Abstract
Cell cycle progression through each phase is regulated by heterodimers formed by cyclin-dependent kinases (CDKs) and their regulatory partner proteins, the cyclins. Together they coordinate the cellular events through cell cycle. De-regulation of cell-cycle control due to aberrant CDK activity is a common feature of most cancer types.
Intensive research on small molecules that target cell cycle regulatory proteins has led to the identification of many candidate inhibitors that are able to arrest proliferation and induce apoptosis in neoplastic cells as a promising strategy to treat cancer. Interestingly, cyclin-dependent kinases (CDKs) have also been proposed as therapeutic targets for Multiple Myeloma (MM). Overexpression and aberrant expression of the cyclins, specifically the D cyclins is seen in the majority of MM underscoring the value of exploring CDK inhibition in MM which currently remains an incurable neoplastic plasma-cell disorder. It is characterized by clonal proliferation of malignant plasma cells in the bone marrow microenviroment and associated organ dysfunction. Recent preclinical and early clinical data explore several CDK inhibitors in the context of MM.
This review will provide an overview of the main classes of CDK inhibitors with a focus on their mechanism of action and discuss clinical and pharmacological implications of CDK inhibitors as possible therapeutic approaches for the treatment of cancer with specific consideration to MM.
Introduction
Cyclin-dependent kinases (CDKs) are specific serine/threonine kinases that play an essential role in cell cycle regulation allowing transition between its different phases.Citation1 Many of the genes involved in cell cycle progression are frequently mutated in human cancers leading to uncontrolled cell division and tumor growth. Furthermore, several components of the CDK machinery are deregulated in different malignancies.Citation2 This knowledge provides a rationale for considering the cell cycle and its complex regulation system as potential targets for new drug development in cancer therapeutics. Hence, in the last decade there has been increasing interest in the development of selective inhibitors of CDKs and mitotic kinases.Citation3 A number of CDK inhibitors (CDKIs) with different mechanisms of action have been evaluated. Yet, it is not clear which CDK or spectrum of CDKs should be targeted. Based on preclinical data it seems clear that compensatory roles of certain CDKs in cancer cell types may influence the biology of specific cancers that would be considered good targets for these compounds.Citation1
Limited clinical activity has been observed in most-single agent studies with CDKIs along with remarkable toxicity.Citation1 Newer molecules with more favorable pharmacokinetics, a better understanding of the biology and mechanisms of action of these drugs and the use of CDKIs in combination with conventional cytotoxics seem promising areas that are being currently explored. In addition, new kinases involved in cell-cycle regulation have been recently identified and represent promising alternative therapeutic targets. Results from ongoing trials, the incorporation of more selective targeted agents and a more profound understanding of the cell cycle and its regulatory mechanisms, will hopefully bring some light to this complex field of anticancer drug development.Citation4,Citation5
Here we review the role of CDKIs in malignancy, with particular attention to multiple myeloma (MM) and we discuss the most relevant CDKIs in clinical development as possible therapeutic approaches in cancer therapy.
Cyclin dependent kinases: from the bench to the clinic
The cell cycle is an ordered series of events required for the faithful duplication of one eukaryotic cell into two genetically identical daughter cells. It is now well-established that, although growth and protein synthesis occur almost constantly throughout the cycle, DNA synthesis takes place only at determinate times. The Gap intervals (G1, G2) between the S phase and M phase are no longer considered idle periods, but rather represent critical regulatory phases where information from the extracellular environment is integrated along with all intracellular changes.Citation1,Citation6 The coordinated transitions between cell cycle phases depend on one family of evolutionarily conserved proteins, called CDKs. These are binary proline-directed serine/threonine-specific kinases that consist of a catalytic subunit (the CDK) and a regulatory subunit (the cyclin) as shown in .
Figure 1. Cyclin-dependent kinase Inhibitors as possible therapeutic approach for the treatment of cancer. Cell cycle progression through each phase is regulated by cyclin-dependent kinases (CDK) and their regulatory partner proteins, the cyclins. De-regulation of cell-cycle control, due to aberrant CDK activity is a common feature of most cancer types. Specific and selective CDK Inhibitors has been developed as a potential anti-tumor therapeutic approach.
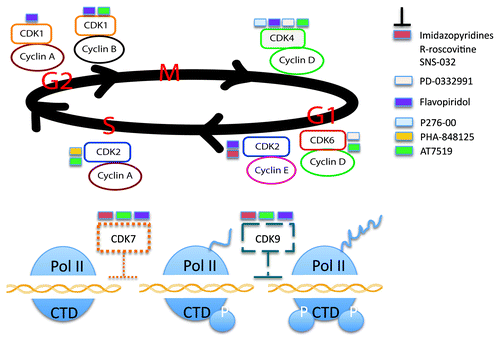
The mammalian genome has 12 loci encoding CDKs, although only five of them—CDK1, CDK2, CDK3, CDK4 and CDK6—have been directly involved in driving the cell cycle. Cyclins, so named because their activity cycles up and down during the cell cycle, restrict the action of their bound kinases to particular substrates. Different families of cyclins have been identified. D-type cyclins are functionally activated from mid to late G1, to direct phosphorylation of the cell cycle inhibitor pRb. Phosphorylation of pRb and related proteins by cyclin D inactivates their regulatory functions, allowing progression into S phase. Cyclin A accumulates at the G1/S phase boundary. It can activate CDK2 and CDK1, promoting progression through the G2 interval. At this point, B-type cyclins, especially cyclin B1 and CDK1 drive cells into mitosis.Citation7,Citation8 Transcription inhibition during mitosis coincides with increased hyperphosphorylation of the RNA polymerase II (RNAPII) carboxyl-terminal domain (CTD), but also with mitosis-specific phosphorylation of transcription activators Sp1, Myc, Myb and of the general transcription factors (GTFs) TFIID and TFIIH.Citation9 Several CTD kinases are members of the cyclin-dependent kinase (CDK) superfamily and associated with transcription initiation complexes. These include CDK7, 8 and 9. The discovery of the multifunctional nature of CDK7 has prompted numerous studies to address the question whether CDK7 provides a functional link between cell cycle regulation and the general transcription machinery. The idea of a functional connection between the regulation of cell cycle progression and RNAPII CTD phosphorylation, through the CDK7-cyclin H-MAT1 complex, is further reinforced by recent studies of the INK4 protein p16INK4A.10 These studies may indicate that CTD phosphorylation by CDK7 directly contributes to cell cycle progression at the G1 to S phase transition.Citation9 Moreover, a direct role of CDK8 in the control of cell proliferation through negative regulation of CDK7 CTD kinase activity and /or TFIIH transcription activity has recently been shown.Citation11
The third CTD kinase of the CDK superfamily found to be associated with RNAPII transcription complexes is CDK9. Similar to CDK7 and CDK8, CTD kinase activity and protein levels of the CDK9-cyclin T complex appear not to significantly fluctuate during the cell cycle in most eukaryotic cells. However, activation of primary human peripheral blood lymphocytes (PBLs) by treatment with mitogens phorbol 12-myristate 13-acetate (PMA) or phytohemagglutinin (PHA) coincides with a dramatic upregulation of intracellular cyclin T protein levels and a corresponding increase of RNAPII phosphorylation. Thus, PMA/PHA activation of PBLs may provide an example of cell type-specific activation of CDK9-cyclin T1 dependent RNAPII CTD kinase activity via a signaling pathway that promotes cell cycle progression.Citation9
Given the critical role that CDKs play in cell cycle control they have been actively considered as targets for anticancer therapy.Citation5 Cancer cells either do not need mitogenic signals to proliferate or their requirements are less stringent than those of normal cells. Similarly, cancer cells are less likely to exit the cell cycle to reach a dormant state in response to growth inhibitory signals. These properties are the result of genetic and/or epigenetic alterations in a variety of molecules and growth regulatory pathways either directly or indirectly involved in cell cycle control. Some of the molecules most frequently altered in cancer are those involved in controlling G1/S transition of the cell cycle. Specifically, the CDK-cyclinD/INK4/pRb/E2F pathway has been found to be abnormal in the majority of cases, either by mutations within the genes encoding these proteins or in their upstream regulators.Citation2 Different human cancers are characterized by cyclin D overexpression, CDK4 and CDK2 hyper-activation and hyper-expression of anti-apoptotic transcripts.Citation12-Citation14
Therefore CDKs represent an interesting therapeutic target, and their pharmacological inhibitors have been proposed for cancer treatment.Citation15 So far, 11 classes of CDK ATP competitive inhibitors have been developed: Staurosporine, Flavonoid, Purine, Indole, Pyrine, Pyrimidine, Indirubin, Pyrazole, Thiazole, Paullone and Hymenialdisine derivatives.Citation16 A brief overview of different CDKIs currently in clinical development is summarized in Table.1.
When attempting to modulate CDK activity, several strategies have been considered.Citation1 In the last decades, small-molecules active as CDKIs for anticancer therapies have been developed starting from the linear sequences and from the crystal structures of the single protein and their complexes.Citation17 Such molecules act as ATP inhibitors.Citation16 Interfering with CDK catalytic activity through ATP-competitive ligands has proved a successful strategy, but a number of protein kinases with sequence similarity within the active site exist, resulting in loss of selectivity. Crystallography has revealed that the ATP-binding site of CDK2 can accommodate different molecular frameworks, exploiting various sites of interaction. In addition, residues outside the main ATP-binding cleft have been identified that could be targeted to increase specificity and potency. These results suggest that it may be possible to design pharmacologically relevant ligands that act as specific and potent inhibitors of CDK activity.Citation16
A growing number of CDK inhibitors representing multiple chemical classes currently are in clinical trials. These drugs may be classified based on their effects against the cell cycle CDKs as either pan-CDKIs or more selective CDKIs, with varying potency against the transcriptional CDKs. Results on the group of compounds currently under study will determine whether multi-targeted CDK inhibition is preferable to selective CDK inhibition. For example, both Seliciclib and BMS-387032 are relatively selective for CDK2. However, these and the majority of molecules that inhibit CDK2 also inhibit CDK1; compounds more equipotent for CDK2 and CDK1 may be superior. Whether a selective CDK2/CDK1 inhibitor will be therapeutically better compared with a CDK2/CDK1/CDK4/6 inhibitor remains to be determined.Citation10
Furthermore, some practical limitations have been noted. For instance, some CDKs may have functions not directly related to cell cycle progression and others, which were believed to be essential for this process, may actually be dispensable. One example to support the latter theory is CDK2. Recent research in animal models suggests that this kinase is not required for mitotic cell division and could be dispensable for cancer cell progression, rendering this CDK unsuitable as a treatment target.Citation18 This is probably due to redundancy of CDKs in the cell and cyclin E may partner with another CDK target when CDK2 is not available.Citation19,Citation20 Limited clinical activity has been observed in most single-agent studies with CDKIs along with remarkable toxicity. These findings precluded further evaluation in some cases and led to the search of different strategies. Newer molecules with more favorable pharmacokinetics and the use of CDKIs in combination with conventional cytotoxics, seem promising areas that are being currently explored. In addition, new kinases involved in cell-cycle regulation have been recently identified representing promising alternative therapeutic targets.Citation1
Results from some ongoing clinical trials of first and second generation CDKIs (listed in .) are summarized next.
Table 1. First and second generation CDK Inhibitors in ongoing clinical trials
Preclinical and clinical studies of first generation CDK inhibitors in cancer therapy
Most of the first generation compounds inhibit multiple CDKs with CDK2 being a common target in drug discovery programs.Citation21 One example is represented by Flavopiridol which is a semisynthetic flavonoid derived from rohitukine, an Indian plant. It was the first potent CDKI to enter in clinical trials.Citation22 In human chronic lymphocytic leukemia (CLL) cells, in vitro Flavopiridol effectively induces apoptosis at clinically achievable concentrations and also decreases expression of Mcl-1 and XIAP, proteins that mediate resistance to apoptosis in CLL cells.Citation23,Citation24 In xenograft models, the most pronounced antitumor effects were seen after prolonged exposure to the drug, prompting the evaluation of a 72 h i.v. continuous infusion every 2 weeks in two phase I trials in humans.Citation25,Citation26 Despite promising preclinical results, clinical activity observed with Flavopiridol has been generally disappointing in a variety of solid tumors when used as a single agent.Citation27-Citation31 However, Flavopiridol and docetaxel have shown synergy both pre-clinically and in the clinical setting, the schedule of this doublet therapy appears to be critical with the optimal sequence being docetaxel followed by Flavopiridol.Citation32 Based on this data, combination studies have been launched in patients with solid tumors.Citation33 There is also clinical data showing that the combination of Flavopiridol with other cytotoxic agents, such as irinotecan or platinum compounds, is feasible but efficacy is limited.Citation34-Citation36 Another example of a pan-CDK inhibitor can be represented by UCN-01 which is a staurosporine analog isolated from the culture broth of Streptomyces species. UCN-01 is associated with G1/S cell cycle arrest, induction of p21CIP/Waf1 and dephosphorylation of both CDK2 and pRb.Citation37,Citation38 Preclinical models have demonstrated synergistic activity of UCN-01 with a number of cytotoxic drugs, more often with topoisomerase inhibiting agents. Several phase I studies have been conducted with UCN-01, both as a single agent and in combination with cytotoxic chemotherapy.Citation39-Citation41 UCN-01 has been evaluated in combination with topotecan in relapsed ovarian cancer, demonstrating no significant clinical activity. In addition, evaluation of UCN-01 and topotecan is also currently ongoing in small-cell lung cancer.Citation37 R-Rescovitine (Seliciclib) is another highly selective, orally bio-available, small molecule inhibitor of several CDKs, competing at their ATP-binding sites.Citation42 The antitumor efficacy of Seliciclib has been demonstrated in human tumor xenografts when administrated at high doses in the range of 500 mg/kg twice a day or 200 mg/kg three times a day. Benson and colleagues performed a phase I trial of Seliciclib in 22 patients with refractory advanced solid tumors.Citation43 A newer bio-isoster of Roscovitine has recently been characterized namely N-&-N1. This compound displays improved anti-tumor properties and shows exquisite selectivity for CDKs with two to 3-fold enhanced potency compared with R-Roscovitine. Inhibition of pRb phosphorylation and RNA pol II in cell lines exposed to N-&-N1 indicates its ability to inhibit CDKs in a cellular contest.Citation44
Preclinical and clinical studies of second generation CDK inhibitors in cancer therapy
The second-generation of CDK inhibitors exhibit more potent activity against their targets and are more selective with preferential inhibition of certain subtypes of CDKs.Citation1 A good example is represented by SNS-032 which is a potent and selective inhibitor of CDKs 2,7 and 9: pre-clinically has shown both cell cycle and transcription inhibition activity.Citation45,Citation46 The in vivo activity of SNS-032 was confirmed in several animal models, including ovarian and colon cancer among others. Oral administration was also studied, showing an average bioavailability of nearly 20%. Future studies evaluating different schedules are required and current phase I evaluation is ongoing in different refractory solid tumors and B lymphoid malignancies.Citation47 Other examples are represented by ZK-304709 and PD-0332991: ZK-304709 is a first-in-class, oral multi target tumor growth inhibitor that blocks tumor cell proliferation via potent inhibition of CDK1,2,4,7,9, but also with anti-angiogenic proprieties due to the blockade of VEGF-R1,2,3 and PDGF-Rβ. Antitumor efficacy of this drug has been tested in human xenografts and also in orthotopic human pancreatic carcinoma models.Citation48 PD-0332991 is a highly reversible specific inhibitor of CDK4 (IC50, 0.011 μmol/L) and CDK6 (IC50, 0.016 μmol/L). It is a potent anti-proliferative agent against Rb-positive tumor cells in vitro, inducing an exclusive G1 arrest.Citation49 Oral administration of this drug to colon cancer xenografts produces marked tumor regression. Therapeutic doses of PD-0332991 cause decrease of phosphor-Rb and Ki-67 in tumor tissue and downregulation of genes under the transcriptional control of E2F. Interestingly, when administrated in preclinical models, in combination with bortezomib, tumor suppression and significant improvement in survival was observed in myeloma cell lines.Citation50
CDK Inhibitors and Multiple Myeloma: Role and Clinical Implication
As described above, CDKs are legitimate targets for therapeutic intervention against different cancers, including MM, and several CDKIs are currently being evaluated pre-clinically and clinically.Citation51
The overexpression of D type cyclins have been implicated in MM pathogenesis and progressionCitation52,Citation53 and the aberrant co-activation of CDK4/cyclin D1 and CDK6/cyclin D2 represents an important factor in myeloma cell proliferation and de-regulation of cell cycle control.Citation54 Consequently, CDKIs such as Flavopiridol, R-Roscovitine as well as P276-00 have been validated in pre-clinical studies in MM and PD-0332991, a specific CDK4/6 inhibitor and P276-00, a CDK4/Cyclin D1 inhibitor, are currently in phase I clinical trials in MM.Citation50 It is known that the above compounds inhibit CDK4/6-specific phosphorylation of Rb and cell cycle progression through G1 in MM cells. Flavopiridol and R-Roscovitine have shown broader activity against CDK2,7,9 and RNA polymerase II CTD, leading to the inhibition of transcription.Citation55-Citation58 Although selective CDKIs have shown potent cytotoxic activity in MM cells, the underlying mechanism is still unclear.Citation12 Interestingly, we have examined the anti-MM activity of RGB 286638Citation59 a novel multi-targeted small molecule inhibitor, originally designed to induce broad cell cycle suppression via multiple CDK inhibition. We demonstrated that treatment with RGB 286638 triggered a dose-dependent cytotoxicity in conventional drug-sensitive and resistant MM cell lines, as well as primary tumor cells from MM patients. Based on sufficient in vitro cytotoxicity, we examined in vivo anti-tumor activity, using a human MM cell xenograft model in SCID mice and they demonstrated that RGB 286638 inhibited tumor growth and prolonged survival. Taken together our results proved preclinical activity and provide the rational to test RGB 286638 in the treatment of MM.Citation59
Furthermore, another study conducted by McMillin and colleagues demonstrated that LCQ195 triggered cell cycle arrest and eventual apoptotic cell death of MM cells, even at sub-umol/l concentrations, while sparing non-malignant cells and overcoming the protective effects conferred to MM cells by stromal cells or major cytokines of the bone marrow milieu. Importantly, LCQ195 (NVP-LCQ195/AT9311) triggered a distinct pattern of molecular sequelae hallmarked by decrease in amplitude of various transcriptional signatures associated with activation of key transcription factors for MM cells (e.g., myc, HIF-1α, IRF4). The authors also observed subsequent decreases in signatures of other molecular pathways associated with oncogenesis, drug resistance and biological aggressiveness of tumor cells. Bortezomib-treated MM patients whose tumors had high baseline expression of genes suppressed by LCQ195 had significantly shorter progression-free and overall survival than those patients whose tumors had low levels of these transcripts. Therefore, these observations indicate that compounds with different patterns of inhibition of individual CDKs can induce distinct networks of transcriptional changes in MM cells. The correlation of these changes with clinical outcome provides insight into the biological relevance of multi-targeted CDK Inhibition.Citation51
Moreover, there are recent studies that have highlighted the role of GSK-3β in different oncogenic pathways, such as PI3K/AKT, Wnt βcatenin and NF-kB-signaling cascades,Citation60 but its role in MM remains to be elucidated. Interestingly, we have studied the GSK-3β pathway (known to have a crucial role in several signaling cascades relevant to MM biology), in the context of CDK inhibition, exploring the pharmacology of AT7519, a multi-targeted CDK inhibitor, that potently inhibits CDK1,2,4,6,7,9.Citation12,Citation61,Citation62 The drug has shown potent anti-MM activity both in vitro and in vivo. In addition, molecular studies of AT7519 revealed that GSK-3β has a crucial role in AT7519-mediated anti-myeloma effect. Moreover, AT7519 in combination with bortezomib has shown synergistic anti-MM activity in vitro. These results have provided the rationale for an ongoing clinical trial of this agent in combination with bortezomib in refractory and relapsed MM patients.Citation12 Another novel multi-kinase inhibitor, sorafenib, exhibited potent in-vitro anti-MM activity, overcoming the proliferative advantage of co-culture with stromal cells or with IL-6, vascular endothelial growth factor (VEGF) and IGF. Examination of cellular signaling pathways demonstrated downregulation of induced myeloid leukemia cell differentiation protein and decreased phosphorylation of STAT3 and MEK/ERK, as potential mechanisms of sorafenib antitumor effect, complemented by anti-angiogenic activity. Promising preclinical data have resulted in the clinical evaluation and development of novel sorafenib combinations. A good example is represented by an ongoing phase I/II study which will test whether sorafenib and lenalidomide have anti-MM activity in patients with relapsed/refractory MM.Citation63
Conclusion
CDKs are essentials for driving each cell cycle phase, therefore therapeutic intervention based on CDK inhibition represents an attractive strategy to treat cancer patients. Based on the literature available and in part discussed in this review, it emerges that mono-specific pharmacological inhibitors are better applicable for the treatment of pathological conditions in which one kinase is de-regulated, whereas multitasking CDKIs seem more likely to increase the possibility of hitting the target in the case of cancer. The pleiotropic effects of the latter warrant the simultaneous interference with the pathways altered in cancer cells thereby reducing the risk of multidrug resistance.Citation16 In fact, while proliferative rates of MM cells are low in early stage of the disease, they are increased when the disease becomes resistant to various conventional or novel anti-MM agents.Citation51
Small molecular weight CDKIs exhibit preclinical anti-tumor activity in diverse neoplasias, including MMCitation10,Citation55,Citation57 with different patterns of inhibition. This led to the conclusion that a multi-targeted CDK inhibitor with a different profile of kinase inhibitory activity, compared with existing CDKIs could trigger a distinct pattern of molecular sequelae in MM cells, which could in turn provide insight into the biology of MM with distinct therapeutic applications.Citation51
In conclusion, there is evidence that novel therapies have increased responses and prolonged survival in patients with cancer, including MM. However, further studies are needed to provide a rational basis for designing more focused clinical trials and, consequently, for tailoring specific therapies to improve treatment for distinctive types of cancer.
| Abbreviations: | ||
| CDK | = | cyclin-dependent kinases |
| CDKI | = | CDK Inhibitor |
| MM | = | multiple myeloma |
References
- Diaz-Padilla I, Siu LL, Duran I. Cyclin-dependent kinase inhibitors as potential targeted anticancer agents. Invest New Drugs 2009; 27:586 - 94; http://dx.doi.org/10.1007/s10637-009-9236-6; PMID: 19262992
- Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 2002; 1602:73 - 87; PMID: 11960696
- Sausville EA. Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends Mol Med 2002; 8:Suppl S32 - 7; http://dx.doi.org/10.1016/S1471-4914(02)02308-0; PMID: 11927285
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008; 14:159 - 69; http://dx.doi.org/10.1016/j.devcel.2008.01.013; PMID: 18267085
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8:547 - 66; http://dx.doi.org/10.1038/nrd2907; PMID: 19568282
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005; 30:630 - 41; http://dx.doi.org/10.1016/j.tibs.2005.09.005; PMID: 16236519
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, et al. Cyclin E ablation in the mouse. Cell 2003; 114:431 - 43; http://dx.doi.org/10.1016/S0092-8674(03)00645-7; PMID: 12941272
- Bagella L, Sun A, Tonini T, Abbadessa G, Cottone G, Paggi MG, et al. A small molecule based on the pRb2/p130 spacer domain leads to inhibition of cdk2 activity, cell cycle arrest and tumor growth reduction in vivo. Oncogene 2007; 26:1829 - 39; http://dx.doi.org/10.1038/sj.onc.1209987; PMID: 17043661
- Oelgeschläger T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol 2002; 190:160 - 9; http://dx.doi.org/10.1002/jcp.10058; PMID: 11807820
- Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24:1770 - 83; http://dx.doi.org/10.1200/JCO.2005.03.7689; PMID: 16603719
- Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000; 407:102 - 6; http://dx.doi.org/10.1038/35024111; PMID: 10993082
- Santo L, Vallet S, Hideshima T, Cirstea D, Ikeda H, Pozzi S, et al. AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 2010; 29:2325 - 36; http://dx.doi.org/10.1038/onc.2009.510; PMID: 20101221
- Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene 2005; 24:2909 - 15; http://dx.doi.org/10.1038/sj.onc.1208618; PMID: 15838524
- Santamaria D, Ortega S. Cyclins and CDKS in development and cancer: lessons from genetically modified mice. Front Biosci 2006; 11:1164 - 88; http://dx.doi.org/10.2741/1871; PMID: 16146805
- McInnes C. Progress in the evaluation of CDK inhibitors as anti-tumor agents. Drug Discov Today 2008; 13:875 - 81; http://dx.doi.org/10.1016/j.drudis.2008.06.012; PMID: 18639646
- Cirillo D, Pentimalli F, Giordano A. Peptides or small molecules? Different approaches to develop more effective CDK inhibitors. Curr Med Chem 2011; 18:2854 - 66; http://dx.doi.org/10.2174/092986711796150496; PMID: 21651493
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res 1996; 68:67 - 108; http://dx.doi.org/10.1016/S0065-230X(08)60352-8; PMID: 8712071
- Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003; 3:233 - 45; http://dx.doi.org/10.1016/S1535-6108(03)00053-9; PMID: 12676582
- Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol 2005; 23:9408 - 21; http://dx.doi.org/10.1200/JCO.2005.01.5594; PMID: 16361640
- Collins I, Garrett MD. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr Opin Pharmacol 2005; 5:366 - 73; http://dx.doi.org/10.1016/j.coph.2005.04.009; PMID: 15964238
- Ortega S, Prieto I, Odajima J, Martín A, Dubus P, Sotillo R, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 2003; 35:25 - 31; http://dx.doi.org/10.1038/ng1232; PMID: 12923533
- Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle 2004; 3:1537 - 42; http://dx.doi.org/10.4161/cc.3.12.1278; PMID: 15539947
- Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, et al. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood 1998; 92:3804 - 16; PMID: 9808574
- Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood 2000; 96:393 - 7; PMID: 10887097
- Thomas JP, Tutsch KD, Cleary JF, Bailey HH, Arzoomanian R, Alberti D, et al. Phase I clinical and pharmacokinetic trial of the cyclin-dependent kinase inhibitor flavopiridol. Cancer Chemother Pharmacol 2002; 50:465 - 72; http://dx.doi.org/10.1007/s00280-002-0527-2; PMID: 12451473
- Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil N, Villalba L, et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol 1998; 16:2986 - 99; PMID: 9738567
- Aklilu M, Kindler HL, Donehower RC, Mani S, Vokes EE. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann Oncol 2003; 14:1270 - 3; http://dx.doi.org/10.1093/annonc/mdg343; PMID: 12881391
- Burdette-Radoux S, Tozer RG, Lohmann RC, Quirt I, Ernst DS, Walsh W, et al. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest New Drugs 2004; 22:315 - 22; http://dx.doi.org/10.1023/B:DRUG.0000026258.02846.1c; PMID: 15122079
- Shapiro GI, Supko JG, Patterson A, Lynch C, Lucca J, Zacarola PF, et al. A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res 2001; 7:1590 - 9; PMID: 11410495
- Liu G, Gandara DR, Lara PN Jr., Raghavan D, Doroshow JH, Twardowski P, et al. A Phase II trial of flavopiridol (NSC #649890) in patients with previously untreated metastatic androgen-independent prostate cancer. Clin Cancer Res 2004; 10:924 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-03-0050; PMID: 14871968
- Schwartz GK, Ilson D, Saltz L, O’Reilly E, Tong W, Maslak P, et al. Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol 2001; 19:1985 - 92; PMID: 11283131
- Motwani M, Rizzo C, Sirotnak F, She Y, Schwartz GK. Flavopiridol enhances the effect of docetaxel in vitro and in vivo in human gastric cancer cells. Mol Cancer Ther 2003; 2:549 - 55; PMID: 12813134
- Fornier MN, Rathkopf D, Shah M, Patil S, O’Reilly E, Tse AN, et al. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res 2007; 13:5841 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-07-1218; PMID: 17908977
- Shah MA, Kortmansky J, Motwani M, Drobnjak M, Gonen M, Yi S, et al. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clin Cancer Res 2005; 11:3836 - 45; http://dx.doi.org/10.1158/1078-0432.CCR-04-2651; PMID: 15897584
- Bible KC, Lensing JL, Nelson SA, Lee YK, Reid JM, Ames MM, et al. Phase 1 trial of flavopiridol combined with cisplatin or carboplatin in patients with advanced malignancies with the assessment of pharmacokinetic and pharmacodynamic end points. Clin Cancer Res 2005; 11:5935 - 41; http://dx.doi.org/10.1158/1078-0432.CCR-04-2566; PMID: 16115936
- George S, Kasimis BS, Cogswell J, Schwarzenberger P, Shapiro GI, Fidias P, et al. Phase I study of flavopiridol in combination with Paclitaxel and Carboplatin in patients with non-small-cell lung cancer. Clin Lung Cancer 2008; 9:160 - 5; http://dx.doi.org/10.3816/CLC.2008.n.024; PMID: 18621626
- Welch S, Hirte HW, Carey MS, Hotte SJ, Tsao MS, Brown S, et al. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: a study of the Princess Margaret Hospital Phase II consortium. Gynecol Oncol 2007; 106:305 - 10; http://dx.doi.org/10.1016/j.ygyno.2007.02.018; PMID: 17537491
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 2000; 60:2108 - 12; PMID: 10786669
- Monks A, Harris ED, Vaigro-Wolff A, Hose CD, Connelly JW, Sausville EA. UCN-01 enhances the in vitro toxicity of clinical agents in human tumor cell lines. Invest New Drugs 2000; 18:95 - 107; http://dx.doi.org/10.1023/A:1006313611677; PMID: 10857990
- Kortmansky J, Shah MA, Kaubisch A, Weyerbacher A, Yi S, Tong W, et al. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J Clin Oncol 2005; 23:1875 - 84; http://dx.doi.org/10.1200/JCO.2005.03.116; PMID: 15699481
- Sausville EA, Arbuck SG, Messmann R, Headlee D, Bauer KS, Lush RM, et al. Phase I trial of 72-hour continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 2001; 19:2319 - 33; PMID: 11304786
- Meijer L, Raymond E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc Chem Res 2003; 36:417 - 25; http://dx.doi.org/10.1021/ar0201198; PMID: 12809528
- Benson C, White J, De Bono J, O’Donnell A, Raynaud F, Cruickshank C, et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer 2007; 96:29 - 37; http://dx.doi.org/10.1038/sj.bjc.6603509; PMID: 17179992
- Bettayeb K, Sallam H, Ferandin Y, Popowycz F, Fournet G, Hassan M, et al. N-&-N, a new class of cell death-inducing kinase inhibitors derived from the purine roscovitine. Mol Cancer Ther 2008; 7:2713 - 24; http://dx.doi.org/10.1158/1535-7163.MCT-08-0080; PMID: 18790752
- Müller-Tidow C, Metzger R, Kügler K, Diederichs S, Idos G, Thomas M, et al. Cyclin E is the only cyclin-dependent kinase 2-associated cyclin that predicts metastasis and survival in early stage non-small cell lung cancer. Cancer Res 2001; 61:647 - 53; PMID: 11212263
- Hunt KK, Keyomarsi K. Cyclin E as a prognostic and predictive marker in breast cancer. Semin Cancer Biol 2005; 15:319 - 26; http://dx.doi.org/10.1016/j.semcancer.2005.04.007; PMID: 16043362
- Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest New Drugs 2008; 26:59 - 65; http://dx.doi.org/10.1007/s10637-007-9090-3; PMID: 17938863
- Siemeister G, Luecking U, Wagner C, Detjen K, Mc Coy C, Bosslet K. Molecular and pharmacodynamic characteristics of the novel multi-target tumor growth inhibitor ZK 304709. Biomed Pharmacother 2006; 60:269 - 72; http://dx.doi.org/10.1016/j.biopha.2006.06.003; PMID: 16887322
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427 - 38; PMID: 15542782
- Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, et al. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res 2008; 68:5519 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-07-6404; PMID: 18632601
- McMillin DW, Delmore J, Negri J, Buon L, Jacobs HM, Laubach J, et al. Molecular and cellular effects of multi-targeted cyclin-dependent kinase inhibition in myeloma: biological and clinical implications. Br J Haematol 2011; 152:420 - 32; http://dx.doi.org/10.1111/j.1365-2141.2010.08427.x; PMID: 21223249
- Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol 2005; 23:6333 - 8; http://dx.doi.org/10.1200/JCO.2005.05.021; PMID: 16155016
- Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood 2004; 104:607 - 18; http://dx.doi.org/10.1182/blood-2004-01-0037; PMID: 15090448
- Ely S, Di Liberto M, Niesvizky R, Baughn LB, Cho HJ, Hatada EN, et al. Mutually exclusive cyclin-dependent kinase 4/cyclin D1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res 2005; 65:11345 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-05-2159; PMID: 16357141
- Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005; 106:1042 - 7; http://dx.doi.org/10.1182/blood-2005-01-0320; PMID: 15827128
- Raje N, Hideshima T, Mukherjee S, Raab M, Vallet S, Chhetri S, et al. Preclinical activity of P276-00, a novel small-molecule cyclin-dependent kinase inhibitor in the therapy of multiple myeloma. Leukemia 2009; 23:961 - 70; http://dx.doi.org/10.1038/leu.2008.378; PMID: 19151776
- Gojo I, Zhang B, Fenton RG. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res 2002; 8:3527 - 38; PMID: 12429644
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res 2005; 65:5399 - 407; http://dx.doi.org/10.1158/0008-5472.CAN-05-0233; PMID: 15958589
- Cirstea D, Hideshima T, Pozzi S, Vallet S, Ikeda H, Santo L, et al. RGB 286638, a Novel Multi-Targeted Small Molecule Inhibitor, Induces Multiple Myeloma (MM) Cell Death through Abrogation of CDK Dependent and Independent Survival Mechanisms. [ASH Annual Meeting Abstracts] Blood 2008; 112:2759
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004; 29:95 - 102; http://dx.doi.org/10.1016/j.tibs.2003.12.004; PMID: 15102436
- Wyatt PG, Woodhead AJ, Berdini V, Boulstridge JA, Carr MG, Cross DM, et al. Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J Med Chem 2008; 51:4986 - 99; http://dx.doi.org/10.1021/jm800382h; PMID: 18656911
- Squires MS, Feltell RE, Wallis NG, Lewis EJ, Smith DM, Cross DM, et al. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol Cancer Ther 2009; 8:324 - 32; http://dx.doi.org/10.1158/1535-7163.MCT-08-0890; PMID: 19174555
- Cirstea D, Vallet S, Raje N. Future novel single agent and combination therapies. Cancer J 2009; 15:511 - 8; http://dx.doi.org/10.1097/PPO.0b013e3181c51c8e; PMID: 20010171