Abstract
Mutual regulation of expression between p53 and AR has been reported. To further investigate the role of p53 in the regulation of AR expression, an ARE-Luciferase vector was inserted into LNCaP and into LNCaP-sip53 transfectants, and AR activity was quantitatively estimated after treatment with proteasome inhibitors. LNCaP expresses a mutated form of AR. Therefore, to investigate whether p53 can modulate the expression of wild-type (wt) of AR, we transfected PC3-wtAR with a p53 vector together with ARE-Luc and showed that p53 expression decreased DHT-dependent activity of wtAR. Since proteasomes also participate in AR transcriptional activity, we investigated the role of p53 in proteasome-dependent inhibition of AR activity. More than 80% of AR activity was inhibited by 3 μM of lactacystin in LNCaP whereas no inhibition was noted in LN-sip53. We also found that lactacystin decreased AR-DNA binding 3-fold in LNCaP but no binding decrease was observed in LN-sip53. Taken together, our data show that the inhibitory effects of proteasome inhibitors are dependent on p53 status, at least in prostate cancer. Therefore, the role of p53 during treatment with proteasome inhibitors in different tumors should be further investigated.
Keywords: :
Introduction
p53 is a regulator of genotoxic stress that plays an important role in DNA damage response, DNA repair, cell cycle regulation, and in triggering apoptosis after cell injury.Citation1 We have previously shown that there is mutual regulation of expression between p53 and AR.Citation2 By using cDNA microarray gene expression profiling we investigated the role of p53 in AR-dependent gene expression and found a 4-fold increase in PSA mRNA levels in LNCaP after suppression of p53.Citation3 Consistent with this, p53 suppression caused a 4- to 8-fold increase in secretion of PSA protein in culture medium, suggesting that PSA gene expression is under negative control of p53. Since LNCaP is considered a meaningful in vitro model of hormone-dependent prostate cancer, we can presume that the results obtained in vitro may reflect regulation of PSA in naturally occurring tumors. Thus, it appears that one of the most useful diagnostic prostate tumor markers may, in fact, also be a specific indicator of p53 inactivation in prostate cells. Being dependent on p53 inactivation, PSA elevation may therefore be indicative of the ongoing selection of p53-deficient cell variants with the dysregulation of apoptosis. In fact, the loss of functional p53 by LNCaP is accompanied not only by elevated PSA secretion but also by acquisition of high tumorigenecity and resistance to TNF-α-mediated apoptosis.Citation4
It remained unclear however, whether p53 directly regulated PSA expression or indirectly via AR. To answer this question, ARE-Luciferase vector was inserted into LNCaP and into three independent LNCaP-sip53 transfectants. Our data suggest that p53 modifies AR activity and this activity is affected by p53-mediated AR-DNA binding after treatment with proteasome inhibitors.
Results
P53 modulates transactivational activity of AR
To investigate the role of p53 in AR activity, cells were treated with different doses of doxorubicin (DOX) since it is well known that DOX increases the level of p53. We confirmed p53 elevation by western blot analysis () and estimation of AR activity showed that DOX treatment completely inhibits AR activity in LNCaP whereas in LNCaP-sip53 DOX inhibits AR activity only to a limited extent ().
Figure 1. (A) DOX induces p53 in LNCaP to higher level compared with LNCaP-sip53 transfectants. Cells were treated for 24 h with indicated doses of DOX and p53 expression was examined using western blot analysis. (B) Inhibition of p53 diminishes doxorubicin-dependent inhibition of AR activity. Cells were cultured for 3 d in SFC and then treated for 24 h with indicated doses of DOX in the presence of 10 nM DHT. To measure AR-dependent transactivation, pARE-Luc luciferase reporter construct was introduced in prostate cancer cell lines. To estimate luciferase activity, cells were harvested by trypsinization, washed in PBS, and lysed in reporter lysis buffer (200 µL, Promega). Luciferase chemiluminescence activity was measured using the luciferase assay kit (Promega). Sample aliquots (20 µL) were assayed for light emission with luminometer (MLX Dynex Technology, Inc.). The values of the luciferase assay were normalized with respect to protein concentration. (C) p53 decreases AR activity in PC3-AR. PC3-AR cells containing the wild type of AR were obtained from Dr. Theodore Brown, and was previously described.Citation13 PC3-AR cells containing ARE-Luc insert were transfected with pPS-wtp53 vector and 48 h after transfection were treated for 24 h with 10 nM DHT. p53 expression was confirmed by western blot analysis. AR activity was measured as described in legend to (B). (D) Inhibition of p53 decreases bicalutamide-dependent inhibition of AR activity. Cells were cultured for 3 d in SFC and then treated for 24 h with 10 nM DHT in the absence or presence of 50 μM bicalutamide. AR activity was measured as described in legend to (B). The levels of prostate specific antigen (PSA) secreted by LNCaP cells were assessed by PSA enzyme immunoassay kit (MP Biomedicals). (E) Antibody to p53 co-immunoprecipitates AR. Control cells were cultured in FCS or treated with 50 nM of lyposomal siRNA AR for 72 h. Two mg of cell lysates proteins were incubated overnight with goat antibodies to human p53 covalently bound to Sepharose beads (R&D Systems) and bound proteins were eluted using sample buffer. AR and p53 expression was examined using western blot analysis. (F) Pifithrin-α (PFT-α) but not pifithrin-μ (PFT-μ) increases AR activity. LNCaP was treated for 24 h with 10 nM DHT either alone or in the presense of 20 μM PFT-α or PFT-μ. AR activity was measured as described in legend to (B). Each point or column in Figures B–D and F represents mean value of four replicates in one of two separate experiments with similar results.
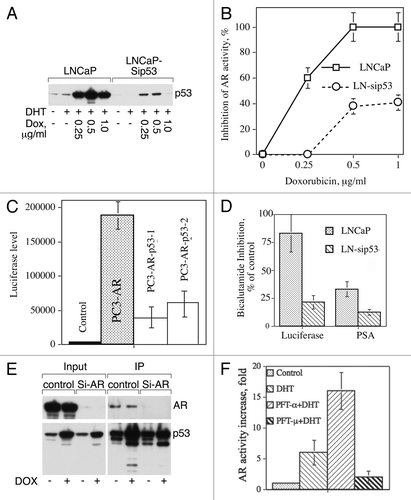
Knowing that LNCaP expresses a mutated form of AR, we investigated whether p53 can modulate the expression of wild-type (wt) AR. We transfected PC3-wtAR with p53 vector together with ARE-Luc and showed that p53 expression decreased DHT-dependent activity of wtAR (). These data suggest that p53 inhibits AR activity, and are in agreement with data of Alimirah, et al.Citation5 and Wang, et al.Citation6 We next asked whether p53 could affect bicalutamide-mediated inhibition of AR. We measured AR activity in these experiments both by luciferase reporter assay and by estimating the level of AR-dependent PSA. As shown in , in the absence of p53 the inhibitory effect of bicalutamide diminished by 2–3 fold. This suggests that p53 participates in the inhibitory effect of bicalutamide, i.e., physically interacts with AR and helps to keep AR in the proper conformation. As shown in , AR indeed co-immunoprecipitated with p53.
To investigate whether p53’s effect on AR activity is dependent on transactivation potential of p53 or on p53 interaction with cytosolic proteins, we used two inhibitors of p53, pifithrin-α and pifithrin-μ. Pifitrin-α was discovered for its ability to block p53-dependent transcriptional activationCitation7 whereas pifithrin-μ is an inhibitor of the p53-BclxL interaction.Citation8 As can be seen from , pifithrin-α increases AR activity, whereas treatment with pifithrin-μ failed to increase AR activity. Apparently, transactivation capacity of p53 plays a major role in modifying AR activity.
P53 modulates inhibitory effect of bicalutamide
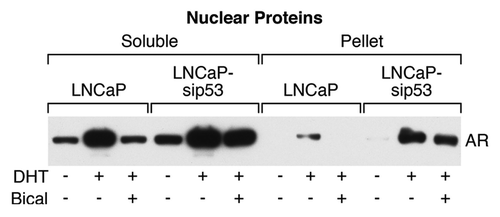
We next investigated the presence of AR in nuclei by dividing nuclear proteins into two fractions: soluble and insoluble (pellet); the latter fraction contains DNA and DNA-binding proteins. As can be seen from , the DHT-dependent part of AR within the soluble fraction was higher in LNCaP-sip53. This observation suggests that bicalutamide prevented translocation of AR into nuclei in LNCaP but failed to do so in LNCaP-sip53. The most prominent difference between LNCaP and LNCaP-sip53 were found in the pellet fraction: DHT induced AR binding in LNCaP but bicalutamide eliminated this binding. In contrast, DHT induced AR DNA binding in LNCaP-sip53 to a higher extent compared with LNCaP and bicalutamide did not prevent this binding.
Figure 2. Expression of AR in different nuclear protein fractions. western blot analysis of AR in nuclear extracts. Nuclear proteins were divided into two fractions: soluble in 1% Triton X-100 buffer and insoluble (pellet). The latter fraction contains DNA and DNA-binding proteins and was dissolved in sample buffer. Equal loading was controlled routinely by reversible staining of the membrane with Ponceau S solution (Sigma).
P53 participates in proteasome inhibition of AR activity
The AR transcription complex, encompassing AR, polymerase II (pol II), and coactivators, on a PSA promoter is a cyclic process involving proteasome function.Citation9 Kang et al., showed that the proteasome inhibitor MG132 did not inhibit occupancy of the PSA promoter by AR but prevented the release of the receptor from the promoter. Proteasomes participate in AR transcriptional activity by regulating the interaction between AR and its coregulators.Citation10 It has also been shown that the proteasome is involved in the dynamic assembly of the AR transcription complex.Citation9 MG132 inhibits AR activity either by inhibiting the interaction between AR and its coregulatorsCitation10 or by preventing the release of the AR from the promoters with ARE.Citation9
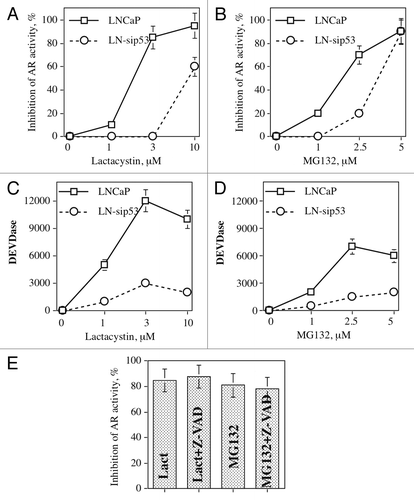
To investigate whether the proteasome pathway participates in p53-mediated activity of AR, we treated LNCaP-ARE-Luc and LN-sip53-ARE-Luc with different doses of two proteasome inhibitors: MG132 and lactacystin. As can be seen from , both inhibitors decreased AR activity in a dose-dependent manner, and LN-sip53 is more resistant to the effect of these inhibitors. Moreover, there are qualitative rather than quantitative differences between these cell lines: more than 80% of AR activity was inhibited by 3 μM of lactacystin in LNCaP whereas no inhibition was noted in LN-sip53. At the same time, both inhibitors induced higher caspase activity () in LNCaP. To investigate whether caspase activity is responsible for the inhibition of AR activity, LNCaP was treated with lactacystin or MG132 in the presence of 50 μM of the pan-caspase inhibitor Z-VAD. As shown in , inhibition of caspase activity did not change the effect of proteasome inhibitors on AR activity.
Figure 3. Proteasome inhibitors decrease AR activity independent of caspase activation. LNCaP-ARE-Luc and LN-sip53-ARE-Luc were treated for 24 h with indicated doses of lactacystin and MG132. AR activity (A, B) and caspase activity (C, D) were measured as described in “Materials and Methods.” (E) LNCaP was treated for 24 h with 3 μM lactacystin and 5 μM MG132 in the presence of 50 μM pan-caspase inhibitor Z-VAD. AR activity was measured as described in legend to .
AR-DNA binding is mediated by p53-dependent proteasome activity
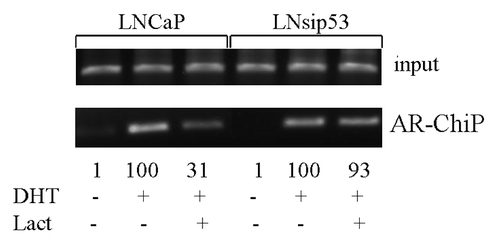
Our data suggested that AR activity was affected by p53-mediated AR-DNA binding after treatment with proteasome inhibitors. The minimal probasin promoter region (PB) (-244/-96 bp) contains androgen receptor binding sites as well as ubiquitous transcription factor binding sites. In order to investigate binding of AR to nuclear DNA, we treated LNCaP-ARE-Luc and LN-sip53-ARE-Luc cells with DHT for 24 h in the presence of lactacystine after culturing in CSS, and performed ChIP through AR antibodies. We then performed PCR using primers for the PB region from the ARE-Luc plasmid. As shown in , lactacystin decreased AR binding 3-fold in LNCaP but no binding decrease was found in LN-sip53. Therefore, in the absence of p53 lactacystin cannot remove AR from ARE-binding sites, which explains the results of the luciferase assay ().
Figure 4. Lactacystin inhibits AR-DNA binding in LNCaP but not in LN-sip53. Cells were cultured for 24 h in CSS and then treated for 24 h with 10 nM DHT in the absence/presence of 3 μM lactacystine. ChiP was performed using antibodies to AR as described in “Materials and Methods.” PCR primers for ARE in probasin promoter were used.
Discussion
We have recently shown that AR inhibition did not lead to growth arrest in the absence of p53 expression (Guseva et al., manuscript in preparation). In this study, we found that inhibition of p53 modulates AR-DNA binding on a genome wide scale. Alimirah, et al.,Citation5 who investigated the link between p53 and AR expression, found that the expression of p53 inversely correlated with the expression level of AR. A search identified a single potential p53 DNA-binding site (nucleotides 5290–5309; GenBank no. X78592) in the 5′-regulatory region of the human AR gene. This potential p53 DNA-binding site is located 469 bp upstream of the mRNA start site in the promoter region of the human AR gene. Interestingly, a systematic study on the role of specific nucleotides within the p53 responsive elements (RE) revealed a specific dinucleotide core combination within the CWWG motif of the p53RE to be the key factor that determines whether p53 transcriptionally activates or represses a target gene. A significant proportion of p53REs (39/162) were found to be repressive, including in the AR gene.Citation6
Proteasome activity is required for AR transcriptional activity via regulation of interaction between AR and its coregulators.Citation10 Involvement of the proteasome in the dynamic assembly of the AR transcription complex has also been shown.Citation9 Proteasome inhibitors effect AR activity either by inhibition of interaction between AR and its coregulatorsCitation10 or by preventing the release of the AR from the promoters with ARE.Citation9 LNCaP and LN-sip53 treated with proteasome inhibitors showed more than 80% of inhibition AR activity was inhibited by lactacystin in LNCaP, whereas no inhibition was noted in LN-sip53. We suggest that AR activity can be affected by p53-mediated AR-DNA binding after treatment with proteasome inhibitors. Indeed, using ChIP with antibodies to AR and PCR with primers for the probasin region, we found that lactacystin decreased AR binding 3-fold in LNCaP but no binding decrease was found in LN-sip53. Therefore, in the absence of p53 proteasome inhibition cannot remove AR from ARE-binding sites, which explains the enhanced activity of AR after p53 suppression.
The role of p53 in PC progression is still poorly understood. Mutations of p53 are uncommon in primary PC but occur frequently in advanced disease.Citation16 Alterations in the p53 tumor suppressor are clearly associated with progressive disease, like metastases to bone- and androgen-independent growth in PC.Citation16-Citation18 In fact, recent studies suggest that mutations in p53 lead to CRPC. Burchardt et al. (2001) have clearly shown that LNCaP cells with reduced p53 expression or function were able to form tumors in castrated male nude mice whereas parental LNCaP did not. There is increasing evidence that p53 directly regulates androgen-AR signaling and that this regulation can exist at multiple levels due to functional complexity of both proteins. In addition, mutual regulation of AR and p53 has been reported.Citation2,Citation19 Both p53 and AR are transcription factors and can affect a broad array of gene expression. It is possible that alteration in p53 can affect transcriptional function of AR, and vice versa. Several p53-dependent genes, such as, Wig-1 and PTK2 are involved in control of cancer progression.Citation20,Citation21
The accepted model of steroid hormone receptor (SHR) activity (reviewed in ref. Citation11) states that the ligand-activated SHR continually samples DNA across the vast excess of nonspecific weak binding sites until a proper high affinity site is reached. Other proteins are often involved in the site-specific binding and this suggests structure-modifying effects of the heterologous proteins on the SHR. It is conceivable that p53 interacts with AR and restricts AR binding to a particular ARE site whereas in the absence of p53 AR acquires the capability to interact with non-canonical AREs and therefore activates additional genes leading to steroid-independent growth of prostate cancer cells. We will test this hypothesis in future experiments.
Finally, the proteasome inhibitor bortezomib is approved by the FDA for the treatment of different tumors and several other proteasome inhibitors are currently in phase I–III of clinical trials.Citation12 Our data show that the inhibitory effects of proteasome inhibitors are dependent on p53 status, at least in prostate cancer. Therefore, the role of p53 during treatment with proteasome inhibitors in different tumors should be further investigated.
Materials and Methods
Cell lines, reagents, and estimation of caspase activity and cell viability
Human prostatic cancer cell lines were cultured in RPMI 1640 as previously described.2 To culture cells in steroid-free condition (SFC) RPMI 1640 was supplemented with 10% charcoal stripped serum (HyClone). PC3-AR cells containing the wild type of AR were obtained from Dr. Theodore Brown, and was previously described.Citation13 Cell viability was measured by CellTiter-BlueTM cell viability assay (Promega) and by trypan blue exclusion counting of live/dead cells. Ac-DEVD-AMC was purchased from BioMol. MG132, and lactacystin were purchased from Calbiochem. Bicalutamide (Casodex) was a generous gift from AstraZeneca Pharmaceutical. The levels of prostate specific antigen (PSA) secreted by cells were assessed by PSA enzyme immunoassay kit (MP Biomedicals).
Knocking down expression of p53
Expression of endogenous p53 was inhibited by infection with a recombinant lentivirus construct pLSL-puro expressing siRNA hairpin under control of the RNA H1 promoter, as previously described.Citation2 The structure of the 19 bp siRNA complementary to human p53 mRNA was as follows: 5′-GACTCCAGTGGTAATCTAC. The structure of the control siRNA derived from the HPV18 E6 gene was as follows: 5′-CTAACACTGGGTTATACAA. LNCaP was infected with lentivirus with siE6 or si-p53 followed by puromycin selection.
Western blot analysis
Western blot detection of proteins was performed as previously described.Citation2 Equal loading in all western blot experiments was controlled by reversible staining of the membrane with Ponceau S solution (Sigma).
Caspase activity measurement in cell lysates and in live cells
Caspase activity in cell lysates and in intact (living) cells was measured with the fluorogenic substrate Ac-DEVD-AMC as previously described.Citation2
Luciferase reporter assay
To measure AR-dependent transactivation, we introduced a pARE-Luc luciferase reporter construct containing three copies of androgen-responsive elements (ARE) with Neo gene,Citation14 which was provided by Dr. Katerina Gurova (Roswell Park Cancer Institute, Buffalo, NY) in prostate cancer cell lines, and selected permanent transfectants using G418 as the selection agent. This reporter consists of a cassette of three AREs from the rat probasin promoter followed by a Hsp70 minimal promoter, producing almost zero background expression per se in AR-negative prostate cell lines. To estimate luciferase activity, cells were harvested by trypsinization, washed in PBS, and lysed in reporter lysis buffer (200 µL, Promega). Luciferase chemiluminescence activity was measured using the luciferase assay kit (Promega). Sample aliquots (20 µL) were assayed for light emission with luminometer (MLX Dynex Technology, Inc.). The values of the luciferase assay were normalized with respect to protein concentration.
Chromatin immunoprecipitation (ChiP)
ChiP was performed as previously described.Citation15 Briefly, cells were cultured in CSS for 24 h, then treated for 24 with 10 nM DHT in the absence/presence of 3 μM lactacystin. Samples were prepared using ChiP IT Express Magnetic Chromatin Immunoprecipitation kit (Active Motive) following manufactures instructions. Cells were fixed with formaldehyde (1% final concentration) for 10 min at room temperature, harvested with scraper in lysis buffer and pellets were sonicated. Immunoprecipitation was performed overnight using monoclonal antibodies to AR. Washing steps, reverse-crosslink and DNA clean up were performed as described in ChiP IT kit protocol. PCR primers for ARE in probasin promoter were as follows: Forward 5′-TGATAGCATCTTGTTCTTAGTC and Reverse -5′-GTGTCATCTAGTATACAAATAG.
Statistical analysis
Statistical analysis was performed using the Students t-test. The statistical significance was determined at p < 0.05. Points in and show mean values for four replicates in one of two or three separate experiments, which gave similar results; error bars represent standard error of the mean (SEM).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol 2010; 80:724 - 30; http://dx.doi.org/10.1016/j.bcp.2010.04.031; PMID: 20450892
- Rokhlin OW, Taghiyev AF, Guseva NV, Glover RA, Chumakov PM, Kravchenko JE, et al. Androgen regulates apoptosis induced by TNFR family ligands via multiple signaling pathways in LNCaP. Oncogene 2005; 24:6773 - 84; http://dx.doi.org/10.1038/sj.onc.1208833; PMID: 16007156
- Gurova KV, Roklin OW, Krivokrysenko VI, Chumakov PM, Cohen MB, Feinstein E, et al. Expression of prostate specific antigen (PSA) is negatively regulated by p53. Oncogene 2002; 21:153 - 7; http://dx.doi.org/10.1038/sj.onc.1205001; PMID: 11791186
- Rokhlin OW, Gudkov AV, Kwek S, Glover RA, Gewies AS, Cohen MB. p53 is involved in tumor necrosis factor-alpha-induced apoptosis in the human prostatic carcinoma cell line LNCaP. Oncogene 2000; 19:1959 - 68; http://dx.doi.org/10.1038/sj.onc.1203453; PMID: 10773886
- Alimirah F, Panchanathan R, Chen J, Zhang X, Ho S-M, Choubey D. Expression of androgen receptor is negatively regulated by p53. Neoplasia 2007; 9:1152 - 9; http://dx.doi.org/10.1593/neo.07769; PMID: 18084622
- Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci U S A 2009; 106:14373 - 8; http://dx.doi.org/10.1073/pnas.0903284106; PMID: 19597154
- Gudkov AV, Komarova EA. Prospective therapeutic applications of p53 inhibitors. Biochem Biophys Res Commun 2005; 331:726 - 36; http://dx.doi.org/10.1016/j.bbrc.2005.03.153; PMID: 15865929
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2006; 2:474 - 9; http://dx.doi.org/10.1038/nchembio809; PMID: 16862141
- Kang Z, Pirskanen A, Jänne OA, Palvimo JJ. Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. J Biol Chem 2002; 277:48366 - 71; http://dx.doi.org/10.1074/jbc.M209074200; PMID: 12376534
- Lin H-K, Altuwaijri S, Lin W-J, Kan P-Y, Collins LL, Chang C. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem 2002; 277:36570 - 6; http://dx.doi.org/10.1074/jbc.M204751200; PMID: 12119296
- Hilser VJ, Thompson EB. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J Biol Chem 2011; 286:39675 - 82; http://dx.doi.org/10.1074/jbc.R111.278929; PMID: 21937423
- Ping Dou Q. Proteasome inhibition and cancer therapy. Nat Rev Cancer 2011; 11:848 - 9
- Rokhlin OW, Glover RB, Guseva NV, Taghiyev AF, Kohlgraf KG, Cohen MB. Mechanisms of cell death induced by histone deacetylase inhibitors in androgen receptor-positive prostate cancer cells. Mol Cancer Res 2006; 4:113 - 23; http://dx.doi.org/10.1158/1541-7786.MCR-05-0085; PMID: 16513842
- Tararova ND, Narizhneva N, Krivokrisenko V, Gudkov AV, Gurova KV. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate 2007; 67:1801 - 15; http://dx.doi.org/10.1002/pros.20662; PMID: 17935158
- Guseva NV, Rokhlin OV, Glover RA, Cohen MB. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biol Ther 2011; 12:80 - 5; http://dx.doi.org/10.4161/cbt.12.1.15721; PMID: 21525791
- Burchardt M, Burchardt T, Shabsigh A, Ghafar M, Chen MW, Anastasiadis A, et al. Reduction of wild type p53 function confers a hormone resistant phenotype on LNCaP prostate cancer cells. Prostate 2001; 48:225 - 30; http://dx.doi.org/10.1002/pros.1101; PMID: 11536301
- Meyers FJ, Gumerlock PH, Chi SG, Borchers H, Deitch AD, deVere White RW. Very frequent p53 mutations in metastatic prostate carcinoma and in matched primary tumors. Cancer 1998; 83:2534 - 9; http://dx.doi.org/10.1002/(SICI)1097-0142(19981215)83:12<2534::AID-CNCR19>3.0.CO;2-V; PMID: 9874460
- Eastham JA, Stapleton AM, Gousse AE, Timme TL, Yang G, Slawin KM, et al. Association of p53 mutations with metastatic prostate cancer. Clin Cancer Res 1995; 1:1111 - 8; PMID: 9815901
- Alimirah F, Panchanathan R, Chen J, Zhang X, Ho SM, Choubey D. Expression of androgen receptor is negatively regulated by p53. Neoplasia 2007; 9:1152 - 9; http://dx.doi.org/10.1593/neo.07769; PMID: 18084622
- Anaganti S, Fernández-Cuesta L, Langerød A, Hainaut P, Olivier M. p53-Dependent repression of focal adhesion kinase in response to estradiol in breast cancer cell-lines. Cancer Lett 2011; 300:215 - 24; http://dx.doi.org/10.1016/j.canlet.2010.10.008; PMID: 21071137
- Golubovskaya VM, Zheng M, Zhang L, Li JL, Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer 2009; 9:280; http://dx.doi.org/10.1186/1471-2407-9-280; PMID: 19671193