Abstract
Accumulating evidence suggests that chemopreventive effects of some dietary polyphenols may in part be mediated by their ability to influence epigenetic mechanisms in cancer cells. Boswellic acids, derived from the plant Boswellia serrata, have long been used for the treatment of various inflammatory diseases due to their potent anti-inflammatory activities. Recent preclinical studies have also suggested that this compound has anti-cancer potential against various malignancies. However, the precise molecular mechanisms underlying their anti-cancer effects remain elusive. Herein, we report that boswellic acids modulate DNA methylation status of several tumor suppressor genes in colorectal cancer (CRC) cells. We treated RKO, SW48 and SW480 CRC cell lines with the active principle present in boswellic acids, acetyl-keto-β-boswellic acid (AKBA). Using genome-wide DNA methylation and gene expression microarray analyses, we discovered that AKBA induced a modest genome-wide demethylation that permitted simultaneous re-activation of the corresponding tumor suppressor genes. The quantitative methylation-specific PCR and RT-PCR validated the gene demethylation and re-expression in several putative tumor suppressor genes including SAMD14 and SMPD3. Furthermore, AKBA inhibited DNMT activity in CRC cells. Taken together, these results lend further support to the growing notion that anti-cancer effect of boswellic acids may in part be due to its ability to demethylate and reactivate methylation-silenced tumor suppressor genes. These results suggest that not only boswellic acid might be a promising epigenetic modulator in the chemoprevention and treatment of CRC, but also provide a rationale for future investigations on the usefulness of such botanicals for epigenetic therapy in other human malignancies.
Introduction
Boswellia extracts, derived from the plant Boswellia serrata, have long been known for their anti-inflammatory effects in the treatment of arthritis, ulcerative colitis and Crohn disease.Citation1-Citation5 Acetyl-11-keto-β-boswellic acid (AKBA), one of the major active boswellic acids present in boswellia extracts, is a pentacyclic terpenoid, and has also been shown to have anti-tumor effects in different types of tumor cells including colon,Citation6,Citation7 prostate,Citation8,Citation9 leukocytes,Citation10 liver,Citation11 and brain.Citation12 Several earlier studies have reported inhibitory effects of AKBA on the nuclear factor kappa-B (NF-κB)Citation13 and the signal transducer and activator of transcription 3 (STAT-3)-related pathways,Citation14 which potentiates apoptosis and inhibits angiogenesis in neoplastic cells. However, the precise molecular mechanisms underlying the anti-tumor effects of AKBA on colorectal cancer (CRC) still remain elusive.
DNA hypermethylation of CpG islands located within the promoter regions of tumor suppressor genes is a common epigenetic alteration involved in most human cancers. Unlike genomic alterations, aberrant methylation of methylation-silenced genes is potentially reversible with therapeutic agents. A DNA methyltransferase (DNMT) inhibitor, 5-aza-2’-deoxycytidine (DAC), which reverses DNA hypermethylation, has recently been used clinically for treating human malignancies such as myelodysplastic syndromes, leading to an improvement in the overall clinical outcome of patients with these diseases.Citation15,Citation16 Given that epigenetic alterations are often present in the earliest stages of cancer, even in the normal-appearing mucosa, there is a growing enthusiasm that demethylating agents could be more beneficial for cancer prevention instead for use in chemotherapy for advanced cancers.Citation17,Citation18 Indeed, DAC and zebularine, an oral DNA methylation inhibitor, have been shown to prevent tumor formation in the Apc Min/+ mouse model.Citation19,Citation20 In spite of its promise, DAC treatment is often associated with several side effects including myelosuppression, nausea, vomiting and diarrhea,Citation15,Citation16 which limits its clinical usefulness, especially for cancer chemoprevention. Another shortcoming of DAC is the manifestation of non-specific demethylation of non-tumor suppressor genes, and possible re-activation of various methylation-silenced proto-oncogenes in humans.Citation21 In view of this, there is a growing desire to identify demethylating agents that not only lack adverse side effects, but preferentially demethylate tumor suppressor genes that enhance cancer treatment, and particularly, genes involved in cancer prevention.
In this regard, a growing body of literature supports that natural dietary compounds generally have multiple molecular targets within cancer cells, and are ordinarily considered quite safe. Consequently, there is a renewed interest to understand the molecular mechanisms underlying anticancer effects of several promising dietary botanicals, with the hope that some of these may eventually be used for cancer chemoprevention and treatment in future. Although limited, recent evidence indicates that chemopreventive potential of several dietary polyphenols may in part be mediated by their ability to reactivate methylation-silenced tumor suppressor genes in cancer cells.Citation22
In the present study, we hypothesized that AKBA might exert its anti-tumor effects in CRC cells partly by modulating methylation status of various tumor suppressor genes. Using both array-based and candidate-gene approaches for the determination of methylation and gene expression status, herein, we provide novel evidence that in addition to its inhibitory effects on cellular signaling pathways, the anti-cancer effects of AKBA are in part due to its ability to induce demethylation and subsequent reactivation of putative tumor suppressor genes in CRC cells.
Results
AKBA inhibits cell viability and proliferation, and induces apoptosis in CRC cells
To elucidate whether AKBA (the molecular structure: ) has any growth inhibitory effects in CRC cells, we performed MTT assays to determine cell viability in RKO, SW48, and SW480 cell lines treated with boswellic acid. We found that AKBA treatment for 72 h resulted in a dose-dependent growth inhibition in all three CRC cell lines: 1–16% with 5 μM doses, 10–18% with 10 μM, 35–46% with 20 μM, and 89–98% with 40 μM AKBA [in all CRC cell lines vs. control cell lines ()]. To further confirm the inhibitory effects of AKBA on cell proliferation, we performed BrdU assays and observed a similar dose-dependent effect on cell proliferation in all cell lines. There was little effect of AKBA in RKO and SW480 cells treated with 5 or 10 μM concentrations, but we observed a 12–53% inhibition in cell proliferation with the 20 μM doses and a 91–99% inhibition with 40 μM AKBA in all CRC cell lines after 96 h of treatment ().
Figure 1. AKBA exerts anticancer effects in CRC cell lines. (A) The molecular structure of AKBA. (B) AKBA treatment inhibits cell viability in RKO, SW48 and SW480 cells using a MTT assay. (C) AKBA reduces proliferation of CRC cells in a BrdU assay. AKBA induces apoptosis in RKO (D), SW48 (E) and SW480 (F) cells in an immunofluorescence staining assay that uses an anti-active caspase-3 antibody. Data are represented as mean ± standard error of mean (SEM) from three independent experiments.
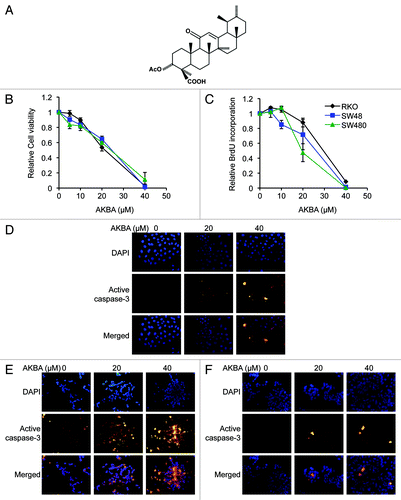
Furthermore, to determine whether the growth inhibition observed following AKBA treatment in CRC cell lines might be due to the induction of apoptosis, we next performed an immunofluorescence assay using an anti-active caspase-3 antibody in RKO, SW48 and SW480 cell lines. As shown in , AKBA treatment increased the number of active caspase-3 positive cells, as evidenced by a greater number of bright yellow cells, in all three cell lines, in a dose-dependent manner. These results suggest that AKBA’s antitumor effects are mediated both by the induction of apoptosis as well as through inhibition of cell proliferation in CRC cells.
AKBA induces demethylation of several CpG loci in CRC cells
To study the effects of AKBA treatment on DNA methylation status in CRC, we next analyzed genome-wide DNA methylation patterns in control and AKBA-treated cell lines using Illumina’s Infinium HumanMethylation27 microarrays. For these experiments, we chose the SW48 CRC cell line for the initial genome-wide methylation analysis, because this cell line possesses a CpG island methylator phenotype (CIMP), which is characterized by aberrant hypermethylation of numerous CpG islands in the promoter regions of many tumor suppressor genes, including the MLH1 gene.Citation23 Methylation patterns of SW48 cells treated with DMSO alone (controls) were compared with cells treated with 2.5 μM DAC or 20 μM AKBA for 6 d. The scatter plots and the histograms illustrate that a large number of CpG sites had reduced methylation levels after both DAC and AKBA treatments compared with control cell lines (). The mean β-values of all CpG loci in SW48 cells treated with AKBA were significantly lower than those of control cells (0.366 for AKBA vs. 0.401 for controls, p < 0.0001 by two-tailed Student’s t-test). These results indicate that AKBA induced demethylation of a large number of CpG sites, although the demethylating activity of AKBA was markedly lower than the global demethylation induced by DAC (0.286; ).
Figure 2. AKBA demethylates CpG islands in CRC cells. (A) A scatter plot comparing the β-values of all CpG loci between control and DAC-treated (2.5 μM) SW48 CRC cells (top panel), and between control and AKBA (20 μM) treated cells (bottom panel). Colored dots represent β-value densities for each CpG locus. The histograms represent mean β-value distribution in SW48 cells treated with DAC (top panel) and those treated with AKBA for 6 d (bottom panel). The horizontal line within the box represents the median values. The upper and lower lines in the box plot represent 75th and 25th percentiles. The horizontal bars above and below the boxes represent the maximum and the minimum values. The small black box within each box plot represents median β-values. The differences in the mean β-values between the control, DAC and AKBA treated SW48 cells were analyzed by use of a two-tailed Student’s t-test. (B) A heat map illustration of the DNA methylation pattern of the control and AKBA treated cells (left panel). The gradient of blue to yellow represents individual β-values, and the degree of methylation from low (blue) to high (yellow; 0 to 1) as indicated. The three clusters of genes that demonstrated the most significant DNA demethylation after AKBA treatment are shown with red boxes. A close-up view of the heat maps corresponding to the subset of demethylated genes in the clusters 1, 2 and 3 are represented in the right panel. (C) The scatter plot of the β-values from the gene clusters 1, 2 and 3 (red, green and blue respectively) in AKBA vs control cells.
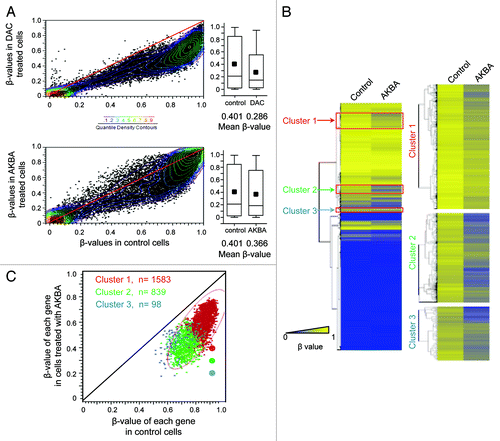
We were interested in elucidating whether a specific subset of genes existed that was preferentially demethylated following AKBA treatment in CRC cell lines. Hierarchical clustering analysis successfully isolated three clusters of genes, “cluster 1” (n = 1583), “cluster 2” (n = 839) and “cluster 3” (n = 98), which were most prominently demethylated in AKBA treated CRC cells (). Our observations for the existence of localized demethylation changes in a significant proportion of genes suggests that perhaps certain subsets of genes are more susceptible to such DNA demethylating activity in AKBA treated cells.
A subset of genes is demethylated and upregulated by AKBA treatment
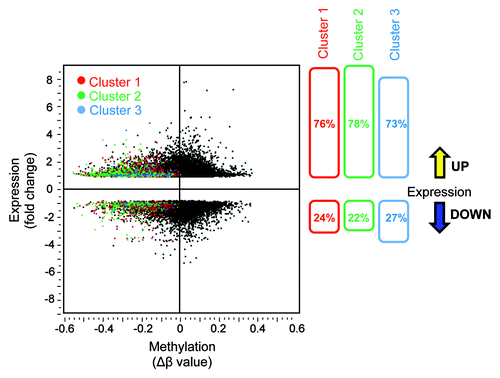
To better understand the biological relevance of AKBA-induced demethylation, and to identify the subsets of genes in which demethylation correlated with their reactivation, we performed genome-wide gene expression analysis using the Illumina’s HumanHT-12 v4 Expression BeadChip microarrays in SW48 cells treated with AKBA. The data output of gene expression arrays was then compared with the β-values from the global methylation arrays by scatter-plot analysis (). When we combined gene methylation and expression array data, 76% of the genes in cluster 1, 78% in cluster 2, and 73% in cluster 3 exhibited a simultaneous decrease in methylation and a corresponding increase in the corresponding gene expression. These data suggest that the expression of the majority of genes in these three clusters were actually upregulated when demethylated.
Figure 3. Correlation between DNA demethylation and re-expression of the corresponding genes in AKBA treated CRC cells. The scatter plot illustration of DNA methylation (β-values) data obtained from genome-wide methylation arrays and the changes in gene expression analyzed by microarray analyses in SW48 cells treated with AKBA. The methylation status after AKBA treatment is shown for each gene from clusters 1, 2 or 3 (colored red, green or blue, respectively, left panel). As shown in the right panel, the majority of demethylated genes within each of the three clusters were upregulated after AKBA treatment (76, 78, and 73% respectively).
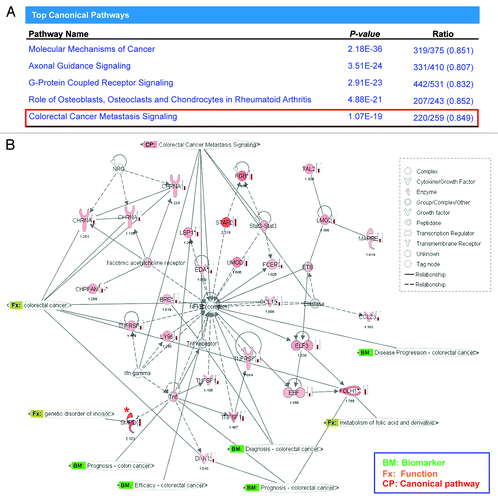
Next, using the gene expression results from the three clusters, we performed Ingenuity Pathway Analysis (IPA) to determine whether there were any specific cell-signaling pathways that are preferentially affected by AKBA treatment. IPA analysis revealed that AKBA treatment of CRC cells modulated five different canonical pathways most profoundly. Based upon the categorical configuration within IPA, these 5 pathways were classified as: (1) molecular mechanisms of cancer, (2) axonal guidance signaling, (3) G-protein coupled receptor signaling, (4) role of osteoblasts, osteoclasts and chondrocytes in rheumatoid arthritis, and (5) CRC metastasis signaling (). Most of the genes modulated by AKBA treatment were listed as part of the CRC metastasis signaling pathway. These genes were related to NF-κB and tumor necrosis factor, both of which have previously been shown to be targets of AKBA in other human cancers ().
Figure 4. The pathway analysis revealed the most affected molecular pathway by AKBA. (A) The pathway analysis revealed that CRC metastasis signaling was among one of the top five canonical pathways that served as a target of AKBA-induced demethylation in CRC cells. (B) An illustration from Ingenuity Pathway Analysis (IPA) demonstrating the biological relationship between demethylated and overexpressed genes from the 3 genes clusters. Green bolded BM represents the genes that are described as biomarkers of CRC, Orange Fx represents the genes that have direct function in CRC, and CP represents the canonical pathway involved in CRC metastasis signaling. One of the genes, SMPD3, which was found within the most demethylated gene group “cluster 1” in the methylation microarray analysis and subsequently confirmed in our validation analysis (expression by qRT-PCR and methylation by qMSP), is marked with “*” in red. Density of red color of icons and the number below the icons represents the fold change of gene expression induced by AKBA obtained in our genome-wide gene expression analysis.
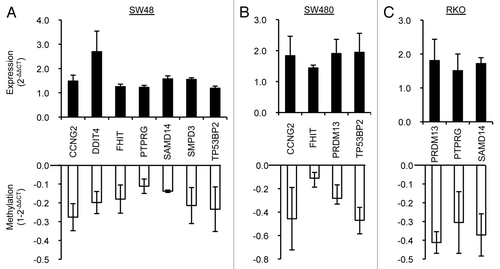
In order to narrow down the list of AKBA-targeted genes in CRC, we focused our attention on those genes that have a confirmed or putative tumor suppressor role in cancer, contain CpG islands in their promoter region, and demonstrated at least a 2-fold increase in gene expression following AKBA treatment. Accordingly, we selected nine putative tumor suppressor genes that met these criteria: cyclin G2 (CCNG2), DNA damage-inducible transcript 4 (DDIT4), death effector domain-containing protein (DEDD), fragile histidine triad gene (FHIT), PR domain containing 13 (PRDM13), protein-tyrosine phosphatase gamma (PTPRG), sterile α motif domain containing 14 (SAMD14), sphingomyelin phosphodiesterase 3 (SMPD3), tumor protein p53-binding protein 2 (TP53BP2). To confirm and validate the demethylation and upregulated gene expression of these nine putative tumor suppressor genes, we analyzed methylation levels by quantitative methylation-specific PCR (MSP) and gene expression levels by real-time RT-PCR in SW48, RKO and SW480 cell lines treated with AKBA. These experiments revealed that six of the nine genes were indeed upregulated and demethylated in two of the three AKBA-treated CRC cells (). Among all nine genes, SAMD14 and SMPD3, both of which were included in cluster 1 in the methylation microarray analysis (), were shown to be demethylated and upregulated in SW48 cells by the validation analysis (). SAMD14 is downregulated in various human lung adenocarcinoma cell lines and its promoter region is frequently hypermethylated in early invasive pulmonary adenocarcinoma.Citation24 Similarly, SMPD3 was one of the genes whose methylation and expression levels were found to be modulated in the CRC metastasis signaling pathway identified by IPA analysis (). Overexpression of SMPD3 leads to an inhibition of cell growth in the human breast cancer cell line MCF7,Citation25 and the promoter region of SMPD3 has been reported to be hypermethylated in human breast cancers.Citation26 The demethylation and upregulation of putative tumor suppressor genes could help explain the anti-cancer effects of AKBA.
Figure 5. Validation of DNA methylation and gene expression status of the selected putative tumor suppressor genes in three CRC cell lines. Re-expression and demethylation of nine putative tumor suppressor genes were confirmed by real-time RT-PCR and quantitative MSP in three CRC cell lines: SW48 (A), SW480 (B) and RKO (C) treated with 20 μM AKBA (SW48 and RKO) or 30 μM of AKBA (SW480) for 6 d. Six of the nine genes (CCNG2, FHIT, PTPRG, PRDM13, SAMD14 and TP53BP2) were upregulated along with demethylation in at least in two of the cell lines. Data are represented as mean ± SEM from two or three independent experiments.
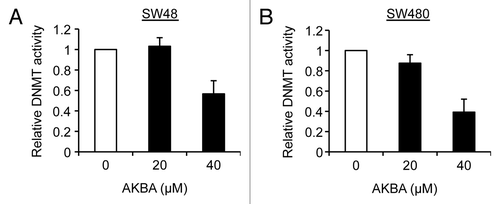
AKBA inhibits DNMT activity
Since DNMT catalyzes the process of methylation, and to further clarify the mechanisms underlying the demethylating activity of AKBA, we aimed to elucidate whether AKBA could inhibit DNMT activity in CRC cells. Use of the DNMT inhibition assay demonstrated that AKBA markedly inhibited DNMT activities in SW48 and SW480 cell lines at 40 μM concentrations, while these effects were modest at 20 μM concentrations (). We also observed that treatment with the classical DNMT inhibitor, DAC (5 μM) resulted in an inhibition (~25%) of DNMT activity in these cell lines (data not shown). Taken together, the results of our experiments show that AKBA seems to have a rather modest inhibitory effect on DNMT activity, which is consistent with previous observations for other botanical compounds.Citation22
Figure 6. AKBA inhibits DNMT activity in CRC cells. DNMT activity was measured in SW48 (A) and SW480 (B) cells treated with DMSO, 20 μM AKBA and 40 μM AKBA for three days. Data are represented as mean ± SEM from three independent experiments.
Discussion
The data presented in this study provides first evidence for the involvement of epigenetic mechanisms underlying the chemopreventative effects of boswellic acids in CRC. Using a series of genome-wide DNA methylation and gene expression analyses, we discovered that AKBA not only induced demethylation of a large number of CpG loci, but the demethylation was associated with a concurrent upregulation of genes that may be involved in CRC carcinogenesis. Our results build upon the existing knowledge for the boswellic acid-induced inhibition of proteins involved in other intracellular signaling pathways, such as NF-κB and STAT-3 that lend additional support in favor of the anticancer effects of this compound in CRC.Citation13,Citation14 Our observations that AKBA caused demethylation and re-activation of putative tumor suppressor genes such as SAMD14 and SMPD3, provides a novel mechanism by which boswellic acid exert its anticancer effects in cancer cells.
DNA hypermethylation of CpG sites in promoter regions of tumor suppressor genes is one of the key mechanisms that cause transcriptional silencing of the corresponding genes in cancer cells. A subset of 30–40% CRC tumors can be characterized by the existence of CIMP, in which several tumor suppressor genes with CpG islands in their promoters are frequent targets of aberrant hypermethylation.Citation27,Citation28 CIMP-positive CRCs associate with proximally located tumors, presence of microsatellite instability (MSI) and existence of BRAF mutations. Although CIMP CRCs significantly associates with sporadic MSI-high tumors due to MLH1 hypermethylation, aberrant methylation is not limited to these tumors and can be observed in other subsets of CRCs including the ones with chromosomal instability (CIN) phenotype.Citation29 In view of this, we studied a panel of CRC cell lines that were representative of all three subsets of CRCs, which included SW48 and RKO cells (CIMP and MSI-high) and SW480 cells (CIN). Our initial hypothesis was that we would perhaps observe most noticeable changes in AKBA-induced reversal of methylation in CIMP cell lines due to the high frequency and levels of methylated genes in these cells. But at the same time, we also included a non-CIMP or CIN cell line for our validation analysis, to ascertain the demethylating potential of AKBA against these cells, which represent about 50–60% of all CRCs. Indeed, our screening results were supportive of our hypothesis, wherein our genome-wide methylation analysis revealed that AKBA induced demethylation of a large number of CpG loci in the CIMP-positive SW48 cells, although the level of demethylation was relatively modest compared with DAC. These findings were further corroborated by our observations that DAC apparently also induced hypomethylation of the LINE-1 elements, which are considered to be markers for global methylation changes, while AKBA did not (data not shown). Another strength of our study is that we successfully validated the preferential demethylation of various tumor suppressor genes along with their upregulation following AKBA treatment in multiple cell lines, in spite of the genetic and epigenetic diversity of these CRC cells. In the SW48 cells, the validation results for most genes (7 of 9) were concordant with our screening array data from independently treated cell lines. More importantly, the majority of genes (6 of 9) were confirmed to be demethylated and upregulated in at least two of the three cell lines following multiple independent treatments. Furthermore, it was interesting to observe that AKBA induced demethylation in some genes even in the non-CIMP SW480 cells, since non-CIMP-related genes are frequently methylated even in CIN phenotype tumors.Citation28,Citation29 Our observations for the definitive demethylation of a subset of genes in the face of a modest generalized demethylation induced by AKBA may be of significance from a cancer chemoprevention perspective, in which long-term drug use may be required, to avoid secondary tumor occurrence resulting from the excess and continuous genome-wide hypomethylation as observed in a DNMT1-deficient mice model.Citation30,Citation31 In addition, we noticed that compared with modest effect seen with 20 μM AKBA, treatment at 40 μM concentrations resulted in marked inhibition of DNMT activity. These results are not entirely surprising considering that in comparison to direct DNMT inhibitors such as DAC, AKBA and other natural dietary compounds may induce DNMT inhibition through an indirect mechanism. Consequently, the precise mechanisms by which AKBA treatment causes demethylation and DNMT inhibition, as well as the optimal doses and/or frequencies of AKBA administration for its demethylating activity in preclinical animal models need to be clarified in future studies.
Although a number of attempts have been made for chemoprevention of CRC, such efforts have met with limited success, and as a result no chemopreventive drugs are widely accepted for clinical use. One of the best-studied drugs for chemopreventive effect on CRC is aspirin. Aspirin’s antitumor effects are primarily attributed to its ability to inhibit cyclooxygenase-2 (COX-2), and some data suggest that it may have a preventive effect on adenoma formation and CRC occurrence in humans in long-term observational studies.Citation32 However, the long-term use of aspirin can also cause some toxic effects including gastroduodenal ulcers and bleeding.Citation32 Selective COX-2 inhibitors, such as celecoxib, have more potent chemopreventive effects and less gastrointestinal toxicities than aspirin, however, these drugs associate with increased risk of cardiovascular events.Citation33 Consequently, specific COX-2 inhibitors have not been accepted for chemopreventive use in general populations either. One of the biggest hurdles for chemoprevention against general populations is the use of a compound that has not toxicity. In this regard, boswellic acids, like several other botanical-derived compounds, seem to extremely safe, and hence may provide a more rational use for cancer chemoprevention.
Moreover, this botanical has the ability to target a wide range of genes/pathways that affect malignant properties in cancer cells including the demethylating activity against a subset of tumor suppressor genes as demonstrated in our present study. Molecular mono-targeted drugs, such as antibodies against EGFR and VEGF, have recently been used for the treatment of patients with metastatic CRC. However, benefits drawn from such mono-targeted agents are limited, and furthermore resistance to these antibodies always occurs with increased duration of treatment.Citation34 CRC arises as a consequence of stepwise accumulation of a multitude of genetic and epigenetic alterations, therefore, multi-targeted therapies that include targeting of both genetic and epigenetic pathways, could be more beneficial to treat this disease compared with a mono-targeted therapy.Citation35,Citation36 In light of accumulating evidence and our findings in this study, clinical validation on the efficacy of boswellic acids for both chemoprevention and therapy against CRC could be promising.
In conclusion, we have shown that AKBA induces demethylation and concurrent upregulation of a subset of tumor suppressor genes, which may contribute to its anticancer effects in CRC cells. Thus, boswellic acid could be useful not only as an epigenetic modulator for the treatment of patients with CRC, it may be effective against other malignancies. Our data provide the rationale for further investigation of this compound, as well as other botanicals, for therapeutic interventions against human cancers.
Materials and Methods
Cell Culture
Human CRC cell lines, RKO, SW48 and SW480 were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Iscove's modified Dulbecco's medium (IMDM, Invitrogen) with 10% fetal bovine serum. Cells were treated with 0.1% DMSO (vehicle controls) or AKBA (Sigma Chemical Co.) at a dose range of 0–40 μM for 6 d. The medium containing DMSO or AKBA was changed every 2 d and in the demethylation experiments, cells were treated with DMSO, AKBA or DAC (2.5 μM) for a total of 72 h.
MTT Assay
The growth inhibitory effect of AKBA on cells was evaluated by the use of 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) assay. Human CRC cell lines were plated in 96-well plates at a density of 5.0 × 103 cells/well in 100 μl of IMDM, and incubated for 24 h. Cells were then treated with AKBA for 72 h with the final concentrations of 5, 10, 20, or 40 μM. Following treatment with AKBA, cells were incubated with 100 μl of 0.5 mg/ml MTT in medium for 3 h at 37°C. The cells were lysed for 12 h at room temperature in a buffer containing 10% SDS and 0.01 N HCl. For each sample, the optical density of the reduced intracellular formazan product was read at 570 nm in a spectrophotometer. Each assay was performed in triplicate in three independent experiments.
BrdU Cell Proliferation Assay
The proliferation index was measured by bromodeoxyuridine (BrdU) incorporation in CRC cells 96 h after AKBA treatment as described above, following the manufacturer's instructions (Cell Proliferation ELISA, BrdU, Roche Diagnositics). Each experiment was performed in triplicate, and the data were calculated based upon three independent experiments.
Immunofluorescence
Cells were cultured on coverslips plated in 12-well plates at a density of 0.5 x 105 cells in 1 ml of IMDM. After attached, cells were treated for an additional 48 h with 0, 20 or 40 μM of AKBA. Cells cultured on coverslips were then fixed with 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.3% Triton X-100 in PBS for 5 min, and blocked with 5% goat serum for 1 h. Cells were incubated with a rabbit anti-active caspase 3 antibody (1:500; Promega) at room temperature for 1 h, washed with PBS for 3 times, followed by a goat anti-rabbit IgG secondary antibody for immunofluorescence staining (1:800) for 30 min at room temperature. Thereafter, slides were washed with PBS three times, followed by nuclear staining with DAPI and mounted in appropriate medium for fluorescence microscopy. The images were taken with an AxioSkop2 multichannel epifluorescence microscope equipped with AxioVision software (Carl Zeiss).
DNA and RNA extraction
DNA was extracted from RKO, SW48 and SW480 cells treated with DMSO alone or with AKBA by use of the QIAmp DNA mini kit (Qiagen). Total RNA was extracted from the cell extracts with the RNeasy Mini kit (Qiagen). The cDNA was obtained using the Advantage RT-for-PCR kit (Clontech).
Genome-wide DNA promoter methylation and gene expression analyses
Genome-wide DNA promoter methylation status in control and AKBA treated SW48 cells were analyzed using the Infinium HumanMethylation27 BeadChip microarrays (Illumina, Inc.). These methylation microarrays span across 28,000 CpG sites that are located within the promoter regions of ~14,475 genes based upon the NCBI Database (Genome Build 36). On an average, two assays are designed for each gene that includes anywhere between 3–20 CpG sites. The methylation level in each CpG site was given a β-value ranging from 0 (indicating no methylation) to 1 (100% methylation). Genome-wide gene expression status was analyzed using Illumina’s HumanHT-12 v4 BeadChips, which included more than 47,000 probes. SW48 cells treated with 2.5 μM DAC (Sigma-Aldrich) were used as positive controls for demethylation.
Quantitative methylation-specific polymerase chain reaction
The DNA methylation status of CCNG2, DDIT4, DEDD, FHIT, PRDM13, PTPRG, SAMD14, SMPD3, TP53BP2, and the collagen, type II, α 1 (COL2A1:reference geneCitation37) were determined by quantitative methylation-specific polymerase chain reaction (MSP) using primers specific for methylated sequences on a bisulfite-modified genomic DNA template (EZ DNA methylation Gold Kit, Zymo Research). Each reaction was performed in duplicate and the results were obtained from two or three independent assays. The 2(-ΔΔCT) method was used to present the data from the genes of interest relative to the controls. shows the primers’ sequences and the putative function of genes.Citation24-Citation26,Citation38-Citation46
Table 1. Primers used in this study
Quantitative real-time reverse transcription-polymerase chain reaction
Expression levels of CCNG2, DDIT4, DEDD, FHIT, PRDM13, PTPRG, SAMD14, SMPD3, TP53BP2 genes, and the reference β-actin gene were determined by use of the SYBR Green based-quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR). Real-time RT-PCR was performed in a 25-μL total volume containing 10 ng of cDNA, 12.5 μl of SYBR Green mix (Applied Biosystems), and 10 pmol of each primer. The following PCR cycle conditions were used: initial denaturation at 95°C for 10 min, followed by 45 cycles at 95°C for 15 sec, and 58°C for 1 min. The relative amounts of the genes of interest and β-actin were measured in two independent assays. The 2(-ΔΔCT) method was used to present the data on the genes of interest in treated cells relative to controls. The primers used are listed in .
DNMT inhibition assay
DNMT activity of CRC cell lines was measured in a DNMT activity/inhibition assay (Active Motif) according to the manufacturer’s protocol. In brief, SW48 and SW480 cells were treated with DMSO alone, 20 μM or 40 μM AKBA for 72 h. The medium with the drug was changed every 24 h. The nuclear extracts were prepared with the Nuclear Extract Kit (Active Motif). Each 10 μg of nuclear extract was incubated with the enzymatic buffer containing AdoMet for 2 h, followed by incubation with His-MBD2 protein (1:200 dilution) for 45 min. After another 45 min incubation with anti-polyHis-HRP antibody (1:1000 dilution), the developing solution was added to the extracts and the absorbance was read on a spectrophotometer at 450 nm. The data was obtained from three independent experiments.
Data Analysis
The Illumina DNA methylation data were analyzed using the open-source programs Cluster 3.0 and Java TreeView. Cluster 3.0 was used to perform an unsupervised hierarchical cluster analysis by applying complete linkage clustering with a Euclidean distance metric. Several analyses including the networks and functional analyses were performed through the use of Ingenuity Pathways Analysis (Ingenuity Systems, http://www.ingenuity.com/). A data set containing gene identifiers and corresponding expression values was uploaded into the application. Each identifier was mapped to its corresponding object in Ingenuity's Knowledge Base Networks of Network Eligible Molecules were then algorithmically generated based on their connectivity. Right-tailed Fisher’s exact test was used to calculate p values determining the probability that each biological function and/or disease assigned to that data set is due to chance alone. The basic statistic and scatter plots were done using JMP, Version 7 (SAS Institute Inc.). All microarray data reported in this article is MIAME compliant and that the raw data has been deposited in the GEO database and can be accessed at http://www.ncbi.nlm.nih.gov/geo/.
Acknowledgments
We thank Esperanza Anguiano and Mamta Sharma, from the Genomics Core Facility, Baylor Research Institute, Dallas, TX for their help with the genome-wide gene expression analysis. The authors owe profound thanks to Margaret Hinshelwood, PhD for her skillful and thorough editing of the manuscript. The present work was supported by grants R01 CA72851 and CA129286 from the National Cancer Institute, National Institutes of Health, and funds from the Baylor Research Institute.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Shah BA, Qazi GN, Taneja SC. Boswellic acids: a group of medicinally important compounds. Nat Prod Rep 2009; 26:72 - 89; http://dx.doi.org/10.1039/b809437n; PMID: 19374123
- Moussaieff A, Mechoulam R. Boswellia resin: from religious ceremonies to medical uses; a review of in-vitro, in-vivo and clinical trials. J Pharm Pharmacol 2009; 61:1281 - 93; PMID: 19814859
- Gupta I, Parihar A, Malhotra P, Singh GB, Lüdtke R, Safayhi H, et al. Effects of Boswellia serrata gum resin in patients with ulcerative colitis. Eur J Med Res 1997; 2:37 - 43; PMID: 9049593
- Sharma ML, Bani S, Singh GB. Anti-arthritic activity of boswellic acids in bovine serum albumin (BSA)-induced arthritis. Int J Immunopharmacol 1989; 11:647 - 52; http://dx.doi.org/10.1016/0192-0561(89)90150-1; PMID: 2807636
- Gerhardt H, Seifert F, Buvari P, Vogelsang H, Repges R. [Therapy of active Crohn disease with Boswellia serrata extract H 15]. Z Gastroenterol 2001; 39:11 - 7; http://dx.doi.org/10.1055/s-2001-10708; PMID: 11215357
- Liu JJ, Nilsson A, Oredsson S, Badmaev V, Zhao WZ, Duan RD. Boswellic acids trigger apoptosis via a pathway dependent on caspase-8 activation but independent on Fas/Fas ligand interaction in colon cancer HT-29 cells. Carcinogenesis 2002; 23:2087 - 93; http://dx.doi.org/10.1093/carcin/23.12.2087; PMID: 12507932
- Liu JJ, Huang B, Hooi SC. Acetyl-keto-beta-boswellic acid inhibits cellular proliferation through a p21-dependent pathway in colon cancer cells. Br J Pharmacol 2006; 148:1099 - 107; http://dx.doi.org/10.1038/sj.bjp.0706817; PMID: 16783403
- Lu M, Xia L, Hua H, Jing Y. Acetyl-keto-beta-boswellic acid induces apoptosis through a death receptor 5-mediated pathway in prostate cancer cells. Cancer Res 2008; 68:1180 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-07-2978; PMID: 18281494
- Pang X, Yi Z, Zhang X, Sung B, Qu W, Lian X, et al. Acetyl-11-keto-beta-boswellic acid inhibits prostate tumor growth by suppressing vascular endothelial growth factor receptor 2-mediated angiogenesis. Cancer Res 2009; 69:5893 - 900; http://dx.doi.org/10.1158/0008-5472.CAN-09-0755; PMID: 19567671
- Hostanska K, Daum G, Saller R. Cytostatic and apoptosis-inducing activity of boswellic acids toward malignant cell lines in vitro. Anticancer Res 2002; 22:2853 - 62; PMID: 12530009
- Liu JJ, Nilsson A, Oredsson S, Badmaev V, Duan RD. Keto- and acetyl-keto-boswellic acids inhibit proliferation and induce apoptosis in Hep G2 cells via a caspase-8 dependent pathway. Int J Mol Med 2002; 10:501 - 5; PMID: 12239601
- Glaser T, Winter S, Groscurth P, Safayhi H, Sailer ER, Ammon HP, et al. Boswellic acids and malignant glioma: induction of apoptosis but no modulation of drug sensitivity. Br J Cancer 1999; 80:756 - 65; http://dx.doi.org/10.1038/sj.bjc.6690419; PMID: 10360653
- Takada Y, Ichikawa H, Badmaev V, Aggarwal BB. Acetyl-11-keto-beta-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-kappa B and NF-kappa B-regulated gene expression. J Immunol 2006; 176:3127 - 40; PMID: 16493072
- Kunnumakkara AB, Nair AS, Sung B, Pandey MK, Aggarwal BB. Boswellic acid blocks signal transducers and activators of transcription 3 signaling, proliferation, and survival of multiple myeloma via the protein tyrosine phosphatase SHP-1. Mol Cancer Res 2009; 7:118 - 28; http://dx.doi.org/10.1158/1541-7786.MCR-08-0154; PMID: 19147543
- Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106:1794 - 803; http://dx.doi.org/10.1002/cncr.21792; PMID: 16532500
- Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007; 109:52 - 7; http://dx.doi.org/10.1182/blood-2006-05-021162; PMID: 16882708
- Kopelovich L, Crowell JA, Fay JR. The epigenome as a target for cancer chemoprevention. J Natl Cancer Inst 2003; 95:1747 - 57; http://dx.doi.org/10.1093/jnci/dig109; PMID: 14652236
- Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res (Phila) 2008; 1:219 - 22; http://dx.doi.org/10.1158/1940-6207.CAPR-08-0029; PMID: 19138962
- Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81:197 - 205; http://dx.doi.org/10.1016/0092-8674(95)90329-1; PMID: 7537636
- Yoo CB, Chuang JC, Byun HM, Egger G, Yang AS, Dubeau L, et al. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev Res (Phila) 2008; 1:233 - 40; http://dx.doi.org/10.1158/1940-6207.CAPR-07-0008; PMID: 19138966
- Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5:37 - 50; http://dx.doi.org/10.1038/nrd1930; PMID: 16485345
- Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol 2010; 80:1771 - 92; http://dx.doi.org/10.1016/j.bcp.2010.06.036; PMID: 20599773
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997; 57:808 - 11; PMID: 9041175
- Sun W, Iijima T, Kano J, Kobayashi H, Li D, Morishita Y, et al. Frequent aberrant methylation of the promoter region of sterile alpha motif domain 14 in pulmonary adenocarcinoma. Cancer Sci 2008; 99:2177 - 84; http://dx.doi.org/10.1111/j.1349-7006.2008.00965.x; PMID: 18823374
- Marchesini N, Luberto C, Hannun YA. Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J Biol Chem 2003; 278:13775 - 83; http://dx.doi.org/10.1074/jbc.M212262200; PMID: 12566438
- Demircan B, Dyer LM, Gerace M, Lobenhofer EK, Robertson KD, Brown KD. Comparative epigenomics of human and mouse mammary tumors. Genes Chromosomes Cancer 2009; 48:83 - 97; http://dx.doi.org/10.1002/gcc.20620; PMID: 18836996
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96:8681 - 6; http://dx.doi.org/10.1073/pnas.96.15.8681; PMID: 10411935
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38:787 - 93; http://dx.doi.org/10.1038/ng1834; PMID: 16804544
- Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology 2007; 132:127 - 38; http://dx.doi.org/10.1053/j.gastro.2006.09.018; PMID: 17087942
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455; http://dx.doi.org/10.1126/science.1083557; PMID: 12702868
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489 - 92; http://dx.doi.org/10.1126/science.1083558; PMID: 12702876
- Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol 2009; 10:501 - 7; http://dx.doi.org/10.1016/S1470-2045(09)70035-X; PMID: 19410194
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, et al, APC Study Investigators. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 2006; 355:873 - 84; http://dx.doi.org/10.1056/NEJMoa061355; PMID: 16943400
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010; 28:1254 - 61; http://dx.doi.org/10.1200/JCO.2009.24.6116; PMID: 20100961
- Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138:2073 - 87, e3; http://dx.doi.org/10.1053/j.gastro.2009.12.064; PMID: 20420947
- van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. J Clin Oncol 2011; 29:1382 - 91; http://dx.doi.org/10.1200/JCO.2010.28.2319; PMID: 21220596
- Kristensen LS, Mikeska T, Krypuy M, Dobrovic A. Sensitive Melting Analysis after Real Time- Methylation Specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. Nucleic Acids Res 2008; 36:e42; http://dx.doi.org/10.1093/nar/gkn113; PMID: 18344521
- Arachchige Don AS, Dallapiazza RF, Bennin DA, Brake T, Cowan CE, Horne MC. Cyclin G2 is a centrosome-associated nucleocytoplasmic shuttling protein that influences microtubule stability and induces a p53-dependent cell cycle arrest. Exp Cell Res 2006; 312:4181 - 204; http://dx.doi.org/10.1016/j.yexcr.2006.09.023; PMID: 17123511
- Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem 2008; 283:31378 - 84; http://dx.doi.org/10.1074/jbc.M803384200; PMID: 18796435
- Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A 2010; 107:4675 - 80; http://dx.doi.org/10.1073/pnas.0907705107; PMID: 20176937
- Schutte B, Henfling M, Ramaekers FC. DEDD association with cytokeratin filaments correlates with sensitivity to apoptosis. Apoptosis 2006; 11:1561 - 72; http://dx.doi.org/10.1007/s10495-006-9113-0; PMID: 16820959
- Arai S, Miyake K, Voit R, Nemoto S, Wakeland EK, Grummt I, et al. Death-effector domain-containing protein DEDD is an inhibitor of mitotic Cdk1/cyclin B1. Proc Natl Acad Sci U S A 2007; 104:2289 - 94; http://dx.doi.org/10.1073/pnas.0611167104; PMID: 17283331
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996; 84:587 - 97; http://dx.doi.org/10.1016/S0092-8674(00)81034-X; PMID: 8598045
- Behrends U, Schneider I, Rössler S, Frauenknecht H, Golbeck A, Lechner B, et al. Novel tumor antigens identified by autologous antibody screening of childhood medulloblastoma cDNA libraries. Int J Cancer 2003; 106:244 - 51; http://dx.doi.org/10.1002/ijc.11208; PMID: 12800201
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004; 304:1164 - 6; http://dx.doi.org/10.1126/science.1096096; PMID: 15155950
- Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8:781 - 94; http://dx.doi.org/10.1016/S1097-2765(01)00367-7; PMID: 11684014