense oligonucleotides inhibit the proliferation of breast carcinoma cells with erbB-2 oncogene amplification. Br J Cancer 1994; 70:819 - 25; http://dx.doi.org/10.1038/bjc.1994.405; PMID: 7947086
- Doody JF, Wang Y, Patel SN, Joynes C, Lee SP, Gerlak J, et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol Cancer Ther 2007; 6:2642 - 51; http://dx.doi.org/10.1158/1535-7163.MCT-06-0506; PMID: 17913857
- Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res 2005; 65:226 - 35; PMID: 15665299
- Yoshida T, Okamoto I, Okabe T, Iwasa T, Satoh T, Nishio K, et al. Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int J Cancer 2008; 122:1530 - 8; http://dx.doi.org/10.1002/ijc.23253; PMID: 18033688
- Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 2007; 97:1139 - 45; http://dx.doi.org/10.1038/sj.bjc.6604009; PMID: 17940504
- Tabernero J, Rojo F, Jimenez E, Montaner I, Santome L, Guix M, et al. A phase I PK and serial tumor and skin pharmacodynamic (PD) study of weekly (q1w), every 2-week (q2w) or every 3-week (q3w) 1-hour (h) infusion EMD72000, a humanized monoclonal anti-epidermal growth factor receptor (EGFR) antibody, in patients (pt) with advanced tumors. Proc Am Soc Clin Oncol 2003; 22:Abstr 770
- Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004; 64:5355 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-04-0562; PMID: 15289342
- Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tabernero J, et al. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res 2004; 10:6487 - 501; http://dx.doi.org/10.1158/1078-0432.CCR-04-0870; PMID: 15475436
- Ramalingam S, Forster J, Naret C, Evans T, Sulecki M, Lu H, et al. Dual inhibition of the epidermal growth factor receptor with cetuximab, an IgG1 monoclonal antibody, and gefitinib, a tyrosine kinase inhibitor, in patients with refractory non-small cell lung cancer (NSCLC): a phase I study. J Thorac Oncol 2008; 3:258 - 64; http://dx.doi.org/10.1097/JTO.0b013e3181653d1b; PMID: 18317068
- Steiner P, Joynes C, Bassi R, Wang S, Tonra JR, Hadari YR, et al. Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wild-type and mutated epidermal growth factor receptor. Clin Cancer Res 2007; 13:1540 - 51; http://dx.doi.org/10.1158/1078-0432.CCR-06-1887; PMID: 17332300
- Ikeda S, Takabe K, Inagaki M, Funakoshi N, Suzuki K, Shibata T. Correlation between EGFR gene mutation pattern and Akt phosphorylation in pulmonary adenocarcinomas. Pathol Int 2007; 57:268 - 75; http://dx.doi.org/10.1111/j.1440-1827.2007.02093.x; PMID: 17493174
- Cicenas J, Urban P, Vuaroqueaux V, Labuhn M, Küng W, Wight E, et al. Increased level of phosphorylated akt measured by chemiluminescence-linked immunosorbent assay is a predictor of poor prognosis in primary breast cancer overexpressing ErbB-2. Breast Cancer Res 2005; 7:R394 - 401; http://dx.doi.org/10.1186/bcr1015; PMID: 15987444
- Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005; 65:1642 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-04-4235; PMID: 15753357