Abstract
Renal cell carcinoma is resistant to chemotherapy and radiotherapy. STAT1 is overexpressed in human RCC tissue. Downregulation of STAT1 expression could significantly increase the radiosensitivity in RCC cell lines. To further investigate the function of STAT1 in RCC resistance to chemoradiotherapy, a stable STAT1 knockdown cell line was established. Knockdown of STAT1 led to significant growth suppression in vitro and in vivo. Inhibition of STAT1 sensitized 786-O cells to radiotherapy and Taxol treatment. Cells with low STAT1 expression accumulated more strongly in the G2/M phase after treatment with chemotherapy and radiotherapy. The Human Cell Cycle and DNA Damage Signaling Pathway Real-time PCR arrays were performed and 3 genes upregulated and 16 genes downregulated after STAT1 knockdown were selected. Functional gene grouping showed that genes involved in the M phase, S phase and DNA replication did not differ between the two cell lines. G1 phase related genes ANAPC2, CCNE1, CUL1 were downregulated, and G2/M checkpoint genes p21, GADD45A and Rb were strongly reduced by STAT1 knockdown. DNA damage-related genes GADD45A, MAP2K6, were significantly downregulated. The results prove that overexpression of STAT1 in human RCC is associated with the chemoradioresistance. Targeting of STAT1 might be a potential strategy to sensitize RCC to chemotherapy and radiotherapy.
Introduction
STATs (signal transducers and activators of transcription) are transcriptional factors and can be activated by various extracellular stimuli, including growth factors and cytokines.Citation1 When cytokines bind to an appropriate receptor, the signal cascade transduce to the cytoplasm and the Janus tyrosine kinase (Jak) family becomes activated, resulting in STAT phosphorylation. STATs can then dimerize and translocate to the nucleus. Once in the nucleus, the STATs bind to specific DNA response elements and promote transcription.Citation2,Citation3 Different STATs are activated by specific cytokines for different functions. STAT1, the first discovered member of the STATs family, is the key effector of interferon (IFN)γ ανδ ΙΦΝ‴α/β signaling.Citation4 STAT1 can form a homodimer or heterodimer with other family members. The target genes of STAT1 include caspase 3, 6, 8, Bcl-XL, FAS/FASL, p21waf1, c-myc and others.Citation1,Citation5-Citation7
As it is an important mediator of IFN signaling, the normal functions of STAT1 are mostly related to the biological effects of IFNs. STAT1 plays a critical role in cell death induced by various stimulating factors,Citation5 and is known to be a tumor suppressor gene. It has been found to be inappropriately activated or even suffer a loss of expression in many type of human cancers, such as breast cancer, melanoma, leukemia, and lymphoma.Citation8 In breast cancer cells, STAT1 activation in response to IFNγ, combined with the anti-cancer drug doxorubicin, has resulted in enhanced apoptosis.Citation9
Recent research has found inconsistent data in some types of cancers. In cervical cancer, expression of STAT1 gene rose in CIN1/2, fell in CIN3/CIS, and then significantly rose in invasive cancers.Citation10,Citation11 In a study of highly invasive and weakly invasive cell clones from a melanoma cell line, STAT1 was found to be strongly upregulated in the highly invasive cell clone.Citation12 Our previous study confirmed the overexpression of STAT1 in both renal cancer cell lines and renal cancer tissues. High STAT1 expression and activation were associated with a greater Fuhrman tumor grade or the presence of sarcomatoid features in RCC.Citation13 STAT1 was also associated with tumor radioresistance and chemoresistance. In research into paired chemosensitive and chemoresistant cell lines, STAT1 was found to be closely correlated with chemoresistance.Citation14 STAT1 overexpression was associated with decreased sensitivity to cisplatin in human ovarian cancer cell lines.Citation15 In radioresistant cancer cells, STAT1 was significantly overexpressed. Upregulation of STAT1 expression in radiosensitive cells confers protection from radiation.Citation16,Citation17
RCC is traditionally considered to be resistant to chemotherapy and radiotherapy.Citation18 Patients with metastatic renal cancer have a median survival time of < 12 mo. Recent advances in immunotherapy, targeted therapy with small molecule kinase inhibitors, vascular endothelial antibodies, and combination treatment modalities have shown promising trends.Citation19 We found overexpression of STAT1 in human clear cell and papillary RCC tissues. Downregulation of STAT1 expression by both fludarabine and siRNA significantly increased the radiosensitivity of RCC cell lines.Citation13 Howerver, the exact mechanism of STAT1 in renal cancer is still not clear. In this study, we knocked down the expression of STAT1 in human RCC clear cell line 786-O, and explored the phenotype and molecular alteration on STAT1 knockdown. We studied the effect of STAT1 on chemosensitivity and radiosensitivity, and the possible molecular mechanism through which STAT1 affects the cell’s response.
Results
STAT1 knockdown in 786-O cells inhibited cell growth in vitro and in vivo
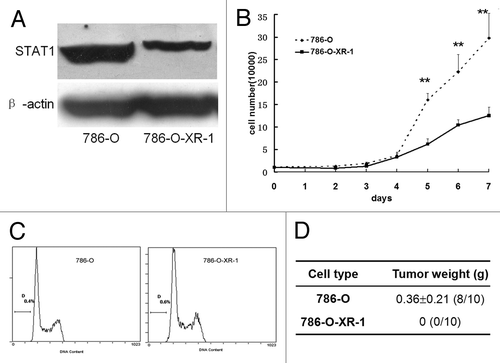
We knocked down the STAT1 expression in 786-O cells by RNAi technology. The expression plasmid was transfected into 786-O cells and selected with 5 μg/ml blasticidin S HCl for 2 weeks. The efficiency of STAT1 reduction was confirmed by western blot. The stable cell line expressing a reduced STAT-1 level was selected and designated as 786-O-XR-1. STAT1 expression was reduced by about 55% compared with the parental cells (). The 786-O-XR-1 stable cell lines were maintained in 5 μg/ml blasticidin S HCl. Growth curves were then constructed by cell counting. The growth rate of 786-O-XR-1 cells was slower than that of 786-O cells (). Cell cycle distribution was also examined by propidium iodide staining and analyzed by FACS. The percentage of cells in each phase had no significant difference between 786-O-XR-1 and 786-O (). Then we subcutaneously injected an equal number of cells into nude mice. Tumor formation was seen in 80% (8/10) mice injected with 786-O but not in the mice injected with 786-O-XR-1 (). The results indicated that reduced STAT1 expression slowed the growth rate of 786-O cells and completely inhibited tumor formation in nude mice.
Figure 1. Knockdown of STAT1 in renal 786-O cells inhibited cell growth in vitro and in vivo. (A) Expression of STAT1 in 786-O and 786-O-XR-1 cells. After transfection with pcDNA6.2-GW/EmGFPmiR, 786-O-XR-1 cells were selected by 5 μg/ml blasticidin S HCl pressure. Total cell proteins were extracted and subjected to SDS-PAGE and immunoblotted using antibody against STAT1. STAT1 protein in 786-O-XR-1 cells was about 45% of that of control cells. (B) Growth curve of 786-O and 786-O-XR-1 cells. Cells (1 × 104) were seeded onto a 24-well plate and counted daily. (C) Cell cycle distribution of 786-O and 786-O-XR-1 cells. (D) Tumor formation in nude mice. Five nude mice were injected subcutaneously with 786-O and 786-O-XR-1 cells and were sacrificed 2 mo after injection. Similar results were observed in two independent experiments. The weights of the tumors were recorded and expressed as means ± SD.
Response of 786-O cells to chemotherapy and radiotherapy after STAT1 downregulation
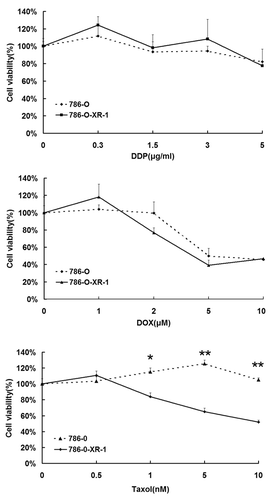
RCC is resistant to both chemotherapy and radiotherapy. We have previously demonstrated that 786-O cells can be radiosensitized by inhibition of STAT1 signaling by fludarabine and STAT1 siRNA.Citation13 Here, we examined the response to chemotherapy after STAT1 inhibition. 786-O-XR-1 and 786-O cells were treated with Taxol (Bristol-Myers Squibb Caribbean Co., USA), cisplatin (Qilu Pharma, China) or doxorubicin hydrochloride (Wanle Pharma, China), at different concentrations for 72 h. Cell viability was measured by MTT assay. With cisplatin and doxorubicin hydrochloride, 786-O-XR-1 and 786-O cells showed no differences in cell viability, but 786-O-XR-1 was more sensitive to Taxol than 786-O cells (). The results indicated that STAT1 downregulation could sensitize cells only to Taxol treatment, but had little effect on cisplatin and doxorubicin hydrochloride treatment.
Figure 2. Response of 786-O and 786-O-XR-1 cells to anti-cancer drugs. Cells were treated with anti-cancer drugs for 72 h. Cell viability was measured by MTT assay. (A) Cisplatin (PPD). (B) Doxorubicin hydrochloride (DOX). C. Taxol. All experiments were performed in triplicate and expressed as means ± SD.
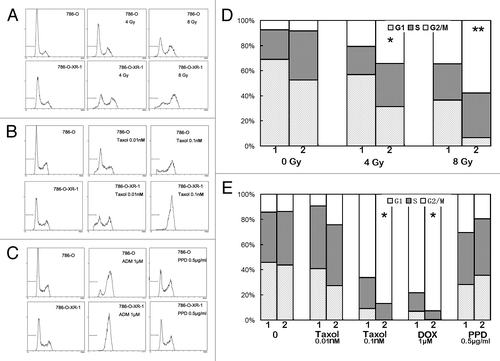
We then analyzed the response of the cell cycle to chemotherapy and radiotherapy using propidium iodide staining and FACS. As previously reported, cells were blocked in the G2/M phase and cell death occurred after X-ray radiation.Citation20 After STAT1 knockdown, upon X-ray radiation, a greater accumulation of 786-O-XR-1 cells was found in the G2/M phase than was found in the control cells ( and ). Similar results were also seen in Taxol and doxorubicin hydrochloride treated 786-O-XR-1 cells (, and ). But in cells treated with cisplatin, the cell cycle distribution of 786-O-XR-1 and 786-O cells had no significant difference ( and ). Thus, downregulation of STAT1 in renal cancer cells pushed accumulation of more cells in the G2/M phase after radiotherapy and chemotherapy.
Figure 3. Analysis of cell cycle progression of 786-O and 786-O-XR-1 cells treated with chemotherapy or radiotherapy. (A) Cells treated with X-ray radiation and cultured for 48 h. (B) Cells treated with Taxol for 24 h. (C) Cells treated with cisplatin (PPD) or doxorubicin hydrochloride (DOX) for 24 h. (D) Cell cycle distribution of 786-O and 786-O-XR-1 cells treated with X-ray radiation. E. Cell cycle distribution of 786-O and 786-O-XR-1 cells treated with anti-cancer drugs. Cells were harvested and stained by propidium iodide and analyzed by a flow cytometer. Data represent the results of three independent experiments.
Gene expression profile assay of 786-O and 786-O-XR
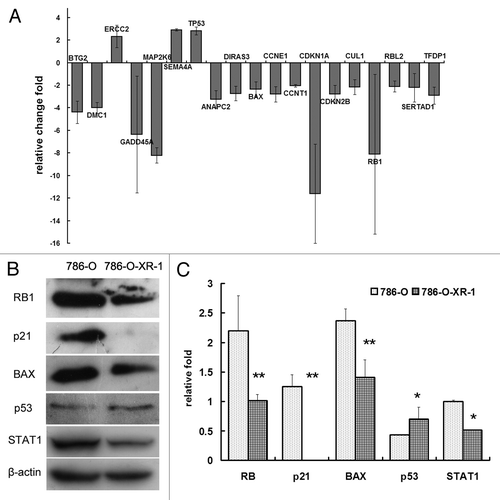
To investigate the molecular mechanism of STAT1 in radioresistance and chemoresistance, we performed a PCR array focusing on the cell cycle and DNA damage to study the different gene expression profile of STAT1 downregulation. Using filtering criteria of a 2-fold or greater fold-change in expression, 3 genes upregulated and 16 genes downregulated upon STAT1 knockdown were selected (). Alterations in the level of protein expression were detected by Western Blotting ( and ). Functional gene grouping showed that genes involved in the M phase, S phase and DNA replication did not differ between the two cell lines. G1 phase related genes ANAPC2, CCNE1, CUL1 were downregulated, and G2/M checkpoint genes p21, GADD45A and Rb were strongly reduced by STAT1 knockdown. DNA damage-related genes GADD45A, MAP2K6, were significantly downregulated. The gene p21(CDKN1A) is a well-known target gene of STAT1, and STAT1 activation is associated with upregulation of p21.Citation7 In our system, p21 RNA was downregulated more than 10-fold, and the protein expression level was also significantly lower in 786-O-XR-1 cells than in control 786-O cells. By PCR array, we screened the DNA damage and cell cycle related genes affected by STAT1. These genes may contribute to the radioresistance and chemoresistance of renal cancer cells.
Figure 4. Alteration of gene expression by STAT1 knockdown. (A) Quantitative comparison of transcript levels in786-O-XR-1 and 786-O cells using the RTCitation2 Profiler™ PCR Array (SABiosciences). (B) western blot analysis showed that the levels of protein expression altered in some of the genes. (C) Protein expression normalized to β-actin.
Discussion
Many patients with RCC are already in a very advanced stage of the disease at diagnosis, and need to be treated with chemotherapy and radiotherapy. But RCC is resistant to this treatment. Patients with metastatic RCC have a median survival time of < 12 mo,Citation19 and so it is imperative to understand the molecular mechanism of resistance and determine which genes prevent or interfere with resistance. STAT1 has been regarded as a key modulator of cell death.Citation5 The exact role of STAT1 in different types of cancer tissue is still controversial. STAT1 has been regarded as a tumor suppressor gene in some cancers because of its inappropriate activation and even loss of expression in malignant cells.Citation8 Our previous study found STAT1 overexpression in human renal carcinoma tissues and RCC cell lines. Inhibition of STAT1 signaling by fludarabine or siRNA might sensitize RCC cell lines to radiotherapyCitation13; Similar results have been obtained by other groups.Citation16,Citation21 To further investigate the function of STAT1 in renal cancer cells, we constructed a renal cell line with STAT1 knockdown. After knockdown of STAT1, the renal cancer cells grew more slowly and lost their potential to form tumors in nude mice. STAT1 may have an important role in cell growth and tumor formation in nude mice. Pitroda et al. knocked down STAT1 in humans in the SCC61 human squamous cell carcinoma cell line, which led to a significant suppression of tumor volume relative to STAT1 wild-type tumors.Citation22
Since STAT1 was associated with radioresistance in renal cancer cells, we then used the model to examine whether STAT1 was involved in the chemoresistance of renal cancer. We treated the 786-O-XR-1 cells with the commonly used anti-cancer drugs: Taxol, cisplatin and doxorubicin hydrochloride. Unexpectedly, STAT1 knockdown sensitized 786-O only to Taxol treatment, but had no significant effect on cisplatin and doxorubicin hydrochloride.. We then analyzed the cell cycle distribution of cells in a number of ways. Although by completely different mechanisms, X-ray radiation and Taxol treatment both blocked cells in the G2/M phase, then progressed to cell death. STAT1 could facilitate cells entering the G2/M phase and postpone or prevent cells from leaving. Our data indicated that STAT1 might participate in chemoresistance/radioresistance through its function in the cell cycle progression. For cisplatin and doxorubicin hydrochloride, they mainly inhibited the biosynthesis of DNA, RNA and protein.
To test our hypothesis, we performed a PCR array focusing on cell cycle and DNA damage to study the expression profile after STAT1 downregulation. Using filtering criteria of a 2-fold or greater fold-change in expression, six genes upregulated, and 20 genes downregulated, upon STAT1 knockdown were selected. By functional gene grouping, we found that there were no differences in the two cell lines for genes involved in the M phase, S phase and DNA replication. G1-phase related genes ANAPC2, CCNE1, CUL1 were downregulated in 786-O-XR-1 cells. APC2 (ANAPC2) and CUL1 form part of the anaphase-promoting complex (APC), which is responsible for the metaphase to anaphase transition and the exit from mitosis.Citation23 So, STAT1 knockdown might prevent cells from leaving mitosis by inhibiting the function of the anaphase-promoting complex. On the other hand, G2/M checkpoint genes p21, GADD45A and Rb were strongly reduced by STAT1 knockdown. These genes mediate sustained G2/M arrest and delay cell cycle progression in order to allow additional time for repair of DNA damage.Citation24,Citation25 Based on this principle, STAT1 knockdown would promote the passage of cells through the G2/M checkpoint quickly and thus cells would have insufficient time to complete DNA repair. So, cells accumulating in the G2/M phase were not blocked at the G2/M checkpoint. They passed the checkpoint quickly and were blocked in the transition from metaphase to anaphase, and then died. Townsend et al. reported that STAT-1-deficient cells had defects both in the intra-S-phase and G2/M checkpoints in response to DNA damage owing to defective Chk2-T68, NBS1-S343 and ATM-S1981 phosphorylation.Citation26 The inconsistency could be explained by the different cell types, but the most important difference was still STAT1 itself. The cell model used by Townsend et al. was STAT-1−/− mouse embryonic fibroblasts,Citation27 which were completely STAT1 defective. In our experiment STAT1 expression in human RCC cells was just knocked down, but STAT1 protein was still detectable.
Of the genes associated with DNA damage, GADD45A and MAP2K6 were significantly downregulated in 786-O-XR-1 cells. MAP2K6 (MAPKK6, MEK6) functions as a mitogen-activated protein (MAP) kinase kinase, and phosphorylates and activates p38 MAP kinase.Citation28,Citation29 p38 was reported to have an essential function in DNA repair.Citation30 So the reduction of MAP2K6 would restrict its activation of the target protein p38 MAP kinase, and consequently disturb the DNA repair.
STAT1 is a transcriptional factor activating many target genes, such as p21, Bcl-XL, caspase,Citation5 and suppressing skp2.Citation31 The STAT1 pathway participates in many physiological processes, including cell growth, proliferation, apoptosis, and functions of the immune system. STAT1 is regarded as a tumor suppressor.Citation8 But its role in chemoresistance and radioresistance seemed to be dependent not only on its transcription activity. Pitroda et al. explored the STAT1-dependent expression of energy metabolic pathways and its relation with radioresistance.Citation22 They found, as in our study, that most resistance-related genes were not the typical target genes of STAT1. The molecular mechanism of STAT1 regulation of genes needs further investigation.
Our results proved that overexpression of STAT1 in human RCC was associated with chemoradioresistance. Targeting of STAT1 might be a potential strategy to sensitize RCC to chemotherapy and radiotherapy.
Materials and Methods
Cell culture and construction of STAT1 knockdown cell line
The human RCC clear cell line (CRL-1932 [786-O]) was obtained from the American Type Tissue Collection and cultured in RPMI-1640 supplemented with 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. STAT1 knockdown was achieved by RNA interference. The BLOCK-iT™ Pol II miR RNAi Technology was provided by Invitrogen Service, China. Briefly, oligo F: TGCTGAATACAGGCGCTCTGCTGTCTGTTTTGGCCACTGACTGACAGACAGCAGCGCCCTGTATT; Oligo R: CCTCAATACAGGCGCTGCTGTCTGTCAGTCAGTGGCCAAAACAGACAGCAGAGCGCCTGTATTC were cloned into pcDNA6.2-GW/EmGFPmiR using a BLOCK-iT™ Pol II miR RNAi expression vector kit with EmGFP (Invitrogen, Cat: K4936–00). The expression plasmid was transfected into 786-O cells and selected with 5 μg/ml blasticidin S HCl (Invitrogen, Cat: 210–01) for 2 weeks. The stable cell line expressing a reduced STAT-1 level was designated as 786-O-XR-1.
Viability assay
To determine the effect of STAT1 on chemotherapy, cells were treated with different types of anti-cancer drugs for 72 h, then the medium was aspirated, and 20 μL of MTT (5 mg/mL) was added to the cells. After 4 h of incubation (37°C, 5% CO2), the media were aspirated and 150 μL of dimethyl sulfoxide was added. The plates were placed on a shaking table at room temperature for 10 min. The cells were then measured by a microplate reader (Bio-Rad, Hercules, CA, USA) at a wavelength of 570 nm.
Cell cycle assay
Cells were harvested 24 or 48 h after treatment, washed in cold sterile phosphate-buffered saline and fixed with 70% ethanol for 1 h. Cells were stained with propidium iodide then underwent flow cytometric analysis (Becton-Dickinson, Franklin Lakes, NJ, USA).
PCR array assay
The expression profiles of 786-O and 786-O-XR-1 were analyzed using an RTCitation2 Profiler™ PCR array human cell cycle (PAHS-020A) and HUMAN DNA damage signaling pathway (PAHS-029A) (SABiosciences, CA, USA) by KangChen Bio-tech Inc., Shanghai, China. Total RNA was isolated from the 786-O and 786-O-XR-1 cells by using the TRIZOL reagent (Invitrogen, cat:15596026) and RNeasy MinElute Cleanup kit (Qiagen, cat:74204). The concentration and purity of RNA were determined by the NanoDrop ND-1000. cDNA was synthesized from 1.5 μg of RNA using a SuperScript III reverse transcriptase(Invitrogen, cat: 18080044). PCR was performed with the RTCitation2 Profiler™ PCR array system according to the manufacturer’s instructions and the data shown represent the average of two replicates. The expression levels of the mRNA were normalized using the expression of GAPDH and ACTB. The p values were calculated based on a Student’s t-test of the replicate 2^(- Delta Ct) values for each gene in the control group and treatment groups, and p values less than 0.05 were considered statistically significant. The results were confirmed by RT-PCR or Western Blot.
Western Blot
Cells were harvested and lysed in RIPA buffer, and equal amounts of protein were separated by SDS-PAGE and transferred to an EC membrane. Membranes were incubated with primary antibody at 4°C overnight, washed with 0.05% Tween 20 in Tris-buffered saline) and incubated with horseradish peroxidase conjugated secondary antibody (Zhongshan, Beijing, China) for 1 h at room temperature and developed with the Luminal Detection System (Santa Cruz, CA, USA). Antibodies against STAT-1 (cat# 9175, Cell Signaling) were purchased from Cell Signaling, Inc. Anti-β-actin (Sigma, cat:A5441) antibody was obtained from Sigma.
Tumor growth in nude mice
Equal numbers (2 × 106) of 786-O and 786-O-XR-1 cells were harvested and subcutaneously injected into the Balb/C nude mice. The mice were kept in pathogen-free environments. After 2 mo, mice were sacrificed and the weights of tumors were measured at the end of the experiment. This experiment was repeated twice.
Statistical analysis
Data were expressed as the means of three different experiments ± SD. The results were analyzed by Student’s t-test and p < 0.05 was considered statistically significant.
| Abbreviations: | ||
| IFN | = | interferon |
| RCC | = | renal cell carcinoma |
| STAT1 | = | signal transducer and activator of transcription 1 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Natural Science Foundation (30800280, 81021061), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, P.R. China.
References
- Darnell JE Jr.. STATs and gene regulation. Science 1997; 277:1630 - 5; http://dx.doi.org/10.1126/science.277.5332.1630; PMID: 9287210
- Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651 - 62; http://dx.doi.org/10.1038/nrm909; PMID: 12209125
- Brierley MM, Fish EN. Stats: multifaceted regulators of transcription. J Interferon Cytokine Res 2005; 25:733 - 44; http://dx.doi.org/10.1089/jir.2005.25.733; PMID: 16375601
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996; 84:431 - 42; http://dx.doi.org/10.1016/S0092-8674(00)81288-X; PMID: 8608597
- Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal 2007; 19:454 - 65; http://dx.doi.org/10.1016/j.cellsig.2006.09.003; PMID: 17085014
- Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, et al. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J 2000; 19:263 - 72; http://dx.doi.org/10.1093/emboj/19.2.263; PMID: 10637230
- Chin YE, Kitagawa M, Su WC, You ZH, Iwamoto Y, Fu XY. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996; 272:719 - 22; http://dx.doi.org/10.1126/science.272.5262.719; PMID: 8614832
- Adámková L, Soucková K, Kovarík J. Transcription protein STAT1: biology and relation to cancer. Folia Biol (Praha) 2007; 53:1 - 6; PMID: 17328836
- Thomas M, Finnegan CE, Rogers KM, Purcell JW, Trimble A, Johnston PG, et al. STAT1: a modulator of chemotherapy-induced apoptosis. Cancer Res 2004; 64:8357 - 64; http://dx.doi.org/10.1158/0008-5472.CAN-04-1864; PMID: 15548705
- Hudelist G, Czerwenka K, Singer C, Pischinger K, Kubista E, Manavi M. cDNA array analysis of cytobrush-collected normal and malignant cervical epithelial cells: a feasibility study. Cancer Genet Cytogenet 2005; 158:35 - 42; http://dx.doi.org/10.1016/j.cancergencyto.2004.08.007; PMID: 15771902
- Rajkumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, Gopal G, et al. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011; 11:80; http://dx.doi.org/10.1186/1471-2407-11-80; PMID: 21338529
- Gütgemann A, Golob M, Müller S, Buettner R, Bosserhoff AK. Isolation of invasion-associated cDNAs in melanoma. Arch Dermatol Res 2001; 293:283 - 90; http://dx.doi.org/10.1007/s004030100232; PMID: 11480587
- Hui Z, Tretiakova M, Zhang Z, Li Y, Wang X, Zhu JX, et al. Radiosensitization by inhibiting STAT1 in renal cell carcinoma. Int J Radiat Oncol Biol Phys 2009; 73:288 - 95; http://dx.doi.org/10.1016/j.ijrobp.2008.08.043; PMID: 19100922
- Rickardson L, Fryknäs M, Dhar S, Lövborg H, Gullbo J, Rydåker M, et al. Identification of molecular mechanisms for cellular drug resistance by combining drug activity and gene expression profiles. Br J Cancer 2005; 93:483 - 92; http://dx.doi.org/10.1038/sj.bjc.6602699; PMID: 16012520
- Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, et al. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer 2005; 92:1149 - 58; http://dx.doi.org/10.1038/sj.bjc.6602447; PMID: 15726096
- Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A 2004; 101:1714 - 9; http://dx.doi.org/10.1073/pnas.0308102100; PMID: 14755057
- Stronach EA, Alfraidi AM, Rama NR, Datler C, Studd JB, Agarwal R, et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res 2011; 71:4412 - 22; http://dx.doi.org/10.1158/0008-5472.CAN-10-4111; PMID: 21571862
- Yagoda A, Abi-Rached B, Petrylak D. Chemotherapy for advanced renal-cell carcinoma: 1983-1993. Semin Oncol 1995; 22:42 - 60; PMID: 7855619
- Ather MH, Masood N, Siddiqui T. Current management of advanced and metastatic renal cell carcinoma. Urol J 2010; 7:1 - 9; PMID: 20209445
- Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol 1994; 31:1 - 13; http://dx.doi.org/10.1016/0167-8140(94)90408-1; PMID: 8041894
- Khodarev NN, Minn AJ, Efimova EV, Darga TE, Labay E, Beckett M, et al. Signal transducer and activator of transcription 1 regulates both cytotoxic and prosurvival functions in tumor cells. Cancer Res 2007; 67:9214 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-07-1019; PMID: 17909027
- Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, et al. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med 2009; 7:68; http://dx.doi.org/10.1186/1741-7015-7-68; PMID: 19891767
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 2006; 6:369 - 81; http://dx.doi.org/10.1038/nrc1881; PMID: 16633365
- Maeda T, Hanna AN, Sim AB, Chua PP, Chong MT, Tron VA. GADD45 regulates G2/M arrest, DNA repair, and cell death in keratinocytes following ultraviolet exposure. J Invest Dermatol 2002; 119:22 - 6; http://dx.doi.org/10.1046/j.1523-1747.2002.01781.x; PMID: 12164919
- Chan TA, Hwang PM, Hermeking H, Kinzler KW, Vogelstein B. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev 2000; 14:1584 - 8; PMID: 10887152
- Townsend PA, Cragg MS, Davidson SM, McCormick J, Barry S, Lawrence KM, et al. STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage. J Cell Sci 2005; 118:1629 - 39; http://dx.doi.org/10.1242/jcs.01728; PMID: 15784679
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996; 84:443 - 50; http://dx.doi.org/10.1016/S0092-8674(00)81289-1; PMID: 8608598
- Stein B, Brady H, Yang MX, Young DB, Barbosa MS. Cloning and characterization of MEK6, a novel member of the mitogen-activated protein kinase kinase cascade. J Biol Chem 1996; 271:11427 - 33; http://dx.doi.org/10.1074/jbc.271.19.11427; PMID: 8626699
- McMullen ME, Bryant PW, Glembotski CC, Vincent PA, Pumiglia KM. Activation of p38 has opposing effects on the proliferation and migration of endothelial cells. J Biol Chem 2005; 280:20995 - 1003; http://dx.doi.org/10.1074/jbc.M407060200; PMID: 15790570
- Phong MS, Van Horn RD, Li S, Tucker-Kellogg G, Surana U, Ye XS. p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells. Mol Cell Biol 2010; 30:3816 - 26; http://dx.doi.org/10.1128/MCB.00949-09; PMID: 20516219
- Wang S, Raven JF, Koromilas AE. STAT1 represses Skp2 gene transcription to promote p27Kip1 stabilization in Ras-transformed cells. Mol Cancer Res 2010; 8:798 - 805; http://dx.doi.org/10.1158/1541-7786.MCR-10-0027; PMID: 20407011