Abstract
We examined the effect of gefitinib (ZD1839), a selective epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor, on cytotoxicity to cisplatin, EGFR downstream signaling, apoptosis and the association between the inhibition of DNA repair by gefitinib and the expression of DNA-dependent protein kinase (DNA-PK) using three ovarian cancer cell lines. In the presence of gefitinib, cisplatin-induced growth inhibition and apoptosis were significantly enhanced in Caov-3 and RMG-1 cells, which express EGFR, and in A2780, which lacks EGFR but expresses HER-2. Gefitinib significantly inhibited the cisplatin-induced ERK and Akt activation in Caov-3 and RMG-1 cells but not in A2780 cells. In all three cell lines, there was delayed repair of DNA intrastrand cross-links damaged by cisplatin used in combination with gefitinib, compared with cisplatin alone. The reduction in DNA-PK levels persisted when cells were exposed to combinations of cisplatin and gefitinib in all cell lines. Moreover, the delayed repair was cancelled by anti-HER2 small-interfering RNA transfection in A2780 cells. These results suggest that combination therapy with cisplatin and gefitinib may increase the therapeutic efficacy of cisplatin by blocking EGFR downstream signaling and/or inhibiting DNA repair in ovarian cancer.
Introduction
The epidermal growth factor receptor (EGFR) is involved in many cellular processes, including cell proliferation and invasion, via the activation of the extracellular signal regulated protein kinase (ERK) cascade and the phosphatidylinositol 3-kinase (PI3K)-Akt cascade, both of which are the two main EGFR-signaling cascades.Citation1,Citation2 EGFR is expressed in a variety of human cancers, including breast, ovary, non-small cell lung, bladder, prostate, and head and neck.Citation3 In addition, many studies have indicated that overexpression of EGFR correlates with the development and progression of several human cancers and with poor prognosis.Citation4 Thus, EGFR is a promising target for cancer therapy.
Gefitinib is an EGFR-tyrosine kinase inhibitor that competitively inhibits binding of ATP at the ATP site on EGFR. In the non-small cell lung cancer, phase III trials combining gefitinib with a variety of agents were negative, and there was no association between EGFR expression and the effect of gefitinib.Citation5,Citation6 However, the combination of cetuximab and irinotecan can resensitize advanced colon cancer refractory to irinotecan.Citation7 EGFR is reported to be present in 33% to 75% of ovarian cancers,Citation8,Citation9 and ovarian cancers that express increased concentrations of EGF receptors are associated with a poor survival.Citation10 Evidence for both autocrine and paracrine regulation of growth by TGF (transforming growth factor) α/EGFR activation has been reported.Citation11 Although gefitinib had limited clinical benefit and responses in a Phase II trial with gefitinib monotherapy in ovarian cancer patients,Citation12 a Phase II trial with gefitinib in combination with paclitaxel and carboplatin as a second-line treatment for advanced ovarian adenocarcinoma, in which the combination therapy revealed a high rate of overall response (63%).Citation13 Therefore, it may be possible to improve the prognosis of ovarian cancer by a combination of EGFR inhibitors with chemotherapeutic agents.
Although the majority of patients with ovarian cancers respond to initial chemotherapy (the combination of paclitaxel with either cisplatin or carboplatin), most eventually relapse, and improved therapeutic strategies are needed for the recurrent disease. The sensitivity of cells to chemotherapeutic drug-induced apoptosis appears to depend on the balance between proapoptotic and antiapoptotic signals. We found that both the ERK and Akt cascades are involved in the resistance to cisplatinCitation14,Citation15 and paclitaxel,Citation16 indicating that these cascades are promising new targets for the development of chemotherapeutic drugs. Because the ERK and Akt cascades cross-talk at BAD (Bcl-2 associated death protein), inhibition of BAD using gene transfection is a more effective method than inhibition of either of these cascades for blocking resistance to cisplatinCitation15 and paclitaxel.Citation16 However, the small molecular inhibitor that blocks both cascades has not been identified. It was reported that gefitinib inhibited EGF-induced activation of both ERK and AktCitation17 in human non-small cell lung cancer cells. In addition, gefitinib is reported to decrease the growth and invasion of ovarian clear cell adenocarcinoma cells, which are commonly resistant to chemotherapy.Citation18
The anti-tumor activity of cisplatin is attributed to the formation of a variety of DNA adducts, including monoadducts, and intrastrand and interstrand cross-links.Citation19 Cisplatin enhances the expression of a serine/threonine kinase, DNA-dependent protein kinase (DNA-PK), which is associated with resistance to cisplatin in various ovarian cancer cell lines.Citation20 Therefore, DNA-PK is potentially a key enzyme in determining the response to cisplatin through the capacity to repair the damaged DNA. The repair of DNA interstrand cross-links was delayed when cisplatin and gefitinib were combined in a human breast cancer cell line, and there is an association between EGFR and DNA-PK, which was increased following gefitinib treatment and was determined by the quantity of EGFR expression.Citation21,Citation22
These considerations led us to examine the molecular mechanism by which gefitinib enhances cisplatin-induced cytotoxicity and apoptosis using a cisplatin-resistant mucinous cystadenocarcinoma cell line (Caov-3) and a clear cell adenocarcinoma cell line (RMG-1), both of which express EGFR; in addition, we used a cisplatin-sensitive human ovarian cancer cell line (A2780) that does not express EGFR.
In the present study, we show that gefitinib enhanced cisplatin-induced cytotoxicity and apoptosis via both the ERK and Akt cascades in Caov-3 and RMG-I cells and that gefitinib delayed the repair of DNA damage induced by cisplatin in all cell lines.
Results
Gefitinib increases cisplatin-induced cytotoxicity and inhibition of intraabdominal dissemination of ovarian cancer
Expression of EGFR, HER2, and HER3 was examined by western blotting in Caov-3, RMG-1, and A2780 cells. SKOV-3 cells which are reported to express all of the receptorsCitation23 were used as a positive control. Caov-3 cells express EGFR and HER3, RMG-1 cells express all of these receptors, and A2780 cells lack EGFR and HER3 but express HER2 ().
Figure 1. Expression of EGFR, HER2, and HER3, and enhancement of cisplatin-induced inhibition of cell proliferation by gefitinib. (A) western blot analysis was performed using anti-EGFR, anti-HER2 and anti-HER3 antibodies. SKOV-3, reportedCitation24 to express all erbB family receptors, was used as a positive control. (B) Cells were seeded in Dulbecco’s modified Eagle’s medium, followed by treatment with the indicated concentrations of cisplatin (CDDP) with or without 5 μM gefitinib (Gefitinib) for 72 h, and tested by the MTS assay (8 wells in each group). Cytotoxicity was expressed as a ratio of the absorbance of cells treated with various concentrations of cisplatin with or without gefitinib to that cultured without these reagents (control group) (mean ± S.E; n = 8). *, p < 0.01 vs cells treated with cisplatin alone. Experiments were repeated at least three times with consistent results, and a representative result is shown. (C) Athymic nude mice inoculated (ip) with Caov-3 cells were randomly placed into four groups that described in “Materials and Methods.” At autopsy, pathological examination was performed to determine the extent of intraabdominal dissemination. The arrow indicates the the intraabdominal dissemination. (D) The disseminated tumor was removed and its weight was measured. Values shown represent the mean ± SE. Different letters above the bar indicate significant difference at p < 0.01.
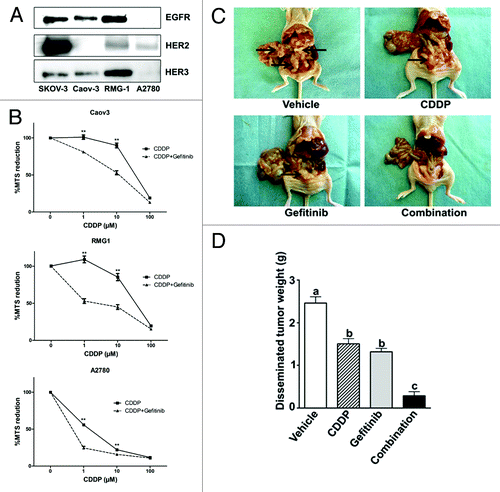
We next investigated the effect of the combination of cisplatin and gefitinib on cytotoxicity in all cell lines using the MTS assay. Cells were treated with cisplatin alone or in combination with gefitinib for 72 h. Treatment with gefitinib and cisplatin enhanced growth inhibition compared with each reagent alone in the three cell lines. (). Moreover, we examined whether combination therapy would increase the therapeutic efficacy of each agent in vivo. The appearance of the mice is shown in . Intraabdominal dissemination was clearly detected in athymic nude mice treated with vehicle PBS. Cisplatin alone or gefitinib alone apparently diminished the extent of intraabdominal dissemination. The combination with cisplatin and gefitinib further enhanced the inhibitory effect on intraabdominal dissemination. The tumors that had disseminated within the abdomen were measured by weight (). Treatment with cisplatin alone and gefitinib alone significantly decreased the extent of intraabdominal disseminated tumor compared with vehicle control. The combination further attenuated the intraabdominal dissemination. These results suggested that combination therapy of cisplatin with gefitinib can increase the therapeutic efficacy of cisplatin.
Gefitinib enhances cisplatin-induced apoptosis via EGFR, ERK, and Akt cascades in EGFR-expressing cell lines
We next investigated the phosphorylation of EGFR and the downstream signaling of EGFR. Cisplatin activates EGFR in human glioma and human breast tumor cells.Citation25 We also observed that cisplatin induced the phosphorylation of EGFR in Caov-3 and RMG-1 cells and that gefitinib significantly inhibited the level of both basal and cisplatin-induced EGFR phosphorylation ().
Figure 2. Gefitinib inhibited EGFR downstream signaling events in the presence of EGFR, whereas cisplatin-induced apoptosis was enhanced by gefitinib independent of EGFR status. (A) Caov-3 (column a) or RMG-1 (column b) cells were pretreated with drug-free medium (lanes “Vehicle” and “CDDP”), or pretreated with 5 μM gefitinib (lanes “Gefitinib” and “Combination”) for 3 h, followed by treatment with 200 μM cisplatin alone (lane “CDDP”), 5 μM gefitinib alone (lane “Gefitinib”), or cisplatin with 5 μM gefitinib (lane “Combination”) for 3 h. Lysates were subjected to western blotting using anti-phospho-EGFR or anti-α-tubulin antibody. (B, C) Caov-3, RMG-1, or A2780 cells were pretreated with drug-free medium (lanes “Vehicle” and “CDDP”), or with 5 μM gefitinib for 3 h (lanes “Gefitinb” and “Combination”), followed by treatment with 200 μM cisplatin alone (lane “CDDP”), cisplatin with 5 μM gefitinib (lane “Combination”), cisplatin with 20 μM MEK inhibitor (PD98052) (lane “PD+CDDP,” panel B), or cisplatin with 50 μM PI3K/Akt inhibitor (LY294002) (lane “LY+CDDP,” panel C) for 3 h. Lysates were subjected to western blotting using anti-phospho-ERK or anti-ERK antibody (B), or using anti-phospho-Akt or anti-Akt antibody (C). (D) Gefitinib enhanced cisplatin induced apoptosis in all three cell lines. Caov-3, RMG-1, or A2780 cells were treated with drug-free medium (lane “Vehicle”), 25 μM cisplatin alone (lane “CDDP”), 5 μM gefitinib alone (lane “Gefitinib”), cisplatin + gefitinib (lane “Combination”), 20 μM MEK inhibitor (PD98052) + cisplatin (lane “PD+CDDP”), or 50 μM PI3K/Akt inhibitor (LY294002) + cisplatin (lane “LY+CDDP”) for 24 h. Lysates (250 μg of protein) were subjected to western blotting using anti-PARP or anti-α-tubulin antibody.
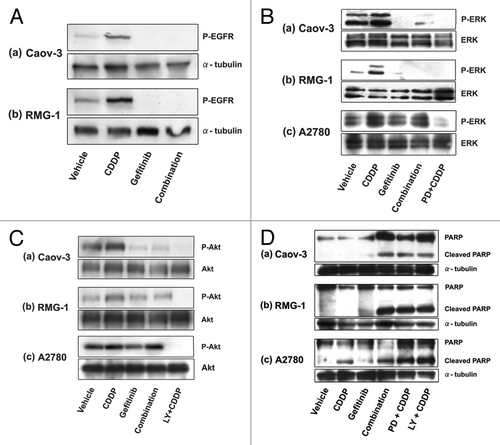
Because the ERK and Akt cascades are the effectors of proliferative and survival signaling downstream of EGFR, we next examined whether gefitinib inhibits the ERK and Akt cascades. Cultured cells were exposed to cisplatin with or without gefitinib, and the phosphorylation of ERK in all cell lines was observed ().Citation14 The increase in phosphorylated ERK by cisplatin treatment was blocked by incubation with gefitinib in Caov-3 and RMG-1cells, but not in A2780 cells (). Cisplatin increased the phosphorylation of Akt in all cell lines (),Citation15 and inhibition of the cisplatin-induced Akt phosphorylation by gefitinib was observed in Caov-3 and RMG-1cells, but not in A2780 cells ().
There have been several reports of increases of chemotherapeutic agent-induced apoptosis via ERK or Akt inactivation,Citation15,Citation16,Citation26,Citation27 leading us to examine whether gefitinib increased efficacy in cisplatin-induced apoptosis. Using anti-PARP antibody, we examined by western blotting the effects of gefitinib on the cisplatin-induced cleavage of PARP (). Gefitinib significantly enhanced the ability of cisplatin to induce cleavage of in all cell lines (, panels a–c, lane “Combination”). Similar results were obtained in combination with PD98059 (, panels a–c, lane “PD + CDDP”) or LY294002 (, panels a–c, lane “LY + CDDP”). These results suggested that gefitinib inhibited cell proliferation and enhanced cisplatin-induced apoptosis by inhibiting activation of the EGFR, ERK, and Akt cascades in Caov-3 and RMG-1, which expressed EGFR. In A2780 cells, which lack EGFR but express HER2, gefitinib inhibited cell proliferation and enhanced the cisplatin-induced apoptosis through pathways other than the inhibition of Akt and ERK.
It was recently reported that an EGFR mutation may predict sensitivity to gefitinib in non-small cell lung cancer.Citation28,Citation29 Therefore, we investigated the presence of mutation in ovarian cancer cell lines using SSPC (single-strand conformational polymorphism). We failed to observe the mutations in all ovarian cancer cell lines (data not shown).
Gefitinib inhibits the repair of cisplatin-induced DNA damage independent of EGFR status
Although gefitinib did not block the phosphorylation of ERK and Akt in A2780 cells without EGFR, gefitinib increased cisplatin-induced cytotoxicity and apotosis in A2780 cells, suggesting this effect did not depend on the EGFR status. In a breast cancer cell line, gefitinib is reported to inhibit the repair of cisplatin-induced DNA damage.Citation21,Citation22 Therefore we hypothesized that combination therapy with cisplatin and gefitinib may increase the therapeutic efficacy of cisplatin by inhibiting DNA repair in the cells without EGFR.
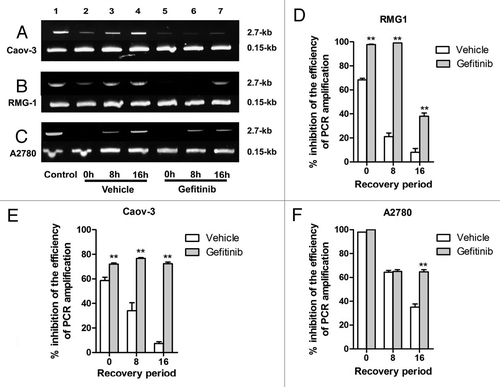
To test if gefitinib inhibits the repair of cisplatin-induced DNA damage in ovarian cancer, we used a PCR stop assay that is sensitive to all modes of DNA damage, including the requirement for replication for repair, i.e., a modification of general assay of Eastman et al.Citation30 In this assay a representative portion of HPRT gene is PCR amplified, and the effects of disruption of the chromatin template by DNA adducts formation are measured as inhibition of PCR amplification efficiency. The ability of PCR amplify a 2.7-kb fragment of the HPRT gene is used to measure repair of cisplatin-DNA adducts because these adducts block the PCR amplification reaction. A 0.15-kb fragment that is too short to suffer cisplatin adduct formation at the doses used here is used as an internal control for PCR amplification. DNA repair is assessed by comparing inhibition of PCR amplification measured immediately after a DNA-damaging event with that after a recovery period during which repair and restored PCR efficiency occur. We incubated cells with cisplatin with or without gefitinib for 1 h, followed by incubation in vehicle or gefitinib for 0, 8 or 16 h. Treatment of Caov-3 cells for 1 h with cisplatin led to a 60% inhibition of PCR product formation (, panel a, lane 2) compared with the control (, panel a, lane 1), which was fully restored after a 16 h “recovery” period (, panel a, lane 4), indicating substantial DNA repair during the recovery period. However, in cells incubated with gefitinib (10 μM) for 0, 8 or 16 h (, panel a, lane 5–7) after 1 h exposure to cisplatin + gefitinib, restoration of PCR product formation after 16 h (, panel a, lane 7) was not observed [73% inhibition compared with the control (, panel a, lane 1)]. Similar experiments were performed in the RMG-1 cell line. Treatment of RMG-1 cells for 6 h with cisplatin led to a 68% inhibition of PCR product formation (, panel b, lane 2) compared with the control (, panel b, lane 1), which showed a 8% inhibition compared with the control (, panel b, lane 1) after 16 h (, panel b, lane 4,). However, cells incubated with gefitinib after treatment with cisplatin + gefitinib showed a 38% inhibition at 16 h (, panel b, lane 7) compared with the control (, panel b, lane 1). In A2780 cells, treatment with cisplatin for 1 h led to a complete inhibition (, panel c, lane 2) compared with control (, panel c, lane 1), and a 35% inhibition at 16 h (, panel c, lane 4) compared with control (, panel c, lane 1). However, cells incubated with gefitinib after treatment with cisplatin + gefitinib showed a 65% inhibition at 16 h (, panel c, lane 7) compared with control (, panel c, lane 1). These results suggested that gefitinib might inhibit repair of cisplatin-induced DNA damage via EGFR and via HER2.
Figure 3. Effect of gefitinib on the repair from cisplatin-induced DNA damage. Cells were treated with 200 μM cisplatin alone (lanes 2–4) or cisplatin with 10 μM gefitinib (lanes 5–7) for an hour (panels a and c, Caov-3 and A2780 cells), for 6 h (panel b, RMG-1), followed by incubation in drug-free medium (lanes 2–4, Vehicle) or 10 μM gefitinib (lanes 5–7, Gefitinib) for 0, 8 or 16 h and subjected to PCR stop assay. In this assay a representative portion of HPRT gene is PCR amplified, and the effects of disruption of the chromatin template by DNA adducts formation are measured as inhibition of PCR amplification efficiency. The ability of PCR amplify a 2.7-kb fragment of the HPRT gene is used to measure repair of cisplatin-DNA adducts because these adducts block the PCR amplification reaction. A 0.15-kb fragment that is too short to suffer cisplatin adduct formation at the doses used here is used as an internal control for PCR amplification. Cells were treated with drug-free medium (lane 1, Control). PCR products were quantified by using Image J System Software (panel d, e, and f). Bars, the results were expressed as inhibition of PCR amplification after normalizing the averaged PCR efficiency of Control set as 1 minus that of a given treatment or a given treatment with recovery period × 100 (% inhibition of the efficiency of PCR amplification). The results are expressed as the averages ± SE of three independent assays. ** indicates p < 0.01 compared the inhibition of PCR amplification measured in Vehicle and that in Gefitinib for 0, 8, 16h each.
Gefitinib reduced the expression of DNA-PK independent of EGFR status
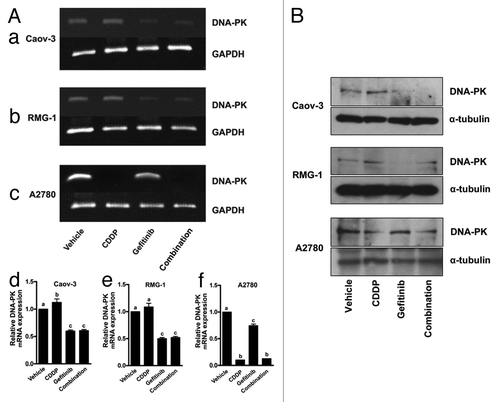
Because of reports that DNA-PK is involved in DNA repair,Citation31 we next examined the expression of DNA-PK using reverse-transcription PCR. After exposure to gefitinib, there was a reduction in DNA-PK at the mRNA level in Caov-3 and RMG-1 cells (, panels a and b, lane “Gefitinib”), both of which are resistant to cisplatin. Treatment of cisplatin slightly increased expression of DNA-PK at the mRNA level (, panels a and b, lane “CDDP”), compared with control (, panels a and b, lane “Vehicle”). The mRNA level of DNA-PK was reduced when cells were exposed to the combination of cisplatin with gefitinib (, panels a and b, lane “Combination”) in Caov-3 and RMG-1 cells. On the other hand, in A2780 cells, which are sensitive to cisplatin, cisplatin treatment results in remarkably decreased expression of DNA-PK (, panel c, lane “CDDP”) compared with the vehicle control (, panel c, lane “Vehicle”). Gefitinib treatment also decreased expression of DNA-PK mRNA (, panel c, “Gefitinib”). Treatment with both cisplatin and gefitinib also remarkably decreased expression of DNA-PK mRNA (, panel c, lane “Combination”). Immunoblot analysis showed that treatment of cisplatin increased the expression of DNA-PK in Caov-3 and RMG-1 but not in A780 cells. Gefitinib decreased the DNA-PK expression in all cell lines. Combination of cisplatin with gefitinib attenuated the expression of DNA-PK compared with cisplatin alone in all cell lines (). These results suggested that gefitinib may reduce expression of DNA-PK, depending not only on EGFR but also on HER2.
Figure 4. Effect of gefitinib on the expression of DNA-PK. (A) Cells were treated with drug-free medium (lane “Vehicle”), 200 μM cisplatin alone (lane “CDDP”), 10μM gefitinib alone (lane “Gefitinib”) or cisplatin + gefitinib (lane “Combination”) for 24 h and subjected to RT–PCR for DNA-PK, as described in “Materials and Methods.” PCR products were quantified by using Image J System Software (panel d, e, and f). Bars, relative DNA-PK mRNA expression, which was quantified by using Image J Imaging System Software, calculated from the ratio of PCR product band to the vehicle band set as 1. Experiments were repeated at least three times with consistent results, and a representative result is shown. Different letters above the bar indicate significant difference at p < 0.01. (B) Expression of DNA-PK at protein levels in ovarian cancer cell lines treated with each reagent alone or combination of cisplatin and gefitinib.
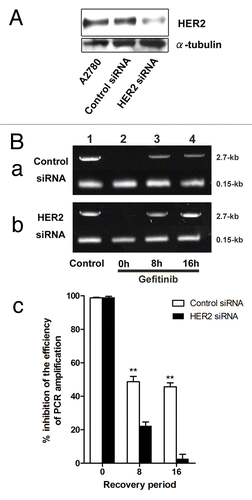
Effect of knock down with HER2 on the inhibition of DNA repair by gefitinib
To further examine the role of HER2 in the effect of gefitinib, we used siRNA to HER2. A2780 cells transfected with anti-HER2 siRNA achieved a 60% reduction in HER2 level at 40 h following transfection compared with that in the control cells transfected with control siRNA (). Using the PCR stop assay, we further examined the effect of anti-HER2 siRNA transfection on DNA repair. A2780 cells transfected with control or anti-HER2 siRNA were treated with cisplatin + gefitinib for 1 h, followed by incubation in gefitinib for 0, 8 or 16 h. A2780 cells, transfected with control or anti-HER2 siRNA, and treated with cisplatin + gefitinib for 1h (, panels a and b, lane 2), led to a remarkable inhibition of PCR product to control cells treated with drug-free medium (, panels a and b, lane 1). Cells transfected with control siRNA, and incubated in gefitinib after treatment by cisplatin + gefitinib for 1 h, remained a 55% inhibition of PCR product at 16 h (, panel a, lane 4), compared with that of control cells (, panel a, lane 1). However, cells transfected with anti-HER2 siRNA almost recovered to the level of that in control cells (, panel b, lane 1) at 16 h (, panel b, lane 4). These results suggested that anti-HER2 siRNA cancelled the effect of gefitinib on the inhibition of DNA repair and that gefitinib may inhibit recovery from cisplatin-induced DNA damage through HER2.
Figure 5. Effect of gefitinib on DNA repair in cells transfected with anti-HER2 siRNA (A) western blot analysis for the expression of HER2 in A2780 cells (lane “A2780”) and cells transfected with control (lane “Control siRNA”) or anti-HER2 (lane “HER2 siRNA”) siRNA. Cells were harvested 40 h after transfection. (C) A2780 cells transfected with control (panel a) or anti-HER2 siRNA (panel b) were treated with 200 μM cisplatin with 10 μM gefitinib (lanes 2–4) for an hour followed by incubation in 10 μM gefitinib for 0, 8 or 16 h and subjected to PCR stop assay. In this assay a representative portion of HPRT gene is PCR amplified, and the effects of disruption of the chromatin template by DNA adducts formation are measured as inhibition of PCR amplification efficiency. The ability of PCR amplify a 2.7-kb fragment of the HPRT gene is used to measure repair of cisplatin-DNA adducts because these adducts block the PCR amplification reaction. A 0.15-kb fragment that is too short to suffer cisplatin adduct formation at the doses used here is used as an internal control for PCR amplification. Cells were treated with drug-free medium (lane 1, Control). PCR products were quantified by using Image J System Software (panel d). Bars, the results were expressed as inhibition of PCR amplification after the normalizing the averaged PCR efficiency of cells transfected with control or anti-HER2 siRNA and treated with drug-free medium set each as 1 minus that of a given treatment or a given treatment with recovery period × 100 (% inhibition of the efficiency of PCR amplification). The results are the averages ± SE of three independent assays. ** indicates p < 0.01 compared the inhibition of PCR amplification measured in cells incubated with gefitinib after the transfection of control siRNA and that in cells incubated with gefitinib after the transfection of HER2 siRNA for 0, 8, 16h each.
Discussion
The results in this study that gefitinib increases the efficiency of cisplatin in ovarian tumor cells in vitro and in vivo were consistent with other works.Citation32,Citation33 The novel finding in our study is that gefitinib enhanced the cisplatin-induced cytotoxicity and apoptosis by delaying the repair of cisplatin-induced DNA damage independent of EGFR status. In clinical studies, interim data from Lacroix et al.Citation13 report that the combination of gefitinib (500 mg per day) plus paclitaxel and carboplatin as a second-line treatment for advanced ovarian adenocarcinoma provides promising anti-tumor activity and is well tolerated. Thus, the data presented in this study support the incoming clinical data regarding ovarian cancers.
Ovarian cancer is the major cause of death from gynecological malignancy. The current standard therapy for patients with advanced ovarian cancer is cytoreductive surgery followed by administration of systematic chemotherapy. First-line therapy consists of platinum (cisplatin or carboplatin) in combination with paclitaxel. Except for some improvement in survival length with the introduction of platinum and paclitaxel therapy, the likelihood of treatment success in women with advanced, recurrent, or persistent ovarian cancer has remained largely unchanged.Citation34 Therefore, there is a need to consider the use of second-line chemotherapeutic options for this cancer. However, the response rates to second-line therapy differ greatly depending on platinum sensitivity. The interval off platinum-based therapy can be a strong predictor of platinum sensitivityCitation35; thus, an important determinant of prognosis seems to be whether recurrent ovarian cancer is sensitive or resistant to platinum. Because the prognosis of patients with relapsed ovarian cancer is poor, it is very important to clarify the mechanisms of platinum refractoriness and to develop molecular-targeting therapies for platinum-refractory ovarian cancer.
Because both the ERKCitation14 and AktCitation15 cascades are involved in resistance to cisplatin, inhibition of both cascades using gene transfection was found to be more effective for blocking cisplatin resistance.Citation15 However, small molecular inhibitors that block both the ERK and Akt cascades have never been discovered. Thus, we thought that gefitinib might hold promise for blocking the mechanisms of platinum-refractory cancer because the ERK and Akt cascades occur downstream of EGFR signaling. In our study, gefitinib inhibited the activation of both ERK and Akt cascades and increased cisplatin-induced apoptosis. We also observed a synergistic effect on cell proliferation in EGFR-expressed cell lines consistent with results in previous reports.Citation32,Citation36
Although inhibition of ERK and Akt phosphorylation by gefitinib was not observed in A2780 cells, which lack EGFR but express HER2, we observed gefitinib increased the cisplatin-induced cytotoxicity and apoptosis in A2780 cells. Several studies reported that heterodimer formation of HER2/HER3 or EGFR/HER3 activates the downstream signaling that plays a pivotal role in drug sensitivity to an EGFR-targeting drug.Citation37,Citation38 Gefitinib did not inhibit the EGFR downstream ERK and Akt signaling downstream EGFR in A2780 cells, which do not express HER3 and express only HER2. We therefore hypothesized that gefitinib might inhibit proliferation and enhance cisplatin-induced apoptosis through pathway other than EGFR downstream signaling via HER2.
Gefitinib is also reported to act through inhibition of repair of cisplatin-induced DNA damage by inhibition of the DNA-PK pathway.Citation21 In our study, gefitinib delayed repair of cisplatin-induced DNA damage and decreased the expression of DNA-PK at the transcriptional and protein levels in all cell lines, including A2780 cells. Although it is reported that the interaction between EGFR and DNA-PK is important,Citation22 we found that the effect of gefitinib on inhibition of was independent of EGFR. Therefore, the mechanism for the inhibition of DNA repair by gefitinib might involve a pathway unrelated to the interaction between EGFR and DNA-PK. Because A2780 cells express only HER2, we thought HER2 might play an important role in this inhibition of DNA repair by gefitinib. We found that gefitinib did not inhibit DNA repair in A2780 cells transfected with anti-HER2 siRNA. However downregulation of HER2 had no effect on DNA-PK expression at protein level (data not shown). Immunoprecipitation of cell exacts demonstrated that after exposure to gefitinib, there was not an association between HER2 and DNK-PK (data not shown). These data suggested that gefitinib might inhibit DNA repair by unknown factor which blocks the interaction between HER2 and DNA-PK. These results may explain in why there was no association between EGFR status and gefitinib effect in clinical studies.Citation5,Citation6 Moreover, the previous study indicated that the antitumor activity of gefitinib in non small lung cancer, alone or in combination with chemotherapy, is tumor-dependent independent of EGFR status,Citation39 and was according with the results in the A2780 in vitro study.
Our results contribute to an understanding of the mechanism of interaction between EGFR inhibitors and chemotherapeutic agents. This enhanced understanding could contribute to the development of new schedule of clinical study and confer a therapeutic benefit on platinum-resistance ovarian cancer patients.
Materials and Methods
Materials
Clinical-grade gefitinib was kindly provided by AstraZeneca. Cisplatin was purchased from Sigma-Aldrich. The anti-phospho-Akt, anti-Akt, anti-phospho ERK, anti-ERK, anti-phospho EGFR, anti-EGFR, anti-HER2, and anti-PARP antibodies were obtained from Cell Signaling. The anti-HER3 antibody was purchased from Upstate Cell Signaling Solutions. The terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL) kit (DeadEndTM Fluorometric TUNEL System®) was obtained from Promega.
Cell cultures
The human ovarian mucinous adenocarcinoma cell line Caov-3 was obtained from American Type Culture Collection. The human ovarian clear cell adenocarcinoma cell line RMG-1 was kindly provided by Dr. S. Nozawa and Dr. D. Aoki (Keio University).Citation40 The human ovarian cancer A2780 cell line, derived from a patient prior to treatment, was kindly provided by Dr. Tsuruo (Institute of Molecular and Cellular Biosciences) and Drs. R.F. Ozols and T.C. Hamilton (NCI, National Institutes of Health).Citation41 The Caov-3 and RMG-1 cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum in a water-saturated atmosphere of 95% air and 5% CO2. The A2780 cells were maintained in RPMI1640 medium (Nissui) with 10% fetal bovine serum.
MTS (3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)- 2H-tetrazolium, inner salt) assay
The number of viable cells was determined by determination of A490 of dissolved formazan product after the addition of MTS for 1 h as described by the manufacturer (Promega).Citation42 Cytotoxicity was assessed by the addition of cisplatin at indicated concentrations with or without gefitinib for 72 h, 1 d after seeding test cells into 96-well plates. All experiments were performed in quadruplicate, and the viability was expressed as the ratio of the number of viable cells with cisplatin treatment to that without treatment.
Western blotting
Cells were incubated without serum for 16 h and then treated with various agents. Cells were washed twice in PBS and scraped into lysis buffer. Western blotting was done as described previously.Citation43 Equal amounts of proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blocking was done in 5% skimmed milk powder in 1X TBS. Western blot analyses were performed with various specific primary antibodies.
In vivo studies
All of the procedures involving animals in this study were approved by the animal care committee of Yamagata University in accordance with institutional and Japanese government guidelines for animal experiments. In vivo study was done as described previously.Citation18,Citation43 One million Caov-3 cells were injected i.p. into 5-week-old female nu/nu athymic mice (n = 20). Two weeks after inoculation, one group of mice (n = 5) was treated with gefitinib (50 μg/kg) plus cisplatin (5 mg/kg) once a week for 4 weeks. A second group of mice (n = 5) was treated with gefitinib alone (50 μg/kg) once a week for 4 weeks. A third group (n = 5) was treated with cisplatin alone (5 mg/kg) once a week for 4 weeks. The remaining mice (n = 5) received vehicle (PBS) alone. The volume of ascites was measured, and tumor tissue was excised and fixed in 4% paraformaldehyde and embedded in paraffin.
Analysis of DNA damage and repair
Cisplatin adduct formation and repair were analyzed by a PCR-based DNA damage assay (PCR stop assay) as described previously.Citation44,Citation45 Briefly, the assay is based on the observation that the efficiency of amplification of cisplatin-treated DNA is inversely proportional to the degree of the platination.Citation30 Genomic DNA was isolated immediately or at the indicated times after treatment of cells for 1 h with cisplatin only or cisplatin + gefitinib, followed by the drug-free medium or 10 μM gefitinib using the DNeasy Tissue Kit (Qiagen) and PCR-amplified using primers complementary to the hypoxanthine phosphoribosyltransferase (HPRT) gene, giving rise to a 2.7-kb product. Polymerase progress and therefore the amount of PCR product formed is reduced if cisplatin adduct formation takes place within this 2.7-kb span. A nested primer giving rise to a 0.15-kb fragment of the hypoxanthine phosphoribosyltransferase gene was also included. The small 0.15-kb segment of genomic DNA sustains undetectable levels of DNA damage under our conditions and served as an internal control, and the PCR values were used for normalization of the values observed for the amplification of the 2.7-kb fragment. The 5′- and 3′- primer sequences were TGG GAT TAC ACG TGT GAA CCA ACC and TGT GAC ACA GGC AGA CTG TGG ATC, respectively. PCR products were analyzed by electrophoresis on 8% acrylamide gels or 1.0% agarose gels stained with ethiduim bromide for 0.15- and 2.7-kb products, respectively. PCR products (2.7-kb) were expressed quantitatively by using Image J Imaging System Software Version 1.3 (National Institutes of Health), with gene expression normalized to internal control (0.15-kb).
RNA isolation and RT- PCR
Total RNA was isolated from cells using ISOGEN (Nippon Gene Co., Ltd.) according to the manufacturer’s directions. Total RNA (5 μg) was reverse-transcribed using a first-stand cDNA synthesis kit following the manufacturer’s instructions (SuperScript First-Stand Synthesis System for RT-PCR; Invitrogen Corp.). An aliquot (1 μg) of the reverse transcription product was used for PCR amplification in a total volume of 50 μl. The PCR primer sets used for DNA-PKCitation30 and GAPDH cDNA amplification were as follows: DNA-PK sense 5′ – AGG GAA GAA GAG TCT CTG GTG G – 3′, anti-sense 5′ – ATT AGG GGA TCT GTT GCC TGG C – 3′; and GAPDH sense 5′ – ACC ACA GTC CAT GCC ATC AC – 3′, anti-sense 5′ – TCC ACC ACC CTG TTG CTG TA -3′. The thermal cycle profile used for DNA-PK and GAPDH was 25 cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 30 sec. PCR fragments were analyzed by electrophoresis on 1.0–1.5% agarose gels and stained with ethidium bromide. The mRNA levels of DNA-PK were expressed quantitatively by using Image J Imaging System Software Version 1.3 (National Institutes of Health), with gene expression normalized to GAPDH.
Small interfering RNA analysis
Cells were seeded at 1 × 105 cells per well into 6-well plates were transfected with either a small interfering RNA (siRNA) specific for HER2 (final concentration of 100 nmol/L; Dharmacon, Inc.) or nontargeted siRNA (siCONTROL Non-targeting siRNA; Dharmacon, Inc.) using DharmaFECT. After 24h, the transfection medium was removed and replaced with serum-free medium. Inhibition of HER2 expression was verified by western blot analysis.
Statistics
Statistical analysis was performed using one-way ANOVA followed by Fisher's least significant difference test, and p < 0.05 was considered significant. Data are expressed as the mean ± SE.
Acknowledgments
This work was supported in part by Grants-in-Aid Scientific Research No. 17390445 (to H.K.) and No.18591822 (to K.T.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and in part by Grants-in-Aid for the 21st Century Center of Excellence (COE) Program from the Japan Society for the Promotion of Science.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Egan SE, Weinberg RA. The pathway to signal achievement. Nature 1993; 365:781 - 3; http://dx.doi.org/10.1038/365781a0; PMID: 8413661
- Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett 1998; 425:145 - 50; http://dx.doi.org/10.1016/S0014-5793(98)00224-5; PMID: 9541025
- Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 1999; 82:241 - 50; http://dx.doi.org/10.1016/S0163-7258(98)00045-X; PMID: 10454201
- Xia W, Lau YK, Zhang HZ, Xiao FY, Johnston DA, Liu AR, et al. Combination of EGFR, HER-2/neu, and HER-3 is a stronger predictor for the outcome of oral squamous cell carcinoma than any individual family members. Clin Cancer Res 1999; 5:4164 - 74; PMID: 10632356
- Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol 2004; 22:777 - 84; http://dx.doi.org/10.1200/JCO.2004.08.001; PMID: 14990632
- Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol 2004; 22:785 - 94; http://dx.doi.org/10.1200/JCO.2004.07.215; PMID: 14990633
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351:337 - 45; http://dx.doi.org/10.1056/NEJMoa033025; PMID: 15269313
- Berchuck A, Rodriguez GC, Kamel A, Dodge RK, Soper JT, Clarke-Pearson DL, et al. Epidermal growth factor receptor expression in normal ovarian epithelium and ovarian cancer. I. Correlation of receptor expression with prognostic factors in patients with ovarian cancer. Am J Obstet Gynecol 1991; 164:669 - 74; PMID: 1992720
- Morishige K, Kurachi H, Amemiya K, Fujita Y, Yamamoto T, Miyake A, et al. Evidence for the involvement of transforming growth factor receptor autcrine growth mechanism in primary ovarian cancers in vitro. Cancer Res 1991; 51:5322 - 8; PMID: 1717146
- Bartlett JM, Langdon SP, Simpson BJ, Stewart M, Katsaros D, Sismondi P, et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br J Cancer 1996; 73:301 - 6; http://dx.doi.org/10.1038/bjc.1996.53; PMID: 8562334
- Morishige K, Kurachi H, Amemiya K, Adachi H, Inoue M, Miyake A, et al. Involvement of transforming growth factor alpha/epidermal growth factor receptor autocrine growth mechanism in an ovarian cancer cell line in vitro. Cancer Res 1991; 51:5951 - 5; PMID: 1718591
- Schilder RJ, Sill MW, Chen X, Darcy KM, Decesare SL, Lewandowski G, et al. Phase II study of gefitinib in patients with relapsed or persistent ovarian or primary peritoneal carcinoma and evaluation of epidermal growth factor receptor mutations and immunohistochemical expression: a Gynecologic Oncology Group Study. Clin Cancer Res 2005; 11:5539 - 48; http://dx.doi.org/10.1158/1078-0432.CCR-05-0462; PMID: 16061871
- Lacroix L, Pautier P, Duvillard P, Motté N, Saulnier P, Bidart JM, et al. Response of ovarian carcinomas to gefitinib-carboplatin-paclitaxel combination is not associated with EGFR kinase domain somatic mutations. Int J Cancer 2006; 118:1068 - 9; http://dx.doi.org/10.1002/ijc.21460; PMID: 16152583
- Hayakawa J, Ohmichi M, Kurachi H, Ikegami H, Kimura A, Matsuoka T, et al. Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem 1999; 274:31648 - 54; http://dx.doi.org/10.1074/jbc.274.44.31648; PMID: 10531373
- Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, et al. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000; 60:5988 - 94; PMID: 11085518
- Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, et al. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem 2002; 277:33490 - 500; http://dx.doi.org/10.1074/jbc.M204042200; PMID: 12087097
- Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res 2003; 9:2316 - 26; PMID: 12796401
- Fujimura M, Hidaka T, Saito S. Selective inhibition of the epidermal growth factor receptor by ZD1839 decreases the growth and invasion of ovarian clear cell adenocarcinoma cells. Clin Cancer Res 2002; 8:2448 - 54; PMID: 12114452
- Eastman A. Separation and characterization of products resulting from the reaction of cis-diamminedichloroplatinum (II) with deoxyribonucleosides. Biochemistry 1982; 21:6732 - 6; http://dx.doi.org/10.1021/bi00269a018; PMID: 6891601
- Shen H, Schultz M, Kruh GD, Tew KD. Increased expression of DNA-dependent protein kinase confers resistance to adriamycin. Biochim Biophys Acta 1998; 1381:131 - 8; http://dx.doi.org/10.1016/S0304-4165(98)00020-8; PMID: 9685611
- Friedmann B, Caplin M, Hartley JA, Hochhauser D. Modulation of DNA repair in vitro after treatment with chemotherapeutic agents by the epidermal growth factor receptor inhibitor gefitinib (ZD1839). Clin Cancer Res 2004; 10:6476 - 86; http://dx.doi.org/10.1158/1078-0432.CCR-04-0586; PMID: 15475435
- Friedmann BJ, Caplin M, Savic B, Shah T, Lord CJ, Ashworth A, et al. Interaction of the epidermal growth factor receptor and the DNA-dependent protein kinase pathway following gefitinib treatment. Mol Cancer Ther 2006; 5:209 - 18; http://dx.doi.org/10.1158/1535-7163.MCT-05-0239; PMID: 16505093
- Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res 2001; 61:7184 - 8; PMID: 11585753
- Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, et al. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995; 82:849 - 56; http://dx.doi.org/10.1016/0092-8674(95)90482-4; PMID: 7671312
- Benhar M, Engelberg D, Levitzki A. Cisplatin-induced activation of the EGF receptor. Oncogene 2002; 21:8723 - 31; http://dx.doi.org/10.1038/sj.onc.1205980; PMID: 12483525
- Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res 1999; 5:1007 - 14; PMID: 10353733
- Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 2002; 62:1087 - 92; PMID: 11861387
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 39; http://dx.doi.org/10.1056/NEJMoa040938; PMID: 15118073
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 500; http://dx.doi.org/10.1126/science.1099314; PMID: 15118125
- Jennerwein MM, Eastman A. A polymerase chain reaction-based method to detect cisplatin adducts in specific genes. Nucleic Acids Res 1991; 19:6209 - 14; http://dx.doi.org/10.1093/nar/19.22.6209; PMID: 1956780
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366 - 74; http://dx.doi.org/10.1038/35077232; PMID: 11357144
- Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 2000; 6:2053 - 63; PMID: 10815932
- Smith JA, Gaikwad A, Yu J, Wolf JK, Brown J, Ramondetta LM, et al. In vitro evaluation of the effects of gefitinib on the modulation of cytotoxic activity of selected anticancer agents in a panel of human ovarian cancer cell lines. Cancer Chemother Pharmacol 2008; 62:51 - 8; http://dx.doi.org/10.1007/s00280-007-0572-y; PMID: 17849119
- Duton CJ. New options for the treatment of advanced ovarian cancer. Semin Oncol. 1997;24(1 Suppl 5): S5-2-S5-11.
- Blackledge G, Lawton F, Redman C, Kelly K. Response of patients in phase II studies of chemotherapy in ovarian cancer: implications for patient treatment and the design of phase II trials. Br J Cancer 1989; 59:650 - 3; http://dx.doi.org/10.1038/bjc.1989.132; PMID: 2713253
- Ciardiello F, Caputo R, Troiani T, Borriello G, Kandimalla ER, Agrawal S, et al. Antisense oligonucleotides targeting the epidermal growth factor receptor inhibit proliferation, induce apoptosis, and cooperate with cytotoxic drugs in human cancer cell lines. Int J Cancer 2001; 93:172 - 8; http://dx.doi.org/10.1002/ijc.1335; PMID: 11410862
- Hirata A, Hosoi F, Miyagawa M, Ueda S, Naito S, Fujii T, et al. HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res 2005; 65:4253 - 60; http://dx.doi.org/10.1158/0008-5472.CAN-04-2748; PMID: 15899817
- Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A 2005; 102:3788 - 93; http://dx.doi.org/10.1073/pnas.0409773102; PMID: 15731348
- Judde JG, Rebucci M, Vogt N, de Cremoux P, Livartowski A, Chapelier A, et al. Gefitinib and chemotherapy combination studies in five novel human non small cell lung cancer xenografts. Evidence linking EGFR signaling to gefitinib antitumor response. Int J Cancer 2007; 120:1579 - 90; http://dx.doi.org/10.1002/ijc.22364; PMID: 17205515
- Nozawa S, Tsukazaki K, Sakayori M, Jeng CH, Iizuka R. Establishment of a human ovarian clear cell carcinoma cell line (RMG-1) and its single cell cloning—with secial reference to the stem cell of the tumor. Hum Cell 1998; 1:426 - 35
- Hamilton TC, Winker MA, Louie KG, Batist G, Behrens BC, Tsuruo T, et al. Augmentation of adriamycin, melphalan, and cisplatin cytotoxicity in drug-resistant and -sensitive human ovarian carcinoma cell lines by buthionine sulfoximine mediated glutathione depletion. Biochem Pharmacol 1985; 34:2583 - 6; http://dx.doi.org/10.1016/0006-2952(85)90551-9; PMID: 4040369
- Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, et al. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem 2004; 279:23477 - 85; http://dx.doi.org/10.1074/jbc.M313709200; PMID: 15026414
- Ohta T, Ohmichi M, Hayasaka T, Mabuchi S, Saitoh M, Kawagoe J, et al. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer model. Endcrinology 2006; 147:1761 - 9; http://dx.doi.org/10.1210/en.2005-1450
- Potapova O, Haghighi A, Bost F, Liu C, Birrer MJ, Gjerset R, et al. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J Biol Chem 1997; 272:14041 - 4; http://dx.doi.org/10.1074/jbc.272.22.14041; PMID: 9162025
- Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem 2003; 278:20582 - 92; http://dx.doi.org/10.1074/jbc.M210992200; PMID: 12663670