Abstract
Histone deacetylase (HDAC) inhibitors, including MGCD0103 and vorinostat, have led to tumor growth inhibition and apoptosis in vivo. However, with limited single-agent activity demonstrated in solid tumor trials, we examined the potential for enhanced effects in combination with topoisomerase I and II inhibitors, a staple for treatment in refractory small cell lung cancer (SCLC). SCLC cell lines were exposed to increasing concentrations of single-agent HDAC inhibitors and topoisomerase inhibitors, in various combinations, to assess for cell viability, additivity or synergy, and apoptosis. We found that MGCD0103 and vorinostat decreased cell viability by at least 60% and 80%, respectively. In the majority of cell lines, the strongest synergism was seen when vorinostat was followed by either etoposide or topotecan; concurrent therapy led to antagonism in most cell lines. Synergistic effects were seen when MGCD0103 was given concurrently or sequentially with both amrubicin and epirubicin. Enhanced additive effects leading to caspase activation were noted for the combination of MGCD0103 or vorinostat with a topoisomerase inhibitor vs. either agent alone. Thus, the combination of HDAC inhibitors and topoisomerase inhibitors showed enhanced cytotoxic effects in SCLC cell lines. Further evaluation in a clinical setting may be warranted.
Introduction
Small cell lung cancer (SCLC) accounts for 13–15% of all new lung cancer cases.Citation1,Citation2 The tumor has a tendency to disseminate early, so that 80–85% of patients are diagnosed with advanced or extensive disease.Citation3 Due to its initial chemo-sensitivity, survival improvements have been obtained with conventional chemotherapy such as platinum and etoposide, a topoisomerase II (topo II)Citation3; nonetheless, most SCLC patients relapse and rarely survive beyond 2 y from time of diagnosis.Citation4
Many relapsed patients remain candidates for additional therapy. However, the sensitivity of relapsed SCLC to further therapies is markedly reduced and varies with the level of response to initial treatment (sensitive vs. refractory relapse). The response rate of any drug or drug regimen in patients with refractory relapse is extremely poor (≤ 10%), and in patients with sensitive relapse, topotecan (Hycamtin), a topoisomerase I (topo I) inhibitor, has been the only approved agent.Citation5,Citation6
Topotecan is associated with a response rate of 24% and median survival of 24 weeks in patients with platinum-sensitive SCLCCitation6; more recent trials have shown that it also prolongs survival compared with best supportive care in all relapsed patients.Citation7 However, the responses are short lived, and disease progression is certain.
Recently, attention has been given to amrubicin, a topo II inhibitor, with hopes that it can improve the frequency of durable responses in patients with relapsed SCLC. In this regard, two similar phase II single-agent studies have been reported. In the first, Onoda et al. treated 60 patients with amrubicin and observed an overall response rate (ORR) of 50% in the refractory group and 52% in the platinum-sensitive group.Citation8 In addition, the progression-free survival (PFS), overall survival (OS), and 1-y survival rates in the platinum-sensitive and platinum-resistant groups were 4.2 vs 2.6 mo, 11.6 vs 10.3 mo, and 46% vs 40%, respectively. In a second study (Caucasian population), the results were somewhat less impressive. The ORR, PFS, and OS rates for amrubicin as second-line therapy for platinum-sensitive disease were 21.3%, 3.2 mo, and 6.0 mo, respectively.Citation9
Compared with single-agent topotecan, a higher efficacy for amrubicin is suggested, and randomized trials support this inference. Jotte et al. initially reported the results of a phase II trial of amrubicin vs topotecan in a platinium-sensitive second-line SCLC.Citation10 In this trial, the amrubicin group demonstrated superior results (ORR: 15% vs 44%, p = 0.021; PFS: 3.3 vs 4.5 mo, and OS: 7.6 vs 9.2 mo), and these results have been reproduced by the North Japan Lung Cancer Study Group Trial 0402.Citation11 In that study, 59 patients with relapsed SCLC were randomized to amrubicin vs topotecan. This group showed ORR and PFS of 38% vs 13% and 3.5 vs 2.2 mo for amrubicin and topotecan arms, respectively. However, the results of the phase III randomized trial comparing amrubicin vs topotecan presented at ASCO 2011 demonstrated a less favorable impact. For those patients with SCLC on second-line therapy, the study failed to meet its primary endpoint. The response rate was 31 vs 17% (p = 0.002), while the OS was 7.5 mo vs 7.8 mo (p = 1.17) for amrubicin vs topotecan. In subgroup analyses, there was a trend toward improved OS for the platinum-refractory group.
Topoisomerases and topoisomerase Inhibitors. Topoisomerase I (topo I) leads to DNA single-strand breaks and resealing, allowing for DNA relaxation and replication. This essential enzyme works via covalent, energy-generating, phosphodiester bonds, which are formed by the catalytic tyrosyl residue from topo at the 3′-end of the broken DNA.Citation12 A break at the 5′ end is also generated, leading to passive rotational unraveling of the double-stranded DNA. This then allows the DNA polymerases to read, replicate (via a primer), and elongate the DNA. Although topo II also leads to interruption of DNA replication, its mechanism of action differs from topo I. Covalent bonds are formed at the 5′ end of the broken DNA and require ATP for strand passage disruption.Citation12 The double-strand breaks caused by topo II allow the unbroken duplex to pass through the gap created to minimize DNA supercoiling. This serves to alleviate torsional stress and allows replication to be maintained.
Topoisomerase inhibitors induce cytotoxicity by disrupting DNA replication and exist in 2 categories: topo I and topo II inhibitors. Topotecan specifically targets DNA topo I (topo I inhibitor) and prevents the elongation phase of DNA replication. It is a semi-synthetic, water-soluble camptothecin analog.Citation11 Resistance to topotecan has been demonstrated in cell lines with topo I point mutationsCitation13 and in cells overexpressing the drug efflux membrane transporters (multi-drug resistant protein) ABCG2 and ABCB1 (Pgp).Citation14-Citation16 Amrubicin, an anthracycline, inhibits topo II by forming complexes with DNA and topo II. It is a fully synthetic 9-amino anthracycline, which is converted by the body to its most active compound, amrubicinol.Citation11 The pathways of resistance for the anthracyclines are similar to those for topo I inhibitors.
HDAC and HDAC inhibitors. Deacetylated histones are strongly negatively charged particles that bind to the DNA backbone. With acetylation, this charge is neutralized and the DNA undergoes a conformational change that allows for expression of genes, such as tumor suppressors. Histone acetylase and histone deacetylase (HDAC) tightly regulate the acetylation and deacetylation processes in normal cells; however, in tumor cells, these processes can be erratic, which may lead to uncontrolled tumor proliferation and progression.Citation17,Citation18
Tumor growth inhibition and apoptosis in response to treatment with HDAC inhibitors are also mediated through changes in acetylation of non-histone proteins. One such example is the chaperone protein heat shock protein 90 (HSP90).Citation19 Many proteins important in cancer proliferation, including Raf and Akt, require HSP90 association. Acetylation blocks the ability of HSP90 to bind to these client proteins, resulting in improper folding and function of client proteins. These misfolded proteins then become targets for degradation in the proteasome. Treatment with HDAC inhibitors leads to the acetylation of HSP90 and blockade of its chaperone function, leading to degradation of the client proteins and apoptosis.
There are 4 classes, multiple subunits or isoforms within a class, and different subclasses of HDACs.Citation20,Citation21 HDAC-1, -2, -3, and -8 are in class 1; HDAC-4 to -7, -9, and -10 are in class II; Sirt-I to -7 are in class III; and HDAC-11 is in class IV. Vorinostat (suberoylanilide hydroxamic acid; Zolinza), a hydroxamic acid, is the first FDA-approved pan-HDAC inhibitor for the treatment of cutaneous T-cell lymphoma. Pre-clinical data have demonstrated that the combination of vorinostat and cytotoxic agents, such as 5-fluorouracil, is synergistic,Citation22 and it has been postulated that this is mainly driven by the modulation of HSP90-dependent proteins that then affect various molecular processes. MGCD0103 (moncetinostat), an aminophenylbenzamide-class of HDAC inhibitors, is an oral isotype-selective HDAC inhibitor that is presently being evaluated in clinical and preclinical studies. MGCD0103 inhibits HDAC isoforms 1, 2, 3 (class I), and 11 (class IV) but no class 2 isoforms.Citation23,Citation24 In addition, it appears to have tolerable side effects and favorable pharmacokinetic and pharmacodynamic profiles, as shown in a phase I study as a single agent for advanced solid tumors.Citation25
Despite some apparent improvements, patients with SCLC continue to relapse rapidly. Thus, novel therapies that have the potential to enhance clinical outcomes are still needed. Given the importance of topo I and II inhibitors in the treatment of SCLC and the potential synergism between them and HDAC inhibitors,Citation26,Citation27in this study, we explored the anti-proliferative activity of vorinostat and MGCD0103 in SCLC cell lines and investigated whether their combination with topo I (topotecan) and II (amrubicin) inhibitors could be considered potentially relevant in the future treatment of SCLC.
Results
Addition of MGCD0103 enhanced the cytotoxic effects of topoisomerase inhibitors in SCLC cell lines. We investigated the inhibitory effects of MDCD0103 in concurrent and sequential combinations with topo II inhibitors (epirubicin and amrubicin). Cell viability assay results (IC50 levels) are shown in .
Table 1. 72-h IC50 cell viability results for study drugs in SCLC cell lines
CellTiter-Blue assays for the single-agent HDAC inhibitors (MGCD0103 and vorinostat) revealed that, for all cell lines, viability was decreased by at least 60% and 80%, respectively (). MGCD0103 demonstrated the lowest IC50 of 0.358 µmol/L (or µM) in the H526 cell line (). For all cell lines, maximal synergistic effects between MGCD0103 and amrubicin occurred when these were used concurrently. Sequential administration was also synergistic, but to a lesser extent than concurrent dosing. When used in combination, epirubicin and MGCD0103 also had a slight to moderate synergistic effect in all cell lines, with the greatest effect seen in DMS 114 cells. For the epirubicin-MGCD0103 combination, maximal synergy was seen when MGCD0103 was added 48 h prior to epirubicin (, Figs. S1 and S2).
Figure 1. Cell viability after 72-h treatment. Indicated SCLC cell lines were plated for 24 h prior to addition of serial diluted concentrations of MGCD0103 or vorinostat. After a 72-h incubation with drugs, the CellTiter-Blue assay was used to monitor viability in treated wells relative to untreated wells (% of untreated).
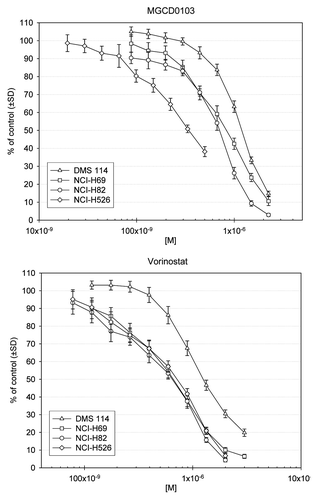
Table 2. Synergistic growth inhibition summary for MGCD0103 combinations with amrubicin or epirubicin in SCLC cell lines
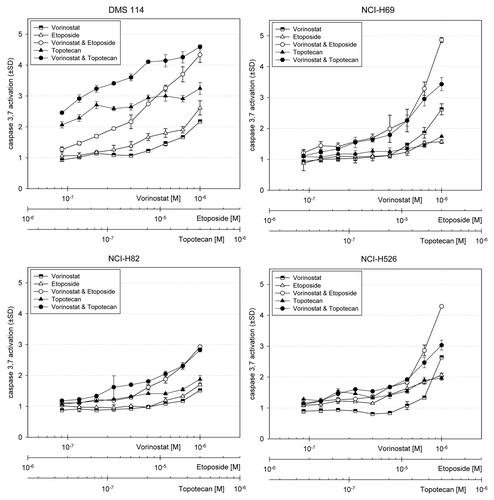
Addition of vorinostat enhanced the cytotoxic effects of topoisomerase inhibitors on SCLC cell lines. In all cell lines, vorinostat as a single agent showed significant activity, with IC50 values of 0.641–2.00 µM. Topotecan as a single agent yielded an IC50 of 21.5–86.0 nM for H82, DMS 114, and H69 cell lines. H526 was particularly sensitive to topotecan. Etoposide also demonstrated single-agent activity in H69, H82, H526, and DMS 114 cell lines, with an IC50 of < 2.38 µM. When topotecan was added concurrently with vorinostat, slight synergism was seen in the H69 cells. For DMS 114, H82, and H526 cell lines, moderate to strong antagonism was seen ( and Fig. S3). Notably, when topotecan was added 48 h after vorinostat, synergism was seen ( and Fig. S4). The concurrent vorinostat-etoposide combination had a similar trend. Similar to the topotecan-vorinostat combination, moderate to strong antagonism was seen with the etoposide-vorinostat combination in DMS 114, H82, and H526 cell lines ( and Fig. S3). At least moderate synergism was demonstrated in the H69, H82, and H526 cell lines when vorinostat was followed by etoposide ( and Fig. S4). Unlike the topotecan-vorinostat combination, only additive results were seen in DMS 114 cells when etoposide followed vorinostat ( and Figs. S3 and S4).
Table 3. Combination index summary for vorinostat combinations in SCLC cell lines
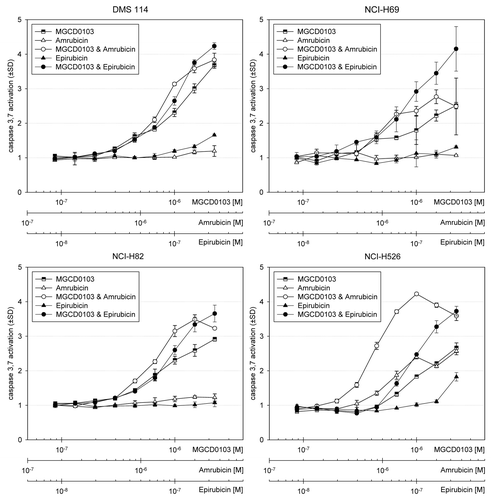
Caspase activation and cleaved PARP as a marker of apoptosis. For activation of caspase 3, there were no meaningful changes in any cell lines exposed to single-agent amrubicin and epirubicin except in H526 cells (). In all cell lines, single-agent MGCD0103 resulted in a 2.4- to 3.7-fold increase in caspase 3 activation, indicating enhanced apoptosis over untreated cells. When MGCD0103 was combined with epirubicin and amrubicin in all cell lines, apoptosis was enhanced. Most notably in H69 cells, a 4-fold increase was seen for the combination with epirubicin; also, in H526 cells, MGCD0103 resulted in a 2.7- to 4-fold increase when combined with amrubicin at concentrations ≥ 1 µM MGCD0103 and ≥ 2 µM amrubicin, respectively. At these higher concentrations of the combination of MGCD0103 and amrubicin, caspase activation was decreased, presumably due to excessive cell lysis and necrosis ().
Figure 2. Apoptosis after 24-h vorinostat and concurrent anthracycline treatment. Indicated SCLC cell lines were plated for 24 h prior to addition of serial diluted concentrations of vorinostat or vorinostat combined with either epirubicin or amrubicin. After a 24-h incubation with drugs, the Caspase-Glo 3,7 assay was used to monitor apoptosis. Vertical axes indicate the relative fold change in caspase activation for treated wells relative to untreated wells.
At concentrations greater than 300–500 µM, single-agent vorinostat showed minimal apoptotic effects in all cell lines. All cell lines demonstrated increased fold changes in caspase activation when vorinostat was combined with etoposide, with a maximum increase of 4.8-fold in H69 cells. Additive apoptotic effects were noted for vorinostat and topotecan in H69, DMS 114, and H82 cell lines (). Confirmatory 24-h experiments of concurrent treatment arms of vorinostat plus etoposide or topotecan as well as MGCD0103 plus epirubicin or amrubicin demonstrated enhanced apoptosis, as measured by increases in cleaved PARP (c-PARP) over controls or single-agent therapy (Fig. S5).
Figure 3. Apoptosis after 24-h MGCD0103 and concurrent anthracycline treatment. Indicated SCLC cell lines were plated for 24 h prior to addition of serial diluted concentrations of MGCD0103 or MGCD0103 combined with either epirubicin or amrubicin. After a 24-h incubation with drugs, the Caspase-Glo 3,7 assay was used to monitor apoptosis as described in .
Discussion
Inhibition of cell growth may be lost when topo I inhibitors are substituted in a non-time-regulated fashion. No suppression of apoptosis has been demonstrated when camptothecin was combined with the antifungal agent trichostatin A, a hydroxamic acid compound that was found to be one of the first HDAC inhibitors.Citation28,Citation29 However, when HDAC inhibitors are given the day before, day of, and day after breast cancer cells were treated with camptothecin, those cells already in G2-M arrest show increased killing.Citation30 In our study, the benefit of topo I inhibitors in combination with HDAC inhibitors has been further elucidated. We evaluated the effects that HDAC inhibitors have on SCLC cell lines and their combination with topo I and II inhibitors (topotecan and amrubicin, respectively).
Preclinical studies have long established an interaction between topo II inhibitors and HDAC inhibitors.Citation29,Citation31 In thyroid cancerCitation32 and breast cancerCitation33 cell lines, combinations of these agents resulted in HDAC inhibitors enhancing the effects of topo II inhibitors. In breast cancer cell lines, these effects were seen regardless of the topo II inhibitor used.Citation33 A complex interaction exists between HDAC inhibitors and topo II. Data published to date indicate that chromatin decondensation and altered gene transcription by HDAC inhibitors help to facilitate topo II inhibitor-mediated DNA damage.Citation29,Citation34 By altering the chromatin structure through remodeling, HDAC inhibitors render the DNA vulnerable to further DNA cytotoxic agents.
In this study, we demonstrated a concentration-dependent downregulation of SCLC cell lines by the HDAC inhibitors, vorinostat and MGCD0103, and topo inhibitors. In all cell lines, single-agent topotecan and epirubicin were the most potent, as demonstrated by the effects seen at nanomolar concentrations. With the other drugs, single-agent effects were shown at micromolar concentrations. This correlates with the clinical responses and toxicity seen when topotecan is applied in the clinical setting.
When vorinostat was used in combination with topotecan or etoposide in SCLC cell lines, we found that cell viability was decreased concomitant with enhanced apoptotic effects. Interestingly, the H526 cell line, which has previously been described as resistant to topoisomerase inhibitors,Citation35 also responded to the combination treatment regimen. Our results are in agreement with these previous findings.Citation35 However, in our cell lines, we found that the sequencing of the therapies may in fact be important. Bruzzese et al.Citation35 observed strong synergistic cytotoxicity when vorinostat was dosed simultaneously with camptothecin or topotecan. Although we did find synergism in one cell line (H69) with simultaneous dosing of vorinostat and etoposide, the three other cell lines demonstrated antagonism with concurrent dosing. Across all of the cell lines that we tested, our strongest synergism was seen with sequential dosing. It has been speculated that HDAC inhibitors are most efficacious when given prior to cytotoxic agents. The premise behind this is that the acetylated histones make the DNA more vulnerable to DNA-damaging agents. Our results may differ from those of Bruzzese et al. because different topo inhibitors and cell lines were used, leading to differences in the intrinsic plasticity and intracellular signaling.
The addition of MGCD0103 to amrubicin or epirubicin also showed synergistic effects with decreased cell viability and enhanced apoptosis in SCLC cell lines. Although the results were similar to the vorinostat data presented here, there were some different responses seen between the cell lines with these different HDAC inhibitors in combination with the topo inhibitors. More specifically, MGCD0103 demonstrated synergism when given concurrently with amrubicin and sequentially when given prior to epirubicin. Dedes et al. have previously demonstrated that cell lines resistant to vorinostat also exhibited resistance to LBH589 and JNJ26481585 (hydroxamate class) and valproic acid (aliphatic acid class) but not to MGCD0103 (benzamide class).Citation36 This supports the differential findings seen between the two HDAC inhibitors across the cell lines used in this study. The origin of these differences is likely multi-factorial. First, each class is marked by a differing chemical structure, which in turn affects the potency and mechanisms of each molecule. Second, the selective nature of MGCD0103, a class I and IV specific inhibitor, compared with the pan-HDAC inhibitor vorinostat, further lends insight into the differences seen between the MGCD0103 and the vorinostat combination findings. Third, it is well established that HDAC inhibitors lead to post-translational modification, including protein acetylation and gene modulation, which affect the function of these proteins.Citation37,Citation38 Fourth, in addition to differences in mechanisms of actions between the two HDAC inhibitors studied, there are differing mechanisms of actions between the topo I inhibitors and topo II inhibitors used in combination with vorinostat and MGCD0103, respectively, which may add to the differential findings.
The importance of scheduling of HDAC inhibitors is still under debate. HDAC inhibitors have been demonstrated to have synergistic effects with topo I and II inhibitors in breast, ovarian, and pancreatic cancer.Citation26,Citation27,Citation30,Citation33,Citation39-Citation41 In breast cancer cell lines, Marchion et al.Citation33 demonstrated an upregulation of DNA binding of topo II inhibitor following treatment with vorinostat, resulting in enhanced DNA damage. Maximal effects occurred after 48-h exposure to vorinostat compared with shorter exposure times. Antagonism was seen when the topo II inhibitor preceded the HDAC inhibitors. These studies have contributed to this controversy that scheduling plays a key role in clinical response.Citation30,Citation33 In preclinical models, to obtain the maximal synergism of HDAC inhibitors in combination with chemotherapy, the topo inhibitors must be delivered prior to the agent used in combination. This rationale is based on the data that the epigenetic modulation of the histones by the HDAC inhibitors will allow a more open chromatin conformation. The chemotherapy agent of interest is thereby allowed more “access” to the DNA and genes of interest to allow more opportunity for damage. Our results support both the concurrent and sequential use of HDAC and topo inhibitors in patients with SCLC but favor the sequential administration. However, whether this translates to maximal efficacy in the patients is not yet proven.
Our results have limitations. First, no further assays were performed to assess for mechanisms of action. These assays could expand on the potential importance of sequencing vs. concurrent administration of medications. Due to the pleiotropic nature of HDAC inhibitors, the mechanisms of action are likely multi-factorial. In vivo efficacy assessment would help to build on this as enhancements in in vitro cytotoxic effects do not necessarily translate to improved in vivo efficacy.
Over the past 20 years, few advances have been made in the treatment of patients with SCLC. Despite these minor achievements, most patients with SCLC still succumb to their disease. In this study, we demonstrated chemosensitization of SCLC cell lines to the combination of HDAC inhibitors and topo inhibitors. Clinical trials exploring this combination are thus reasonable to explore and are currently underway.
Materials and Methods
Compounds. Vorinostat, MGCD0103, and amrubicin were supplied by Merck, MethylGene, and Celgene Corporation, respectively. Etoposide was purchased from Sigma-Aldrich (St. Louis, MO), and topotecan was purchased from Sequoia Research Products (Pangbourne, UK). Stock solutions (10 mM) of the compounds were prepared in DMSO and stored at -80°C or diluted in media for immediate use. Epirubicin was purchased from Sigma.
Cell culture. The cell lines DMS 114, NCI-H69, NCI-H82, and NCI-H526 (obtained from American Type Culture Collection, all p53 mutant) are among the most frequently published cell lines in preclinical SCLC studies. Cells were maintained in RPMI or DMEM (DMS 114 only) with 10% fetal bovine serum. Cells were grown at 37°C and 5% CO2 in an incubator. All cell lines tested free of mycoplasma with MycoAlert tests (Lonza Rockland, Inc., Rockland, ME).
Cell viability assays. The activity levels of drugs alone and in combination were determined by a high-throughput CellTiter-Blue cell viability assay. For these assays, 1.2 x 103 cells were plated in each well of 384-well plates using a Precision XS liquid handling station (Bio-Tek Instruments, Inc., Winooski, VT) and allowed to incubate overnight at 37°C and 5% CO2. A liquid handling station was used to serially dilute all drugs (2:3) in media, and 5 µl of these dilutions were added to appropriate wells at indicated times. Four replicate wells were used for each drug concentration, and an additional four control wells received a diluent control without drug. At the end of the incubation period with drugs, 5 µl CellTiter-Blue reagent (Promega Corp., Madison, WI) were added to each well. Cell viability was assessed by the ability of the remaining viable cells to bioreduce resazurin to resorufin. The fluorescence of sesorufin (579 nm excitation/584nm emission) was measured with a Synergy 4 microplate reader (Bio-Tek Instruments, Inc.). The fluorescence data were transferred to Microsoft Excel to calculate the percent viability relative to the four replicate cell wells that did not receive drug. IC50 values were determined using a sigmoidal equilibrium model regression with XLfit version 5.2 (ID Business Solutions Ltd.). The IC50 was defined as the concentration of drug required for a 50% reduction in growth/viability. The IC50 values obtained from single drug cell viability assays were used to design subsequent drug combination experiments. The concentration ranges employed for each compound were as follows: vorinostat (range: 20 nM to 2.3 µM); MGCD (range 73 nM to 3 µM); amribicin (range: 18 nM to 16 µM); epirubicin (range: 8.8 nM to 16 µM); etoposide (range: 35 nM to 20 µM); and topotecan (range: 2.5 nM to 500 nM).
Analysis of additivity and synergy. For drug combination experiments, the cell viability assays were performed as described above and the results were analyzed for synergistic, additive, or antagonistic effects using the CI method developed by Chou and Talalay.Citation42 For application of this method, the drug concentration dilutions were used at fixed concentration ratios based on the IC50 values of each drug obtained from preliminary experiments (e.g., 50:1, 2:5, 1:250). Briefly, the concentration-effect curve for each drug alone is determined based on experimental observations using the median-effect principle and is compared with the effect achieved with a combination of the two drugs to derive a CI value. This method involves plotting concentration-effect curves, for each agent and their combination, using the median-effect equation: fa/fu = (D/Dm)m, where D is the concentration of the drug, Dm is the concentration required for a 50% effect (equivalent to IC50), fa and fu are the affected and unaffected fractions, respectively (fa = 1-fu), and m is the exponent signifying the sigmoidicity of the concentration-effect curve. The computer software XLfit was used to calculate the values of Dm and m. The CI used for the analysis of the drug combinations was determined by the isobologram equation for mutually nonexclusive drugs that have different modes of action: CI = (D)1/(Dx)1 + (D)2 /(Dx)2 + (D)1(D)2/(Dx)1(Dx)2, where (Dx)1 and (Dx)2 in the denominators are the doses (or concentrations) for D1 (drug 1) and D2 (drug 2) alone that gives x% inhibition, whereas (D)1 and (D)2 in the numerators are the concentrations of drug 1 and drug 2 in combination that also inhibited x% (i.e., isoeffective). CI < 1, CI = 1, and CI > 1 indicate synergism, additive effects, and antagonism, respectively.
Apoptosis assay. Caspase 3/7 activation was measured using a 384-well plate based Caspase-Glo® 3/7 (Promega) luminescent assay. Cells were treated for 24 h with serial dilutions of each compound or a combination of two drugs. Molar drug concentrations are indicated on the horizontal axis and are plotted against the fold increase in caspase 3/7 activation relative to untreated cells.
Cleaved PARP (asp 214) sandwich ELISA. Cleaved PARP was assayed using PathScan cleaved PARP (asp 214) sandwich ELISA kit (Cell Signaling, Danvers, MA). Briefly, the cell lines were grown overnight and then treated for 24 h in 6-well plates. The cells were lysed in 200 µl ice-cold extraction buffer supplied in kit. 100 µl of the soluble fraction was used in each well of the ELISA plate. The ELISA was performed according to the manufacturer’s instructions. Absorbencies were measured with a Synergy 4 microplate reader.
| Abbreviations: | ||
| CI | = | combination index |
| HDAC | = | Histone deacetylase |
| HSP90 | = | heat shock protein 90 |
| ORR | = | overall response rate |
| OS | = | overall survival |
| PFS | = | progression-free survival |
| SCLC | = | small cell lung cancer |
| Topo I | = | topoisomerase I |
Additional material
Download Zip (295.3 KB)Acknowledgments
Vorinostat and research funding were supplied by Merck and Co, Inc. MGCD0103, amrubicin, and research funding were supplied by Celgene Corporation
Note
Supplemental material can be found at www.landesbioscience.com/journals/cbt/article/19848/
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Jänne PA, Freidlin B, Saxman S, Johnson DH, Livingston RB, Shepherd FA, et al. Twenty-five years of clinical research for patients with limited-stage small cell lung carcinoma in North America. Cancer 2002; 95:1528 - 38; http://dx.doi.org/10.1002/cncr.10841; PMID: 12237922
- Simon GR, Wagner H, American College of Chest Physicians. Small cell lung cancer. Chest 2003; 123:Suppl 259S - 71S; http://dx.doi.org/10.1378/chest.123.1_suppl.259S; PMID: 12527584
- Davies AM, Lara PN, Lau DH, Gandara DR. Treatment of extensive small cell lung cancer. Hematol Oncol Clin North Am 2004; 18:373 - 85; http://dx.doi.org/10.1016/j.hoc.2003.12.012; PMID: 15094177
- Ardizzoni A, Hansen H, Dombernowsky P, Gamucci T, Kaplan S, Postmus P, et al. Topotecan, a new active drug in the second-line treatment of small-cell lung cancer: a phase II study in patients with refractory and sensitive disease. The European Organization for Research and Treatment of Cancer Early Clinical Studies Group and New Drug Development Office, and the Lung Cancer Cooperative Group. J Clin Oncol 1997; 15:2090 - 6; PMID: 9164222
- von Pawel J, Schiller JH, Shepherd FA, Fields SZ, Kleisbauer JP, Chrysson NG, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999; 17:658 - 67; PMID: 10080612
- O’Brien ME, Ciuleanu TE, Tsekov H, Shparyk Y, Cuceviá B, Juhasz G, et al. Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J Clin Oncol 2006; 24:5441 - 7; http://dx.doi.org/10.1200/JCO.2006.06.5821; PMID: 17135646
- Onoda S, Masuda N, Seto T, Eguchi K, Takiguchi Y, Isobe H, et al, Thoracic Oncology Research Group Study 0301. Phase II trial of amrubicin for treatment of refractory or relapsed small-cell lung cancer: Thoracic Oncology Research Group Study 0301. J Clin Oncol 2006; 24:5448 - 53; http://dx.doi.org/10.1200/JCO.2006.08.4145; PMID: 17135647
- Ettinger DS, Jotte R, Lorigan P, Gupta V, Garbo L, Alemany C, et al. Phase II study of amrubicin as second-line therapy in patients with platinum-refractory small-cell lung cancer. J Clin Oncol 2010; 28:2598 - 603; http://dx.doi.org/10.1200/JCO.2009.26.7682; PMID: 20385980
- Jotte R, Conkling P, Reynolds C, Galsky MD, Klein L, Fitzgibbons JF, et al. Randomized phase II trial of single-agent amrubicin or topotecan as second-line treatment in patients with small-cell lung cancer sensitive to first-line platinum-based chemotherapy. J Clin Oncol 2011; 29:287 - 93; http://dx.doi.org/10.1200/JCO.2010.29.8851; PMID: 21135284
- Inoue A, Sugawara S, Yamazaki K, Maemondo M, Suzuki T, Gomi K, et al. Randomized phase II trial comparing amrubicin with topotecan in patients with previously treated small-cell lung cancer: North Japan Lung Cancer Study Group Trial 0402. J Clin Oncol 2008; 26:5401 - 6; http://dx.doi.org/10.1200/JCO.2008.18.1974; PMID: 18854562
- Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 2009; 109:2894 - 902; http://dx.doi.org/10.1021/cr900097c; PMID: 19476377
- Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Updat 1999; 2:307 - 18; http://dx.doi.org/10.1054/drup.1999.0102; PMID: 11504505
- Brangi M, Litman T, Ciotti M, Nishiyama K, Kohlhagen G, Takimoto C, et al. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res 1999; 59:5938 - 46; PMID: 10606239
- Bates SE, Medina-Pérez WY, Kohlhagen G, Antony S, Nadjem T, Robey RW, et al. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxycamptothecin) and homocamptothecins. J Pharmacol Exp Ther 2004; 310:836 - 42; http://dx.doi.org/10.1124/jpet.103.063149; PMID: 15075385
- Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, et al. Reduced expression of DNA topoisomerase I in SF295 human glioblastoma cells selected for resistance to homocamptothecin and diflomotecan. Mol Pharmacol 2008; 73:490 - 7; http://dx.doi.org/10.1124/mol.107.041178; PMID: 17984197
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15 - 23; http://dx.doi.org/10.1016/j.gene.2005.09.010; PMID: 16289629
- Kim SC Sr., Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607 - 18; http://dx.doi.org/10.1016/j.molcel.2006.06.026; PMID: 16916647
- Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res 2003; 63:5126 - 35; PMID: 12941844
- Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 2008; 112:981 - 9; http://dx.doi.org/10.1182/blood-2007-10-115873; PMID: 18495956
- Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB. HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol 2009; 20:1293 - 302; http://dx.doi.org/10.1093/annonc/mdn792; PMID: 19515748
- Fazzone W, Wilson PM, Labonte MJ, Lenz H-J, Ladner RD. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int J Cancer 2009; 125:463 - 73; PMID: 19384949
- Li Z, Zhou N, Fournel M, et al. Antitumor Activities of MGCD0103, a novel and isotype-selective histone deacetylase inhibitor. Eur J Cancer Supp Mol Targets Cancer Ther 2004; 2:Abstract 83
- Moradei O, Leit S, Zhou N, Fréchette S, Paquin I, Raeppel S, et al. Substituted N-(2-aminophenyl)-benzamides, (E)-N-(2-aminophenyl)-acrylamides and their analogues: novel classes of histone deacetylase inhibitors. Bioorg Med Chem Lett 2006; 16:4048 - 52; http://dx.doi.org/10.1016/j.bmcl.2006.05.005; PMID: 16713259
- Siu LL, Pili R, Duran I, Messersmith WA, Chen EX, Sullivan R, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol 2008; 26:1940 - 7; http://dx.doi.org/10.1200/JCO.2007.14.5730; PMID: 18421048
- Marchion DC, Bicaku E, Turner JG, Daud AI, Sullivan DM, Munster PN. Synergistic interaction between histone deacetylase and topoisomerase II inhibitors is mediated through topoisomerase IIbeta. Clin Cancer Res 2005; 11:8467 - 75; http://dx.doi.org/10.1158/1078-0432.CCR-05-1073; PMID: 16322310
- Münster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol 2007; 25:1979 - 85; http://dx.doi.org/10.1200/JCO.2006.08.6165; PMID: 17513804
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 1990; 265:17174 - 9; PMID: 2211619
- Johnson CA, Padget K, Austin CA, Turner BM. Deacetylase activity associates with topoisomerase II and is necessary for etoposide-induced apoptosis. J Biol Chem 2001; 276:4539 - 42; http://dx.doi.org/10.1074/jbc.C000824200; PMID: 11136718
- Bevins RL, Zimmer SG. It’s about time: scheduling alters effect of histone deacetylase inhibitors on camptothecin-treated cells. Cancer Res 2005; 65:6957 - 66; http://dx.doi.org/10.1158/0008-5472.CAN-05-0836; PMID: 16061681
- Tsai SC, Valkov N, Yang WM, Gump J, Sullivan D, Seto E. Histone deacetylase interacts directly with DNA topoisomerase II. Nat Genet 2000; 26:349 - 53; http://dx.doi.org/10.1038/81671; PMID: 11062478
- Catalano MG, Fortunati N, Pugliese M, Poli R, Bosco O, Mastrocola R, et al. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J Endocrinol 2006; 191:465 - 72; http://dx.doi.org/10.1677/joe.1.06970; PMID: 17088416
- Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem 2004; 92:223 - 37; http://dx.doi.org/10.1002/jcb.20045; PMID: 15108350
- Marchion DC, Bicaku E, Turner JG, Schmitt ML, Morelli DR, Munster PN. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol Cancer Ther 2009; 8:794 - 801; http://dx.doi.org/10.1158/1535-7163.MCT-08-0985; PMID: 19372552
- Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol Cancer Ther 2009; 8:3075 - 87; http://dx.doi.org/10.1158/1535-7163.MCT-09-0254; PMID: 19887547
- Dedes KJ, Dedes I, Imesch P, von Bueren AO, Fink D, Fedier A. Acquired vorinostat resistance shows partial cross-resistance to ‘second-generation’ HDAC inhibitors and correlates with loss of histone acetylation and apoptosis but not with altered HDAC and HAT activities. Anticancer Drugs 2009; 20:321 - 33; http://dx.doi.org/10.1097/CAD.0b013e3283262a32; PMID: 19322073
- Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol 2005; 23:3971 - 93; http://dx.doi.org/10.1200/JCO.2005.16.600; PMID: 15897549
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90:595 - 606; PMID: 9288740
- Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 2003; 63:7291 - 300; PMID: 14612526
- Piacentini P, Donadelli M, Costanzo C, Moore PS, Palmieri M, Scarpa A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch 2006; 448:797 - 804; http://dx.doi.org/10.1007/s00428-006-0173-x; PMID: 16568310
- Nolan L, Johnson PW, Ganesan A, Packham G, Crabb SJ. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential?. Br J Cancer 2008; 99:689 - 94; http://dx.doi.org/10.1038/sj.bjc.6604557; PMID: 18728657
- Chou T, ed. The median-effect principle and the combination index for quantitation of synergism and antagonism. San Diego: Academic Press; 1991.
- Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621 - 81; http://dx.doi.org/10.1124/pr.58.3.10; PMID: 16968952