Abstract
The present studies were initiated to determine whether inhibitors of MEK1/2 or SRC signaling, respectively, enhance CHK1 inhibitor lethality in primary human glioblastoma cells. Multiple MEK1/2 inhibitors (CI-1040 (PD184352); AZD6244 (ARRY-142886)) interacted with multiple CHK1 inhibitors (UCN-01, AZD7762) to kill multiple primary human glioma cell isolates that have a diverse set of genetic alterations typically found in the disease. Inhibition of SRC family proteins also enhanced CHK1 inhibitor lethality. Combined treatment of glioma cells with (MEK1/2 + CHK1) inhibitors enhanced radiosensitivity. Combined (MEK1/2 + CHK1) inhibitor treatment led to dephosphorylation of ERK1/2 and S6 ribosomal protein, whereas the phosphorylation of JNK and p38 was increased. MEK1/2 + CHK1 inhibitor-stimulated cell death was associated with the cleavage of pro-caspases 3 and 7 as well as the caspase substrate (PARP). We also observed activation of pro-apoptotic BCL-2 effector proteins BAK and BAX and reduced levels of pro-survival BCL-2 family protein BCL-XL. Overexpression of BCL-XL alleviated but did not completely abolish MEK1/2 + CHK1 inhibitor cytotoxicity in GBM cells. These findings argue that multiple inhibitors of the SRC-MEK pathway have the potential to interact with multiple CHK1 inhibitors to kill glioma cells.
Keywords: :
Introduction
In the United States, glioblastoma multiforme (GBM) is diagnosed in ~20,000 patients per annum. High-grade tumors, such as anaplastic astrocytoma and GBM, account for the majority of astrocytic tumors.Citation1,Citation2 Even under optimal circumstances, in which essentially all of the tumor can be surgically removed and the patients are maximally treated with radiation and chemotherapeutic agents such as Temodar® or Gliadel® wafers, the mean survival of this disease is only extended from 2–3 mo to 12 mo.Citation3-Citation6 These depressing statistics accentuate the need to develop more effective therapies against this devastating and invariably fatal disease.
In response to DNA damage or DNA-replication stress, ATM, ATR and CHK1/2 are activated and cell cycle checkpoints become engaged, allowing for cell cycle arrest and DNA damage repair. CHK1 inhibitors including UCN-01 (7-hydroxystaurosporine) and AZD7762 are currently being evaluated as anti-neoplastic agents in clinical trials, both alone and in combination with chemotherapeutic agents and ionizing radiation.Citation7 These agents are postulated to enhance the cytotoxicity of established chemotherapeutic agents by inhibition of CHK1 thereby facilitating inappropriate cell cycle progression after DNA damage.Citation8,Citation9
We have published several studies combining SRC / MEK1/2 inhibitors with CHK1 inhibitors in established tumor cell types, notably breast cancer cells, but not in CNS tumor cells that are generally thought to be much more resistant to many therapeutic modalities, and demonstrated that the lethality of CHK1 inhibitors is enhanced by their combination with inhibitors of the SRC-RAS-MEK1/2-ERK1/2 pathway.Citation9-Citation14 Treatment of tumor cells with CHK1 inhibitors enhances signaling through the SRC-RAS-MEK1/2-ERK1/2 pathway and expression of a dominant negative CHK1 protein elevates basal ERK1/2 activity and blocks CHK1 inhibitor–induced ERK1/2 activation.Citation9-Citation14 Interruption of MEK1/2-ERK1/2 pathway by MEK1/2 inhibitors dramatically potentiated UCN-01-induced cell death.Citation15,Citation16 What is perhaps more of note are findings that non-transformed blood cells e.g., peripheral mononuclear cells, CD34+ stem cells or primary cultures of human mammary epithelial cells or of rodent hepatocytes were resistant to the CHK1 inhibitor based drug combination toxicity.Citation10,Citation17 In myeloma blasts freshly isolated from patients, cells that are essentially in G0 phase of the cell cycle, CHK1 and MEK1/2 inhibitors synergized to kill arguing against the lethal effect being dependent on changes in cell cycle progression.Citation12
The present studies were performed to define whether inhibitors of the SRC-MEK1/2-ERK1/2 pathway interacted with CHK1 inhibitors to kill semi-established isolates of primary human glioblastoma cells. Unlike commercial established cell lines, these isolates exhibit a highly invasive phenotype when grown in the brains of athymic mice that is identical to the disease found in patients. Our studies focused in particular detail on the interactions between CHK1 inhibitors and MEK1/2 inhibitors and demonstrated that this drug combination causes profound levels of glioma cell killing.
Materials and Methods
Materials
Primary human GBM isolates were freely obtained from the Mayo Clinic (Rochester, MN) and were unlinked to any patient information.Citation18 The cells have been characterized in detail previously.Citation19-Citation21 Two human medulloblastoma cell lines, DAOY and HTB-185 (D283 Med) were from the American Type Culture Collection (Menassas, VA, USA). The primary culture (VC312) of medulloblastoma was derived from a tumor of a 4-y-old male patient treated at the Virginia Commonwealth University Health System’s Medical College of Virginia Hospital as described.Citation22,Citation23 Antibiotics-Antimycotics (100 units/ml penicillin, 100 μg/ml streptomycin and 250 μg/ml amphotericin B) and Trypsin-EDTA were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT).PD184352 (CI-1040) and HA14–1 was purchased from Calbiochem/EMD sciences (San Diego, CA). UCN-01 was purchased from Sigma (St. Louis MO). CI-1040 (PD184352), AZD6244 (ARRY-142886), AZD7762 and AZD0530 were purchased from Selleck chemicals (Houston TX). The JNK inhibitor peptide (JNK-IP) was supplied by Calbiochem (San Diego, CA) as a powder, dissolved in sterile DMSO, and stored frozen under light-protected conditions at -80°C. All antibodies were purchased from Cell Signaling Technologies (Worcester, MA). All the secondary antibodies and Immobilon-FL PVDF membrane were purchased from LI-COR Biosciences (Lincoln, Nebraska). Lipofectamine 2000 transfection reagent was purchased Invitrogen Life Technologies, Inc. (Carlsbad, CA).
Methods
Cell culture and in vitro exposure of cells to drugs
All GBM cell lines were maintained in a DMEM medium supplemented with 5% FBS and 1% antibiotic-antimycotic in a humidified incubator under an atmosphere containing 5% CO2 at 37°C. DAOY and HTB-185 cells were grown in GIBCO™ Minimum Essential Medium (MEM) Alpha Medium supplemented with 10% FBS and 1% antibiotic-antimycotic. VC312 cells were maintained in DMEM/F12 supplemented with 10%FBS, 1% antibiotic-antimycotic, 20 ng/mL recombinant human epidermal growth factor, and 10 ng/mL recombinant human basic fibroblast growth factor. In vitro Vehicle / UCN-01 / CI-1040 / AZD6244 / AZD0530 / AZD7762 treatment was from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v).
Cell treatments, SDS-PAGE and western blot analysis
For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 x 103per CmCitation2 and were treated with Vehicle/UCN-01/CI-1040/AZD6244/AZD0530/AZD7762 or their combination for the indicated times. Cells for colony formation assays were plated at 250–4000 cells per well in sextupilcate and for in vitro assays 14 h after plating were treated with the individual or the drug combination(s) for 48h followed by drug removal. Ten-14 d after exposure or tumor isolation, plates were washed in PBS, fixed with 100% methanol and stained with a filtered solution of crystal violet (5% w/v). After washing with tap water, the colonies were counted both manually (by eye) and digitally using a ColCount TM plate reader (Oxford Optronics, Oxford, England).Colony formation was defined as a colony of 50 cells or greater. For SDS PAGE and immunoblotting, cells were plated at 5 x 105 cells/cm and treated with therapeutic drugs at the indicated concentrations and after the indicated time of treatment, lysed with whole-cell lysis buffer (0.5 M TRIS-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue) in the presence of a protease inhibitor cocktail (Sigma, S8830), and the samples were sonicated and boiled for 5 min. The boiled samples were loaded onto 10–14% SDS-PAGE and were fractionated by SDS-PAGE gels in a Bio-Rad Protean II system. After transferring proteins to the Immobilon-FL PVDF membrane, the membrane was blocked with Odyssey Blocking Buffer from LI-COR Biosciences for 60 min at room temperature and incubated with the primary antibody at appropriate dilutions in Odyssey Blocking Buffer at 4°C overnight. After overnight incubation with appropriate primary antibodies, the membrane was washed (3x) with TBS-T for a total of 15 min, probed with fluorescently-labeled secondary antibody (1:5000) for 80 min at room temperature and washed (3x) with TBS-T for a total of 15 min. The immunoblots were visualized by an Odyssey Infrared Imaging System (LI-COR Biosciences).
Short-term cell viability assays after drug exposure
Cells were isolated at the indicated times in the Figure by trypsinization. Cell viability was measured with Vi-CELL™ Series Cell Viability Analyzers (Beckman Coulter, Inc.), which is based on traditional cell viability method of trypan blue exclusion.
Recombinant adenoviral vectors; infection in vitro
We generated and purchased previously noted recombinant adenoviruses to express constitutively active MEK1, constitutively active AKT, constitutively active p70S6K, dominant negative p38 and BCL-XL (Vector Biolabs, Philadelphia, PA). Cells were infected with these adenoviruses at an approximate m.o.i. of 50. As note dabove, cells were further incubated for 24 h prior to irradiation to ensure adequate expression of transduced gene products.Citation20,Citation24
Plasmid transfection
Plasmid DNA (0.5 μg/total plasmid transfected) was diluted into 50 μl of growth media that lacked supplementation with FBS or with penicillin-streptomycin. Lipofectamine 2000 reagent (1 μl) (Invitrogen, Carlsbad, CA) was diluted into 50 μl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The two solutions were then mixed together and incubated at room temperature for 30 min. The total mix was added to each well (6-well plate) containing 200 μl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The cells were incubated for 4–8 h at 37°C, after which time the media was replaced with growth media containing 5% (v/v) FBS and 1x pen-strep.
Data analysis
Comparison of the effects between drug treatments was performed following ANOVA using the Student’s t test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Results
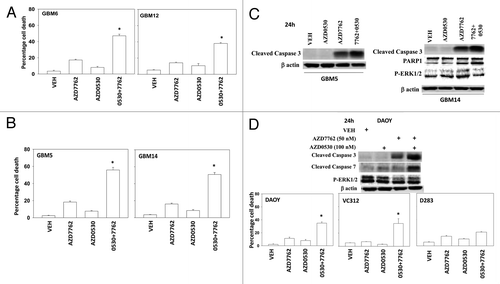
In the present study, we have used four semi-established primary human GBM isolates. GBM5 expresses PDGFR α and contains a mutant active PI3K; GBM6 expresses truncated activated mutant ERBB1 vIII; GBM12 expresses full length mutant activate ERBB1; GBM14 lacks PTEN function. In all of the tested GBM isolates, the toxicity of the CHK1 inhibitors UCN-01 and AZD7762 were enhanced by inhibition of MEK1/2 using the inhibitors CI-1040 (PD184352) and AZD6244 in a dose and time -dependent manner (-). Similar data, showing a greater than additive killing effect, were observed in colony formation assays ( and ). Radiotherapy is a major component of treating glioblastoma.Citation25,Citation26 MEK1/2 and CHK1 inhibitor treatment radiosensitized GBM cells either when irradiation occurred with drug exposure (concomitant) or shortly after drug exposure ( and ). The results are similar to what have been observed in non-CNS tumor cell types.Citation12-Citation14
Figure 1. MEK inhibitors interact with CHK1 inhibitors to kill multiple primary human glioma cell isolates. (A) GBM5 and GBM6 cells were treated with AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0μM), or AZD7762+CI-1040 for 24 h. Floating and attached cells were isolated after drug exposure and cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (B) GBM5 cells were treated with UCN-01 (10 or 50nM), AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0μM), AZD6244 (0.5 or 1.0μM) or UCN-01+CI-1040 or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value; Upper blot: portions of the cell lysates were immunoblotted against cleaved caspases-3, and cleaved caspases-7. (C) GBM6 cells were treated with UCN-01 (10, 25, or 50 nM), CI-1040 (0.5 or 1.0μM), AZD6244 (0.5, 1.0 μM) or UCN-01+CI-1040 or AZD6244+UCN-01 for 24 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (D) GBM6 cells were treated with UCN-01 (10–50nM), AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0 μM), UCN-01+CI-1040 or AZD7762+CI-1040 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (E) GBM12 and GBM14 cells were treated with UCN-01 (10 or 50 nM), CI-1040 (0.5 or 1.0μM) or UCN-01+CI-1040 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (F) GBM5, 6, 12, and 14 cells were treated with AZD7762 (300 nM), AZD6244 (500 nM) or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value.
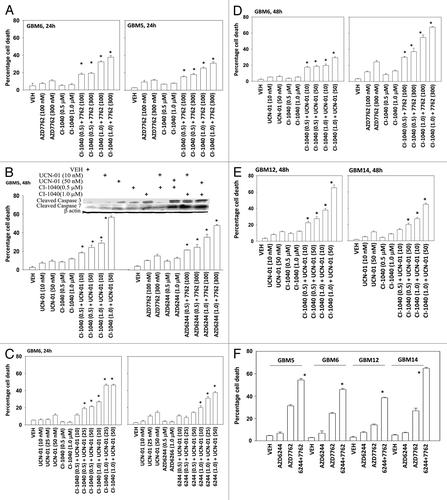
Table 1. Pre-treatment of GBM6 cells with inhibitors of MEK1/2 and CHK1 causes radiosensitization. GBM6 cells, plated as single individual cells in sextuplicate, were treated with AZD6244 (500 nM); AZD7762 (100 nM) or the drug combination for 48h. The drug containing media was removed and fresh media lacking drugs added to the cultures. Cells were then irradiated (1 Gy, 2 Gy) and colonies permitted to form over the following 14 d. Data are the means of all conditions from multiple experiments (n = 3, ± SEM). #p < 0.05 less than CHK1 inhibitor value
Table 2. Concomitant drug treatment and radiation exposure radiosensitizes GBM6 cells. GBM6 cells, plated as single individual cells in sextuplicate, were treated with AZD6244 (500 nM); AZD7762 (100 nM) or the drug combination. Fifteen minutes after drug treatment cells were irradiated (1 Gy, 2 Gy). Forty eight h after drug treatmentthe drug containing media was removed and fresh media lacking drugs added to the cultures. Colonies permitted to form over the following 14 d. Data are the means of all conditions from multiple experiments (n = 3, ± SEM). #p < 0.05 less than CHK1 inhibitor value
Medulloblastoma is the most common cause of CNS tumor in children and although disease free response rates now approach ~80% they do so with considerable morbidity to the child. Clearly, better therapeutic approaches are required to treat this CNS tumor type. AZD7762 and UCN-01 combined with CI-1040 and AZD6244 to kill in a greater than additive fashion the pediatric medulloblastoma / PNET cell lines DAOY, D283 and VC312 (-).
Figure 2. MEK inhibitors interact with CHK1 inhibitors to kill human kill pediatric medulloblastoma cell lines. Medulloblastoma cell lines (A) DAOY (B) VC312; and (C) D283 cells were treated with UCN-01 (50nM), AZD7762 (300 nM), CI-1040 (1.0 μM), AZD6244 (500 nM), UCN-01+CI-1040, AZD7762+CI-1040 or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value.
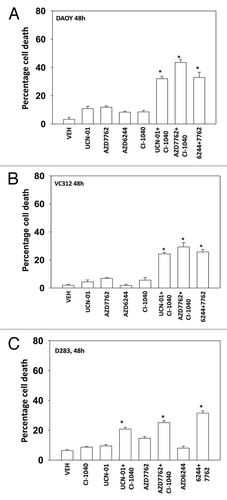
Based on our findings in and we performed immunoblotting studies, 24h after drug treatment. In all cells tested the combination of a MEK1/2 inhibitor with a CHK1 inhibitor caused cleavage of pro-caspase 3 and of PARP1 (-). The activation of ERK1/2 by CHK1 inhibitors at the 24h time-point was still evident in GBM14 and GBM6 cells and to a lesser extent in GBM5 and DAOY cells, an effect that was blocked by co-incubation with MEK1/2 inhibitors (-). Unlike in breast cancer cell lines, drug combination treatment suppressed AKT activity in GBM cells, though not medulloblastoma cells ( and ). The induction of pro-caspase 3 cleavage and PARP1 cleavage correlated with dephosphorylation of S6 protein and with p38 MAPK and / or JNK1–3 activation.
Figure 3. MEK + CHK1 inhibitors cause glioma and medulloblastoma cell death that is associated with dephosphorylation of ERK1/2 and phosphorylation of JNK and p38. (A) and (B) GBM5, 6 and 14 cells were treated with AZD7762 (25 or 50 nM), AZD6244 (500 nM) or AZD7762+AZD6244 for 24 h. Cell lysates were immunoblotted against cleaved caspase-3, PARP1, P-ERK1/2, P-JNK1–3, P-p38, and P-S6. (C) GBM5 and 14 cells were treated with UCN-01 (10 or 50nM), CI-1040 (0.5 or 1.0 mM) or UCN-01+CI-1040 for 24 h. The cell lysates were immunoblotted against P-ERK1/2, P-p38 and P-JNK1–3. (D) DAOY cells were treated with UCN-01 (50 nM), AZD7762 (50 nM), AZD6244 (500 nM), CI-1040 (1.0 μM), UCN-01+CI-1040, AZD7762+CI-1040, or AZD7762+AZD6244 for 24 h. Cell lysates were immunoblotted against caspase-3, cleaved caspase-7, PARP1, P-JNK1–3, P-S6, P-ERK1/2 and P-AKT.
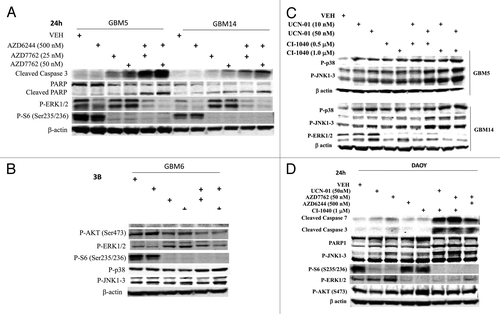
We next evaluated the relative contributions of loss of ERK1/2 and loss of p70 S6K activity after drug combination exposure to cell killing; to perform these studies we used the MEK1/2 inhibitor PD98059, which does not inhibit activated MEK1. Expression of either activated MEK1 or activated p70 S6K protected GBM cells from MEK1/2 and CHK1 inhibitor lethality (). We next evaluated the relative contributions to survival of activation of p38 MAPK and of JNK1–3 activity after drug combination exposure. Expression of either dominant negative p38 MAPK or use of the JNK inhibitory peptide significantly reduced drug combination lethality; inhibition of both pathways abolished cell killing ().
Figure 4. Inactivation of MEK/12 and p70 S6K and activation of JNK1–3/p38 and BAX/BAK are essential for MEK + CHK1 inhibitor killing in glioma and medulloblastoma cells. (A) GBM5, GBM12 and GBM14 cells were transfected with plasmids to express constitutively active p70 S6K, active MEK1 or empty vector CMV control. Twenty four hours after transfection, the cells were treated with PD98059 (25.0 μM) and AZD7762 (300 nM) for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value. (B) GBM5 and 14 cells were infected using a recombinant adenovirus to express dn-p38 in the presence or absence of JNK inhibitory peptide (JNK-IP, 10 μM). Thirty six h after transfection, the cells were treated with CI-1040 (1.0 μM), UCN-01 (50nM) or CI-1040+UCN-01 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value. (C) Top: GBM6 cells were treated with AZD7762 (300 nM) + AZD6244 (500 nM) for 24 or 48 h. Lysates were isolated for blotting of the indicated proteins and for immunoprecipitation of activated BAX and activated BAK. Bottom: GBM6 cells were infected with dn-p38 and 24 h after infection, the cells were treated with AZD7762 (300 nM) + PD98059 (25.0 μM) for 48 h in the presence or absence of JNK-IP (10 uM). The activity of BAK and BAX was determined after immunoprecipitation of the conformationally active BAX and BAK proteins. (D) GBM5 and GBM6 cells were infected with BCL-XL or empty vector control CMV. Twenty four h after transfection, the cells were treated with AZD7762 (300 nM) + AZD6244 (500 nM) in the presence or absence of HA14–1 (HA14–1, 500 nM) for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value.
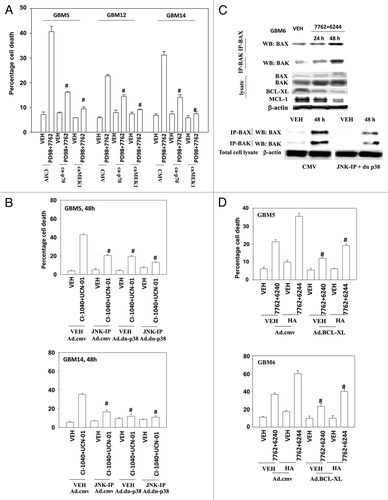
Prior studies in other malignancies have argued that MEK1/2 and CHK1 inhibitors kill cells by activation of the intrinsic apoptosis pathway.Citation27,Citation28 The activity of BAX and BAK, as judged by immunoprecipitation of the activated forms of each protein, increased 24–48h following drug treatment (, upper blots). Activation of both BAX and BAK was suppressed by inhibition of p38 MAPK and JNK1–3 signaling (, lower blots). Overexpression of BCL-XL suppressed drug combination lethality, however of note, we were unable to completely abolish killing using this approach as we have previously seen in breast cancer cells ().Citation16,Citation28 Treatment of cells with the BCL-2/BCL-XL inhibitor HA14–1 significantly enhanced drug combination toxicity and partially reversed the protective effects of BCL-XL overexpression ().Citation29,Citation30
In addition to MEK1/2 inhibitors, we have also observed in other tumor types that inhibitors of SRC family kinases interact with CHK1 inhibitors to kill tumor cells.Citation11,Citation12 SRC kinases in many cell systems are known to play an important role in RAS related ERK1/2 signaling / activation.Citation31 In the present study, we also found that SRC inhibitor, Saracatinib (AZD0530), enhanced CHK1 inhibitor lethality in GBM cells (-). These effects correlated with enhanced cleavage of PARP1, pro-caspase 3 and pro-caspase 7 ( and ). AZD0530 blocked CHK1 inhibitor-induced activation of ERK1/2 in GBM14 cells though had no effect on ERK1/2 signaling in DAOY medulloblastoma cells. Collectively, our findings argue that the combination of CHK1 and MEK1/2 inhibitors and CHK1 and SRC inhibitors could both represent promising therapeutic combinations to investigate in brain tumor patients.
Figure 5. A SRC kinase inhibitor interacts with CHK1 inhibitors to kill primary human glioma cell isolates and pediatric medulloblastoma cell lines. (A) GBM6 and GBM12 cells and (B) GBM5 and GBM14 cells were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion based cell staining (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (C) GBM5 and GBM14 cells were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 24 h and cell lysates were immunoblotted against cleaved caspase 3, PARP1 and P-ERK1/2. (D) Medulloblastoma cell lines DAOY, VC312 and D283 were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. The cell lysates were immunoblotted against cleaved caspases-3, cleaved caspases-7 and P-ERK1/2.
Discussion
Glioblastoma is a fatal malignancy for which there are few therapeutic options, and those treatments that do exist and have been validated only modestly enhance survival. There is a pressing need to develop new approaches that can be translated from the laboratory to the clinic.
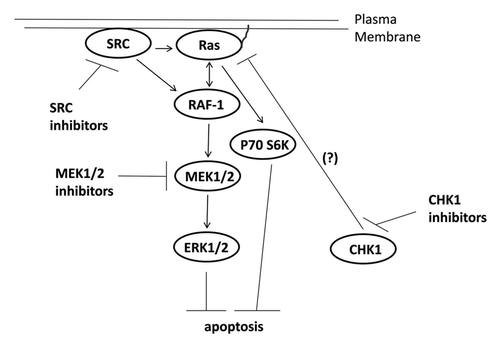
Signaling by the ERK1/2 pathway has been recognized for many years as a key player in tumor cell growth and in the resistance of tumor cells to chemo-/radio-therapy. Almost a decade ago, it was noted that treatment of mammary carcinoma and leukemia cells with low clinically relevant concentrations of UCN-01 caused a rapid and sustained activation of the ERK1/2 pathway, and inhibition of this ERK1/2 pathway activation resulted in cell death ().Citation15,Citation32 Treatment of tumor cells with the chemically unrelated CHK1 inhibitor AZD7762 or transient expression of dominant negative CHK1 also caused ERK1/2 activation.Citation17,Citation33 In developing Drosophila melanogaster pupae loss of CHK1 function has been shown to promote MEK1/2 activation, which provides independent genetic confirmation of these studies.Citation34
Figure 6. Possible signaling pathways by which CHK1 inhibitors activate ERK, and mechanisms by which MEK1/2 and SRC inhibitors potentiate CHK1 inhibitor lethality. CHK1 inhibitors through mechanisms not fully understood causes activation of the ERK1/2 pathway downstream of RAS. SRC kinases can increase ERK1/2 activity both by promoting RAS activation and also downstream of RAS at the level of RAF-1 tyrosine phosphorylation. Inhibition of either MEK1/2 disrupts this activation of ERK1/2 leading to tumor cell death.
Both CHK1 and CHK2 play critical roles in cell cycle arrest driven by cellular stresses and in controlling DNA repair, genomic stability, and apoptosis.Citation35-Citation37 Both kinases translate upstream signals, particularly those transduced from the ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3 (ATR) to downstream effectors such as checkpoint kinases.Citation38 CHK1 and CHK2 share several downstream substrates such as, p53, Mdm2, and Cdc25A/C for cell cycle regulation,Citation35 which may explain their redundant roles in the DNA damage response. However, mounting evidence suggests that CHK1 and CHK2 have respective specific substrates and differential cellular functions. For instance, silencing of CHK1 in the presence of endogenous CHK2 is sufficient to abolish S- and G2-checkpoints in response to double-strand DNA breaks.Citation39 Knockdown of CHK1 but not CHK2 increases sensitivity to toward gemcitabine and 5-fluoro-2’-deoxyuridine in pancreatic and colon cancer cells.Citation40 In contrast, CHK2 silencing failed to induce check point bypass and did not synergize with CHK1 knockdown to promote checkpoint bypass.Citation40-Citation42 Inhibition of CHK2 by VRX0466617, a selective CHK2 inhibitor, does not synergize with anticancer drugs doxorubicin, Taxol, and cisplatin.Citation43 These data strongly argue that CHK1 instead of CHK2 may be the most promising therapeutic target in tumor cells.
Our present findings demonstrated that multiple CHK1 inhibitors interact with multiple MEK1/2 inhibitors to kill a genetically diverse set of primary human glioblastoma isolates. In some of our CNS tumor cells, in contrast to breast cancer cell lines, it was still evident 24h after exposure that treatment of cells with a CHK1 inhibitor activated ERK1/2. In addition, drug treatment of GBM cells resulted in lower levels of ribosomal S6 protein phosphorylation, even in GBM cells that expressed mutant active PI3K or that lacked PTEN function. As loss of PTEN is common in GBM and is a negative indicator for a patient responding to chemotherapy or to radiotherapy, our data argues for the combination of MEK1/2 and CHK1 inhibitors being a useful treatment for many patients. Expression of activated forms of either MEK1 or of p70 S6K suppressed drug toxicity. Thus our data in GBM isolates argue, unlike findings in breast cancer cells, that loss of both ERK1/2 and S6 phosphorylation plays a key role in causing glioma cell death ().
We noted that following drug treatment the level of BCL-XL and MCL-1 declined and BAX and BAK became activated. All of these phenomena are associated with mitochondrial dysfunction. Overexpression of BCL-XL suppressed, though did not abolish, drug combination lethality that could be partially reversed using the BCL-2 / BCL-XL inhibitor HA14–1. Treatment with MEK1/2 + CHK1 inhibitors resulted in a significant increase in the phosphorylation of JNK1–3 and p38 MAPK, two other major MAPK pathways. Activation of the JNK1–3 and p38 MAPK pathways has most often been implicated in translating environmental and genotoxic stresses into signals for tumor cell death in response to a broad spectrum of chemotherapeutic agents.Citation44-Citation46 Suppression of p38 MAPK function or inhibition of JNK1–3 protected cells from drug combination toxicity, and that blocked BAX and BAK activation. Activation of BAX and BAK leads to pore formation in the outer mitochondrial membrane thereby permitting proteins such as cytochrome c to enter the cytoplasm and to trigger / activate the intrinsic apoptosis pathway.
Collectively, together with our prior in vitro and in vivo findings, the data in the present manuscript strongly argue for the clinical translation of the combination of MEK1/2 inhibitors together with CHK1 inhibitors as a cancer therapeutic in GBM tumors.
| Abbreviations: | ||
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| JNK | = | c-Jun NH2-terminal kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| MAPK | = | mitogen activated protein kinase |
| CHK | = | checkpoint kinase |
| ca | = | constitutively active |
| dn | = | dominant negative |
| GX | = | obatoclax |
| EGFR | = | epidermal growth factor receptor |
| CMV | = | empty vector plasmid or virus |
| si | = | small interfering |
| SCR | = | scrambled |
| IP | = | immunoprecipitation |
| Ad | = | adenovirus |
| TUNEL | = | Terminal deoxynucleotidyltransferased UTP nick end labeling |
| ATM | = | Ataxia telangiectasia mutated |
| ATR | = | Ataxia telangiectasia and Rad3-related |
| Chk1 and Chk2 | = | checkpoint kinases 1 and 2 |
| VEH | = | vehicle |
Acknowledgments
Studies in this manuscript were funded by: Department of Defense Idea award W81XWH-10–1-0009; R01 CA100866; R01 CA141703; R01 CA150214, and the Massey Cancer Center training grant T32 CA85159.
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs 2009; 18:1061 - 83; http://dx.doi.org/10.1517/13543780903052764; PMID: 19555299
- Kanu OO, Mehta A, Di C, Lin N, Bortoff K, Bigner DD, et al. Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets 2009; 13:701 - 18; http://dx.doi.org/10.1517/14728220902942348; PMID: 19409033
- Gömöri E, Halbauer JD, Kasza G, Varga D, Horvath Z, Komoly S. Glioblastoma multiforme with an unusual location and clinical course. Clin Neuropathol 2009; 28:165 - 7; PMID: 19537131
- Nieder C, Grosu AL, Molls M. A comparison of treatment results for recurrent malignant gliomas. Cancer Treat Rev 2000; 26:397 - 409; http://dx.doi.org/10.1053/ctrv.2000.0191; PMID: 11139371
- Eley KW, Benedict SH, Chung TD, Kavanagh BD, Broaddus WC, Schmidt-Ullrich RK, et al. The effects of pentoxifylline on the survival of human glioma cells with continuous and intermittent stereotactic radiosurgery irradiation. Int J Radiat Oncol Biol Phys 2002; 54:542 - 50; http://dx.doi.org/10.1016/S0360-3016(02)02983-8; PMID: 12243834
- Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011; 17:88 - 96; http://dx.doi.org/10.1016/j.molmed.2010.10.009; PMID: 21087899
- Ashwell S, Zabludoff S. DNA damage detection and repair pathways--recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res 2008; 14:4032 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-07-5138; PMID: 18593978
- Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv 2011; 11:133 - 40; http://dx.doi.org/10.1124/mi.11.2.11; PMID: 21540473
- Pei XY, Dai Y, Youssefian LE, Chen S, Bodie WW, Takabatake Y, et al. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood 2011; 118:5189 - 200; http://dx.doi.org/10.1182/blood-2011-02-339432; PMID: 21911831
- Mitchell C, Hamed HA, Cruickshanks N, Tang Y, Bareford MD, Hubbard N, et al. Simultaneous exposure of transformed cells to SRC family inhibitors and CHK1 inhibitors causes cell death. Cancer Biol Ther 2011; 12:215 - 28; http://dx.doi.org/10.4161/cbt.12.3.16218; PMID: 21642769
- Dai Y, Chen S, Shah R, Pei XY, Wang L, Almenara JA, et al. Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo. Blood 2011; 117:1947 - 57; http://dx.doi.org/10.1182/blood-2010-06-291146; PMID: 21148814
- Dai Y, Chen S, Pei XY, Almenara JA, Kramer LB, Venditti CA, et al. Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood 2008; 112:2439 - 49; http://dx.doi.org/10.1182/blood-2008-05-159392; PMID: 18614762
- Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther 2005; 4:457 - 70; PMID: 15767555
- Dai Y, Yu C, Singh V, Tang L, Wang Z, McInistry R, et al. Pharmacological inhibitors of the mitogen-activated protein kinase (MAPK) kinase/MAPK cascade interact synergistically with UCN-01 to induce mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res 2001; 61:5106 - 15; PMID: 11431348
- Dai Y, Landowski TH, Rosen ST, Dent P, Grant S. Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood 2002; 100:3333 - 43; http://dx.doi.org/10.1182/blood-2002-03-0940; PMID: 12384435
- Mitchell C, Park M, Eulitt P, Yang C, Yacoub A, Dent P. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in carcinoma cells. Mol Pharmacol 2010; 78:909 - 17; http://dx.doi.org/10.1124/mol.110.067199; PMID: 20696794
- Yacoub A, Mitchell C, Hong Y, Gopalkrishnan RV, Su ZZ, Gupta P, et al. MDA-7 regulates cell growth and radiosensitivity in vitro of primary (non-established) human glioma cells. Cancer Biol Ther 2004; 3:739 - 51; http://dx.doi.org/10.4161/cbt.3.8.968; PMID: 15197348
- Hamed HA, Yacoub A, Park MA, Eulitt PJ, Dash R, Sarkar D, et al. Inhibition of multiple protective signaling pathways and Ad.5/3 delivery enhances mda-7/IL-24 therapy of malignant glioma. Mol Ther 2010; 18:1130 - 42; http://dx.doi.org/10.1038/mt.2010.29; PMID: 20179672
- Yacoub A, Gupta P, Park MA, Rhamani M, Hamed H, Hanna D, et al. Regulation of GST-MDA-7 toxicity in human glioblastoma cells by ERBB1, ERK1/2, PI3K, and JNK1-3 pathway signaling. Mol Cancer Ther 2008; 7:314 - 29; http://dx.doi.org/10.1158/1535-7163.MCT-07-2150; PMID: 18281516
- Yacoub A, Mitchell C, Lebedeva IV, Sarkar D, Su ZZ, McKinstry R, et al. mda-7 (IL-24) Inhibits growth and enhances radiosensitivity of glioma cells in vitro via JNK signaling. Cancer Biol Ther 2003; 2:347 - 53; http://dx.doi.org/10.4161/cbt.2.4.422; PMID: 14508103
- Yang F, Van Meter TE, Buettner R, Hedvat M, Liang W, Kowolik CM, et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol Cancer Ther 2008; 7:3519 - 26; http://dx.doi.org/10.1158/1535-7163.MCT-08-0138; PMID: 19001435
- Yang F, Jove V, Xin H, Hedvat M, Van Meter TE, Yu H. Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Mol Cancer Res 2010; 8:35 - 45; http://dx.doi.org/10.1158/1541-7786.MCR-09-0220; PMID: 20053726
- Yacoub A, Park MA, Gupta P, Rahmani M, Zhang G, Hamed H, et al. Caspase-, cathepsin-, and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells. Mol Cancer Ther 2008; 7:297 - 313; http://dx.doi.org/10.1158/1535-7163.MCT-07-2166; PMID: 18281515
- Keime-Guibert F, Chinot O, Taillandier L, Cartalat-Carel S, Frenay M, Kantor G, et al, Association of French-Speaking Neuro-Oncologists. Radiotherapy for glioblastoma in the elderly. N Engl J Med 2007; 356:1527 - 35; http://dx.doi.org/10.1056/NEJMoa065901; PMID: 17429084
- Rockne R, Rockhill JK, Mrugala M, Spence AM, Kalet I, Hendrickson K, et al. Predicting the efficacy of radiotherapy in individual glioblastoma patients in vivo: a mathematical modeling approach. Phys Med Biol 2010; 55:3271 - 85; http://dx.doi.org/10.1088/0031-9155/55/12/001; PMID: 20484781
- Hamed H, Hawkins W, Mitchell C, Gilfor D, Zhang G, Pei XY, et al. Transient exposure of carcinoma cells to RAS/MEK inhibitors and UCN-01 causes cell death in vitro and in vivo. Mol Cancer Ther 2008; 7:616 - 29; http://dx.doi.org/10.1158/1535-7163.MCT-07-2376; PMID: 18347148
- Pei XY, Dai Y, Tenorio S, Lu J, Harada H, Dent P, et al. MEK1/2 inhibitors potentiate UCN-01 lethality in human multiple myeloma cells through a Bim-dependent mechanism. Blood 2007; 110:2092 - 101; http://dx.doi.org/10.1182/blood-2007-04-083204; PMID: 17540843
- Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P, et al. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther 2010; 10:903 - 17; http://dx.doi.org/10.4161/cbt.10.9.13273; PMID: 20855960
- Martin AP, Park MA, Mitchell C, Walker T, Rahmani M, Thorburn A, et al. BCL-2 family inhibitors enhance histone deacetylase inhibitor and sorafenib lethality via autophagy and overcome blockade of the extrinsic pathway to facilitate killing. Mol Pharmacol 2009; 76:327 - 41; http://dx.doi.org/10.1124/mol.109.056309; PMID: 19483105
- Guarino M. Src signaling in cancer invasion. J Cell Physiol 2010; 223:14 - 26; PMID: 20049846
- McKinstry R, Qiao L, Yacoub A, Dai Y, Decker R, Holt S, et al. Inhibitors of MEK1/2 interact with UCN-01 to induce apoptosis and reduce colony formation in mammary and prostate carcinoma cells. Cancer Biol Ther 2002; 1:243 - 53; PMID: 12432271
- Pei XY, Li W, Dai Y, Dent P, Grant S. Dissecting the roles of checkpoint kinase 1/CDC2 and mitogen-activated protein kinase kinase 1/2/extracellular signal-regulated kinase 1/2 in relation to 7-hydroxystaurosporine-induced apoptosis in human multiple myeloma cells. Mol Pharmacol 2006; 70:1965 - 73; http://dx.doi.org/10.1124/mol.106.028373; PMID: 16940414
- Mogila V, Xia F, Li WX. An intrinsic cell cycle checkpoint pathway mediated by MEK and ERK in Drosophila. Dev Cell 2006; 11:575 - 82; http://dx.doi.org/10.1016/j.devcel.2006.08.010; PMID: 17011495
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3:421 - 9; http://dx.doi.org/10.1016/S1535-6108(03)00110-7; PMID: 12781359
- Ghosh JC, Dohi T, Raskett CM, Kowalik TF, Altieri DC. Activated checkpoint kinase 2 provides a survival signal for tumor cells. Cancer Res 2006; 66:11576 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-3095; PMID: 17178848
- Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, et al. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res 2006; 66:1401 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-3278; PMID: 16452195
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177 - 96; http://dx.doi.org/10.1101/gad.914401; PMID: 11544175
- Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A 2002; 99:14795 - 800; http://dx.doi.org/10.1073/pnas.182557299; PMID: 12399544
- Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle 2006; 5:1983 - 8; http://dx.doi.org/10.4161/cc.5.17.3184; PMID: 16931916
- Carrassa L, Broggini M, Erba E, Damia G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: differential activity in cells expressing or not p53. Cell Cycle 2004; 3:1177 - 81; http://dx.doi.org/10.4161/cc.3.9.1080; PMID: 15326376
- Cho SH, Toouli CD, Fujii GH, Crain C, Parry D. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle 2005; 4:131 - 9; http://dx.doi.org/10.4161/cc.4.1.1299; PMID: 15539958
- Carlessi L, Buscemi G, Larson G, Hong Z, Wu JZ, Delia D. Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther 2007; 6:935 - 44; http://dx.doi.org/10.1158/1535-7163.MCT-06-0567; PMID: 17363488
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem 2000; 275:322 - 7; http://dx.doi.org/10.1074/jbc.275.1.322; PMID: 10617621
- Sampath D, Plunkett W. The role of c-Jun kinase in the apoptotic response to nucleoside analogue-induced DNA damage. Cancer Res 2000; 60:6408 - 15; PMID: 11103806
- Kharbanda S, Ren R, Pandey P, Shafman TD, Feller SM, Weichselbaum RR, et al. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995; 376:785 - 8; http://dx.doi.org/10.1038/376785a0; PMID: 7651539