Abstract
EphB4 is a member of the Eph receptor tyrosine kinase family shown to act in neuronal guidance and mediate venal/arterial separation. In contrast to these more established roles, EphB4’s function in cancer is much less clear. Here we illustrate both tumor promoting as well as suppressing roles of EphB4, by showing that its activation resulted in inhibition of the Ras/ERK pathway in endothelial cells but activation of the same pathway in MCF-7 breast cancer cells. This was true if EphB4 was stimulated with EphrinB2, its natural ligand, or an agonistic monoclonal antibody for EphB4. Correspondingly, EphB4 activation stimulated MCF7 growth while inhibiting HUVEC cell proliferation. The reason for these dramatic differences is due to functional coupling of EphB4 to different downstream effectors. Reduction of p120 RasGAP in HUVEC cells attenuated the inhibitory effect of EphB4 activation on the ERK pathway, whereas knockdown of PP2A in MCF7 cells attenuated EphB4 activation of the ERK pathway. This represents the first time a functional coupling between Eph receptor and PP2A has been demonstrated leading to activation of an oncogenic pathway. Our study illustrates the caveats and potential challenges of targeting EphB4 for cancer therapy due to the conflicting effects on cancer cell and endothelial cell compartments.
Keywords: :
Introduction
The sixteen member vertebrate Eph receptor family represents the largest group in the receptor tyrosine kinase superfamily. They are type-I transmembrane proteins with an extracellular ligand-binding domain and an intracellular tyrosine kinase domain. They have been shown to regulate multiple important biological processes, such as neuronal patterning, organ development, and vascular and muscle remodeling.Citation1,Citation2 They consist of ten EphA receptors and six EphB receptors that bind to Ephrin A and Ephrin B ligands, respectively. The Ephrin family consists of nine members in vertebrate and can be classified into two sub-categories, including Ephrin A, which has six members anchored to the membrane by a glycophosphatidylinositol group, and Ephrin B, which has three type-I transmembrane members.Citation1-Citation3 The most well-established functions of Eph receptors are providing guidance cues during neuronal and vascular development. The interaction between Eph receptors and Ephrin ligands has been classically proposed as repulsive in nature, leading to definable boundaries between two different compartments. For example, knockout of the EphB4 gene in mice results in embryonic lethality due to defects in vascular remodeling.Citation4 Interestingly, EphrinB2 is highly expressed on arteries but not veins, whereas EphB4, its cognate receptor, is much more abundant on veins than arteries. This reciprocal expression profile constitutes the best molecular marker to distinguish venous from arterial endothelial cells and is the primary contributor to the establishment of boundary between arteries and veins. The repulsive interaction between EphB4 and EphrinB2 has also been shown to define tissue segment boundaries in neuronal development. Studies have shown that cell populations differentially expressing EphB4 and EphrinB2 do not intermingle in the same culture dish.Citation1,Citation2,Citation5
In contrast to their well-validated roles in neuronal and vascular development, the function of EphB receptors in cell growth and proliferation has not been firmly established.Citation6 On one hand, EphB receptors have been reported to be tumor suppressors. While this role has been ascribed in indications such as breast cancer and acute lymphoblastic leukemia,Citation7,Citation8 the tumor suppressive effect of EphB receptors in intestinal carcinogenesis provides the best evidence.Citation9 A number of reports have shown that silencing of EphB receptors was important for tumor formation in genetically engineered mouse models of colon cancer and that loss of the receptors frequently occurred in colorectal cancer patients.Citation10-Citation12 The tumor suppressor activity of EphB receptors has been shown to be mediated through a variety of pathways including ERK, AKT, and Abl. In contrast to its putative role as a tumor suppressor, EphB expression has been shown to be significantly associated with advanced stages of ovarian, HNSCC and breast cancers, and its overexpression predicted poor survival.Citation6,Citation13 Additionally, EphB4 is preferentially induced in prostate and colorectal cancer and confers a survival advantage.Citation14,Citation15 Functional relevance as a putative oncogene was demonstrated in several model systems using soluble monomeric EphB4 to inhibit tumor growth by acting as a dominant negative inhibitor through blockade of ligand binding.Citation16,Citation17 The dichotomy between tumor suppressor and oncogene suggests that the function of EphB receptors may be highly context-dependent.
The complexity increases when considering that the impact of EphB receptors can also be mediated through surrounding vasculature. For example, forward signaling through EphB receptors stimulated by EphrinB2 inhibited endothelial cell proliferation and VEGF-induced ERK activation.Citation18 Building upon this finding we show here that EphB4 is the receptor responsible for the EphrinB2-mediated suppression of ERK in HUVECs. Interestingly, we find that the same receptor is responsible for EphrinB2-mediated activation of the ERK pathway in MCF-7 breast cancer cells. The same inverse relationship holds when assessing the impact of EphrinB2 treatment on cell growth in both cell models. The data herein suggests that EphB4 transduces the EphrinB2 signal through different downstream effector molecules to positively or negatively regulate the ERK pathway in the different cell types.
Results
EphB4 is necessary and sufficient to direct EphrinB2-mediated suppression of the Ras/ERK pathway in HUVECs
Soluble dimeric EphrinB2-Fc treatment has been shown to abrogate VEGF or Ang-1-induced phospho-ERK (pERK) signal in HUVEC cells.Citation18 To more accurately mimic the heterogeneous growth factor conditions in vivo, we grew HUVEC cells in full growth medium with a cocktail of growth factors. We then treated the cells for 20 min with EphrinB2-Fc in the presence or absence of anti-Fc antibody to mimic cell surface clustering of the ligand. The clustered ligand almost completely eliminated the high basal pERK signal induced in the full growth media (). We also studied the activation status of EphB4, following stimulation with ligand. The clustered ligand stimulated EphB4 tyrosine-phosphorylation (), suggesting the observed pERK suppression may be mediated by EphB4 signaling. Interestingly, in contrast to the pERK suppression effect, pAKT signal was not perturbed by EphrinB2-Fc treatment, indicating that the inhibition was specifically on the ERK pathway (). We additionally characterized upstream components of the ERK pathway to see if they are also affected by EphrinB2 treatment. Not surprisingly, pMEK was similarly inhibited as pERK, suggesting inhibition of the MEK/ERK pathway. The pERK inhibition effect by cross-linked EphrinB2-Fc was also observed in HUAEC cells, suggesting that the effect is not specific to one cultured model of endothelial cells (, bottom panel).
Figure 1. Activation of EphB4 by EphrinB2-Fc treatment attenuates ERK activation in HUVEC. (A) HUVECs were either untreated (-) or treated with or without EphrinB2-Fc in the presence or absence of goat anti-human Fc IgG (α-Fc) as indicated for 20 min in EGM-2 complete growth medium. Phosphorylated ERK (P-ERK) and total ERK, phosphorylated AKT (P-AKT) and total AKT, phosphorylated MEK (P-MEK) were probed directly by Western Blot while phosphorylated EphB4 (P-EphB4) and total EphB4 were probed by Western Blot following EphB4 immunoprecipitation. HUAEC cells were either untreated (-) or treated with or without EphrinB2-Fc in the presence or absence of goat anti-human Fc IgG (α-Fc) as indicated for 20 min in EGM-2 complete growth medium. Cell lysates were subjected to Western Blot for P-ERK and total ERK analysis. (B) HUVECs were transfected with either non-targeted (Ctrl) or EphB4 siRNA. Three days post-transfection, cells were analyzed by flow cytometry for EphrinB2-Fc binding. (C) EphB4 and Ctrl siRNA transfected cells were treated with clustered EphrinB2-Fc for 20 min. Whole cell lysates were immunoblotted for EphB4, EphB2, pERK and total ERK.
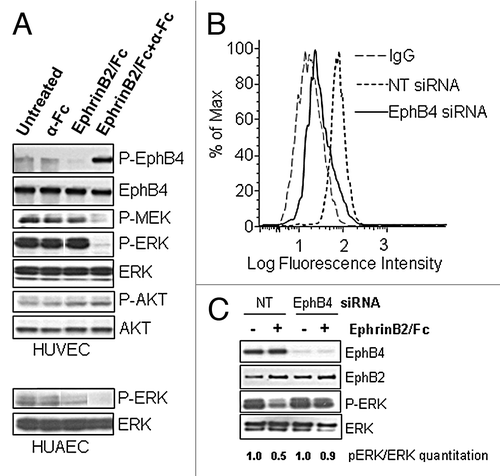
To ascertain whether EphB4 is the primary mediator of the pERK-suppression effect of EphrinB2, we used siRNA to selectively knockdown EphB4 in HUVEC cells. The EphB4 siRNA specifically reduced EphB4 level without perturbing EphB2 level (). The cell-surface binding of EphrinB2-Fc was dramatically reduced after EphB4 knockdown, indicating EphB4 constitutes the major binding site for EphrinB2 in the HUVEC cells (). EphrinB2-Fc again substantially inhibited pERK in control cells (50% reduction) but failed to do so in the EphB4 knockdown cells (10% reduction) (), indicating EphB4 is the major driver for inhibiting activation of the ERK pathway.
A human monoclonal antibody against EphB4, 6C2, acts as an agonist to promote EphB4 activation and ERK inhibition in HUVECs
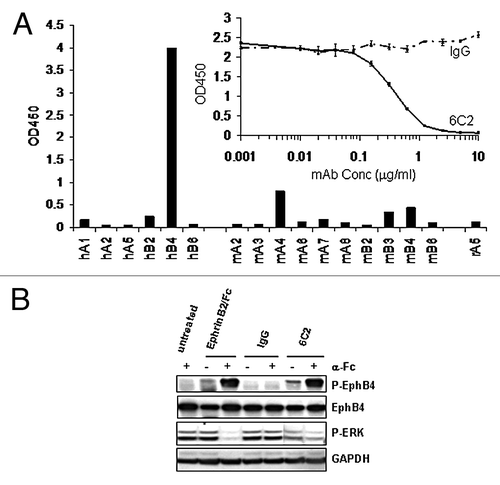
The above finding suggests that an agonistic mAb for EphB4 that mimics the effect of the ligand EphrinB2 may possess anti-angiogenic potential in cancer therapy. Toward this goal, we generated a fully human monoclonal antibody 6C2, that specifically binds to human EphB4 and does not cross-react with any other human or rodent Eph receptor family members (). A competition ELISA binding assay demonstrated that 6C2 blocks EphrinB2-binding to EphB4 in a dose-dependent manner ( inset), indicating that 6C2 shares an overlapping epitope with the ligand. More importantly, the antibody was selected for its ability to activate the EphB4 receptor in cells. Following just 20 min of treatment, the antibody induced a similar level of EphB4 tyrosine phosphorylation as achieved by the clustered ligand (). Similar to the EphrinB2-Fc, the 6C2 antibody required clustering with a secondary anti-Fc antibody to induce maximal activation of the receptor. The clustered EphB4 antibody, but not the control IgG, inhibited pERK in the HUVEC cells (); however, the inhibition was not as complete as that achieved by the ligand. Given the specificity of the antibody for EphB4, these results further support the previous finding that EphB4, not another EphB receptor, is driving most of the inhibitory signal in HUVEC.
Figure 2. A specific EphB4 antibody mimics the effects of ligand. (A) Binding of EphB4 antibody clone 6C2 to human (h), murine (m) or rat (r) Eph receptors was detected by ELISA. The ability of increasing concentrations of 6C2 to compete with EphrinB2-Fc binding to human EphB4 was assessed by ELISA (inset). (B) HUVECs were untreated or treated with EphrinB2-Fc (B2), control IgG (IgG) or 6C2 in the presence (+) or absence (-) of clustering anti-human Fc antibody for 20 min in complete EGM-2 medium. Lysates were either immunoprecipitated for EphB4 and immunoblotted for phosphotyrosine (P-EphB4) or directly immunoblotted for P-ERK. Total EphB4 and GAPDH were immunoblotted as loading controls for P-EphB4 and P-ERK respectively.
EphB4 activates the Ras/ERK pathway in MCF7 breast cancer cells
We next investigated whether stimulation of EphB4 on tumor cells would result in a similar suppression of the ERK pathway. For these studies we used the breast tumor cell line MCF-7, which has been previously shown to express EphB4 and respond to stimulation of clustered ligand.Citation7 Similar to the previous findings, clustered EphrinB2-Fc acutely stimulated tyrosine phosphorylation of EphB4 after only minutes of treatment (). Surprisingly, clustered EphrinB2 induced ERK phosphorylation whereas control IgG had no effect (). In contrast with HUVEC cells, basal ERK phosphorylation was not detectable in MCF-7 cells cultured in complete growth media.
Figure 3. EphrinB2 stimulates ERK activation specifically through EphB4 in MCF-7 breast cancer cells. (A) MCF-7 cells were untreated (-) or treated with human control IgG or EphrinB2-Fc in the presence of clustering anti-human Fc antibody (Fc) for 20 min. EphB4 was immunoprecipitated and immunoblotted for phosphotyrosine (P-EphB4) or total EphB4. (B) MCF7 cells were treated for 20 min with EphrinB2-Fc (B2), 6C2, or human control IgG in the presence (+) or absence (-) of clustering anti-human Fc antibody in complete growth medium. (C) 72 h after transfection with 40 nM of non-targeted (Ctrl), EphB2 or EphB4 siRNA, cells were left untreated (-) or treated with EphrinB2-Fc (+) for 20 min and lysates were immunoblotted for EphB4, EphB2, P-ERK and ERK. Densitometry-based quantitation was performed for pERK (both bands) and total ERK (both bands) using ImageJ software (NIH). pERK readings were then normalized against total ERK readings and the ratios are shown at the bottom of the panel (normalized pERK value of NT siRNA transfected cells without the EphrinB2/Fc stimulation was arbitrarily set as 1.0). EphrinB2/Fc-treatment induced fold of induction in normalized pERK was calculated for cells transfected with NT, EphB4 or EphB2 siRNAs and folds plotted in histogram.
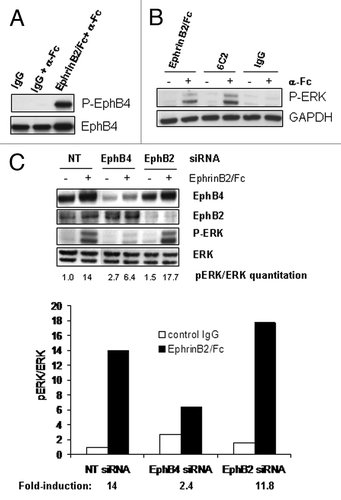
To determine if EphB4 was responsible for the activation of ERK in MCF-7 cells just as it was responsible for the inhibition of ERK in HUVECs, we again took a two-pronged approach. First, the EphB4 specific antibody 6C2 was capable of stimulating the phosphorylation pf ERK when clustered with a secondary antibody (). Since 6C2 binds to EphB4 but not EphB2 or other EphB family members, its successful induction of pERK indicates that EphB4, not EphB2, is the main mediator of the ligand’s effect in MCF7 cells. To further corroborate this selectivity, we knocked down EphB2 or EphB4 expression through specific siRNAs and treated these cells with EphrinB2-Fc. The siRNAs were able to specifically knock down the expression of their respective target without significantly impacting the expression of the other (). EphB2 knockdown did not significantly interfere with the ligand-induced pERK induction (14-fold induction for control cells, 11.8-fold induction in EphB2 knockdown cells), whereas EphB4 knockdown substantially suppressed this induction (2.4-fold induction), confirming it is the dominant receptor mediating EphrinB2-Fc activity.
EphB4 activation regulates cell growth through different effectors depending on the cell type
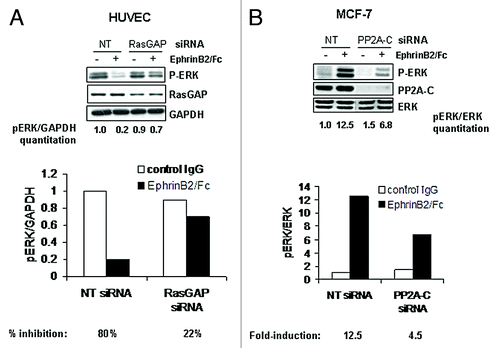
Given the relative importance of the ERK pathway to cell growth and proliferation, we next examined the phenotypic response to EphrinB2-Fc in both MCF-7 breast tumor cells and HUVECs. Consistent with the activation status of ERK in both cell lines, EphrinB2-Fc inhibited HUVEC cell growth by nearly 2-fold compared with control IgG, while it stimulated MCF-7 cell growth by nearly 2-fold compared with control IgG (). These data reinforce the importance of the ERK pathway to cell growth in both of these cultured lines while raising the question of how the same receptor-ligand pair could modulate the same pathway in completely opposite directions depending on the cell type. EphB2 and other Eph-family members such as EphA2 have been shown to inhibit the Ras pathway in neuronal cells through engaging the Ras inhibitor, p120RasGAP.Citation19-Citation21 To determine if EphB4 uses p120RasGAP to modulate the ERK pathway in HUVECs, we transfected the cells with siRNA targeting p120RasGAP prior to treatment with EphrinB2-Fc. The p120RasGAP siRNA only reduced the protein levels 2-fold but was still able to attenuate most of the EphrinB2-mediated ERK inhibition (). HUVECs transfected with non-targeting siRNA reduced pERK by 80% in response to EphrinB2-Fc while p120RasGAP siRNA transfected cells only reduced pERK by 22%. These results suggest that p120RasGAP may be a downstream effector molecule of EphB4 to mediate ERK inhibition in HUVECs. However, this is unlikely to be the case in MCF-7 cells since EphB4 stimulated but did not inhibit Ras/ERK pathway. Correspondingly, siRNA-mediated knockdown of p120RasGAP failed to block EphrinB2-stimulated ERK activation in MCF-7 cells (data not shown). At the same time, we also examined the protein phosphatase PP2A as a possible effector molecule to mediate the inhibition of the ERK pathway in HUVECs, given that Raf and MEK are known substrates. However, knocking down the catalytic subunit PP2A-C in HUVEC cells had no impact on EphrinB2-mediated inhibition of the ERK pathway (data not shown), suggesting that PP2A is not an important effector of EphB4 activity in these cells. To our surprise, knockdown of this same PP2A catalytic subunit in MCF-7 cells significantly blunted the ligand-induced pERK induction (12.5-fold induction in control cells; 4.5-fold in PP2A-C knockdown cells; ). These data suggest that EphB4 may activate Ras/ERK by utilizing a ubiquitous protein phosphatase PP2A.
Figure 4. EphrinB2 suppresses HuVEC cell growth but induces MCF7 cell growth. HuVEC and MCF-7 cells were untreated or treated with control IgG or EphrinB2-Fc (B2) in the presence of clustering anti-human Fc antibody and cell number was determined by Cell Titer-Glo. *p < 0.01 and **p < 0.05 by analysis of variance (ANOVA) followed by Tukey's post hoc test.
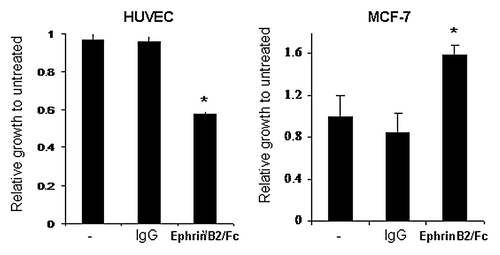
Figure 5. EphB4 transduces a signal to ERK using different downstream effectors depending on the cell type. (A) HUVECs or (B) MCF7 cells were transfected with 40 nM non-targeted (NT), p120 RasGAP or PP2A siRNA and treated with or without clustered EphrinB2-Fc for 20 min. Protein samples were analyzed by immunoblot for P-ERK, p120RasGAP and PP2A with either GAPDH or ERK probed as a loading control. For (A), pERK was normalized against GAPDH and the ratio listed under each lane (normalized pERK value of NT siRNA transfected cells without the EphrinB2/Fc stimulation was arbitrarily set as 1.0). Percentage of inhibition of normalized pERK was then calculated for EphrinB2/Fc treatment in both NT or RasGAP siRNA transfected cells and shown as histogram. For (B), densitometry-based quantitation was performed for pERK (both bands) and total ERK (both bands) using ImageJ software (NIH). pERK readings were normalized against total ERK readings and the ratios are shown below each lane (normalized pERK value of NT siRNA transfected cells without the EphrinB2/Fc stimulation was arbitrarily set as 1.0). EphrinB2/Fc treatment-induced fold of induction in normalized pERK was calculated for both NT and PP2A-C siRNA transfected cells and the folds plotted in histogram.
Discussion
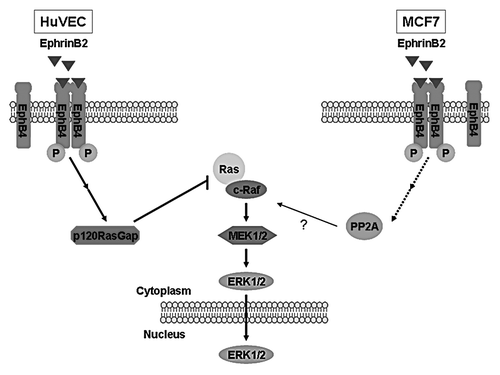
Our current study illustrated an intriguing dichotomy in EphB4 signaling between endothelial cells and breast cancer cells. We hypothesize that these variable effects are the result of EphB4 functionally coupling to distinct downstream effectors (). Our data suggests that p120RasGAP is involved in the EphrinB2-mediated blockade of the ERK pathway in HUVEC cells as has been shown for other Eph receptors in other cell systems. More surprisingly, our result suggests the involvement of the protein phosphatase PP2A in the activation of the ERK pathway in response to EphrinB2 treatment of MCF-7 cells.
Figure 6. Model of bi-functional effects of EphB4 on Ras/ERK pathway in different cellular contexts. Based on the current results, we propose that EphB4 signals to the Ras inhibitor p120RasGAP to suppress the high basal ERK pathway activity in HUVEC cells; whereas it mediates activation of the same pathway in MCF7 breast cancer cells through engaging PP2A.
Examples of the dichotomy in EphB4’s role in cancer progression exist throughout the literature. Noren and Pasquale have nicely reviewed much of the conflicting data supporting the apparent paradox.Citation22 Our data use cells from two different cell origins so the results could be explained by the inherent differences in the way the two cell types regulate signal transduction pathways. Differences in cell cycle stage, normal vs. malignant or mutational background, could all contribute to the divergent impact of EphrinB2 stimulation on EphB4 signaling. Also the bidirectional nature of the Eph-Ephrin signaling axis may play a significant part in driving the divergent signaling and phenotypic responses observed in these studies. Regardless of the reason, therapeutic targeting of this axis is complex. The hope of developing EphB4 therapeutics like the antibody 6C2 into an effective cancer therapy based on its suppression of endothelial cell growth is substantially complicated by its proliferative activity in MCF-7 breast cancer cells. It is even more complicated when considering that the antibody and ligand had to be artificially clustered to drive potent activity as suggested by our observation that cross-linking by anti-Fc is usually required to impact the ERK activity. Mimicking the presentation of membrane-anchored ligands or receptors that are enriched on plasma membrane microdomains will be very difficult with conventional therapeutic approaches. For therapeutic approaches to Eph receptors and ligands to be successful, a better understanding of the signaling pathways underpinning the phenotypic response will be critical.
Herein, we report that EphrinB2 treatment of HUVEC cells caused a rapid inactivation of the elevated pERK signal sustained by full growth medium leading to cell growth suppression. Although both EphB2 and EphB4 are expressed on HUVEC cells and both can bind to EphrinB2, our study showed that EphB4 is the primary receptor and signal transducer of EphrinB2 in HUVEC cells. The factors that govern Eph receptor ligand interactions between cells are still unclear, so it is difficult to speculate why EphrinB2-Fc would not utilize EphB2 when it is clearly expressed in both cell systems. Since the two EphB receptors were detected on the immunoblot with different antibodies, no relative abundance comparison between EphB2 and EphB4 could be drawn. We went on to demonstrate that this effect is achieved through potential EphB4 engagement of p120 RasGAP, previously shown to confer EphB2 or EphA2-mediated suppression of Ras/ERK pathways.Citation19-Citation21 This data implies that the phosphor-tyrosine motif that mediates the p120RasGAP coupling may be well-conserved among the Eph family members.
The most surprising finding here is that EphB4 promotes ERK activity through PP2A, a protein phosphatase that generally inactivates kinase signaling through direct removal of their phosphate groups. PP2A is an essential serine/threonine protein phosphatase. The holoenzyme is a heterotrimeric complex that consists of a structural A, catalytic C, and regulatory B subunits.Citation23 The A and C subunits bind to each other to form a core complex, which then incorporates one of a highly divergent set of B or B’ subunits. Binding of B-type subunits modulates enzymatic activity by controlling the subcellular localization and substrate specificity of the holoenzyme. It is more often known as a negative regulator of pMEK or pERK as a direct result of dephosphorylation of pMEK and pERK.Citation24,Citation25 In contrast, PP2A has also been shown to mediate upregulation of pERK through its dephosphorylation of phospho-c-Raf (S259P), which is known to be an inhibitory phosphorylation on Raf kinase activity and in turn dampens the activity of MEK and ERK.Citation26,Citation27 If this is the case, our data implies that MCF7 cells may overexpress the specific PP2A B-isoform that selectively targets it to phospho-c-Raf but not the B-isoform that preferentially targets it to pMEK or pERK.
While this study opens up new avenues to explore regarding in-depth Eph receptor signaling, much is still unknown. Future research should focus on what molecular switches exist to allow Eph receptors to signal to different pathways, and pharmacological agents that may influence this cellular decision-making process. Given the heterogeneity of tumors, it will be interesting to learn if Eph receptor signaling differs between subpopulations of cells within the same tumor or even if Eph receptor signaling itself serves to modulate the differentiation state of tumor cells. When some of these basic questions are answered, we may be able to better design the appropriate therapy targeting this axis for specific patients.
Materials and Methods
Cell-lines and reagents
HUVEC (Human Umbilical Vein Endothelial Cells) and HUAEC (Human Umbilical Artery Endothelial Cells) cells were obtained from ATCC and grown with EBM-2 medium supplemented with endothelial growth pellet (Clonetics). MCF-7 cells were also obtained from ATCC and maintained in RPMI-1640 medium with 10% FBS. EphrinB2-Fc, the human Fc-fusion to the recombinant ECD domain of murine EphrinB2, was purchased from R&D Systems. Goat-anti-human Fc antibody was purchased from Sigma. Mouse monoclonal and rabbit polyclonal antibodies against P-AKT (S473P), P-ERK1/2, rabbit polyclonal antibody against P-MEK1/2 were purchased from Cell Signaling technologies. Rabbit polyclonal antibody against p120 RasGAP was purchased from Santa Cruz Biotechnologies, GAPDH monoclonal antibody was purchased from Sigma. Anti-Phospho-Tyrosine monoclonal antibody (4G10) and anti-PP2A-C-subunit antibody were purchased from Upstate Biotechnology. EphB2 antibody is obtained from Zymed/Invitrogen; and PP2A antibodies were from Cell Signaling Technology. Smartpool siRNAs against human p120RasGAP, EphB4, EphB2 and PP2A-C were obtained from Thermo Scientific/Dharmacon.
Immunoprecipitation
Cells were treated as indicated in the figure legends. At the end of treatment, cells were washed once with PBS and lysed with Triton-X lysis buffer (Boston Bioproducts) containing protease and phosphatase inhibitors. Cell extracts were immunoprecipitated for EphB4 based on standard protocols established before.Citation14,Citation28 Phospho-EphB4 in the immunoprecipitate was detected by immunoblotting with pan-phospho-Tyr antibody (4G10, Upstate).
FACS-binding of EphrinB2-Fc to HuVECs
Control and siRNA-treated HuVECs were removed from flasks using enzyme-free dissociation buffer (Invitrogen), washed once, and incubated with rmEphrinB2-Fc (R&D Systems), 1 μg/1 million cells on ice for 40min in FACS buffer (PBS + 2%FBS). Cells were then washed in FACS buffer and stained with a goat anti-human IgG (H+L)-Alexa Fluor 488 secondary antibody (1 μg/1 million cells, Molecular Probes) on ice for 40 min. Subsequently, cells were washed twice and resuspended in 200 μl FACS buffer for analysis. Live whole cells were gated using PI staining and 10,000 events were acquired using a BD LSRII flow cytometer (BD Biosciences). Data were analyzed using Flowjo software (Tree Star, Inc.).
HUVEC Proliferation Assay
HUVECs were seeded into a clear-bottomed, white-walled 96-well plate at a density of 2,500 cells per well (50 μl volume), in half-strength EGM-2 media (EBM-2 media with complete bullet kit added, diluted 1:1 with basal EBM media, Lonza). Treatments were prepared at 2X concentration in half-strength EGM-2, 50 μl of each treatment was added to cells for a total volume of 100 μl/well. After 3 d, cell growth was determined with Cell Titer Glo (Promega) and plates were analyzed using a luminometer.
MCF-7 Proliferation Assay
MCF-7 cells were plated at a density of 3,000 cells per well in 100 μl of medium containing 10% heat-inactivated FBS. Costar white-walled tissue culture 96-well plates with clear bottoms (Corning) were used. The next day, the plating medium was removed, cells washed twice with PBS and treatments were added in 100 μl medium containing 0.5% heat-inactivated FBS. After incubation at 37°C for 6 d, equal volumes of CellTiter Glo reagent (Promega) were added to each well. Plates were rocked on a plate shaker for 10 min at room temperature to ensure complete cell lysis. Luminescence was measured using an EnVision 2104 Multilabel Reader (PerkinElmer).
siRNA transfection
HUVEC cells were seeded at 40–50% confluence and allowed to attach overnight. Medium was refreshed the day of transfection. siRNA transfection was performed with lipofectamine RNAiMax transfection reagent (Invitrogen), according to manufacturer’s instructions. Briefly, proper amount of siRNA was added to OptiMEM serum-free medium (Invitrogen) to form 50 μl of solution A, 1ul of RNAiMax transfection reagent was added to OptiMEM medium to form 50 μl of solution B. The two solutions were combined for 15–20 min and then added to cells under 500 μl of growth medium.
MCF-7 cells were transfected with siRNA oligos targeting EphB4, EphB2, and PP2A-C using a Reverse Transfection protocol. Briefly, transfection mixes were prepared as described above and siRNA-lipid complexes were allowed to form in 24-well tissue culture dishes for 15–20 min. Cells were then added to the wells containing the transfection mixes. The seeding density was 75,000 cells in 500 μl complete growth medium. Twenty-four hours post-transfection, transfection medium was removed and replaced. Cells lysates were collected after an additional 48 h incubation.
Fully human EphB4 mAb 6C2 generation through XenoMouse technology
Fully human antibodies to human EphB4 were generated using XenoMouse immunization. Animals were immunized with human EphB4-ECD (R&D Systems). Ten milligrams per mouse was used for the initial injection followed by boosts of 5 mg/mouse every week for 6 weeks. Titers of antibody against EphB4 were monitored by binding to recombinant human EphB4-ECD in an ELISA assay. 6C2 was isolated as a positive hit.
6C2 binding ELISA to Eph family members
96-well ELISA plates were coated with 6C2 and control Mabs overnight at 4°C followed by wash and PBSTB blocking. Blocking was done for 1 h at room temperature followed by washing. While plates were blocking Eph family members were biotinylated. Biotinylation of Ephs was performed at room temperature. Biotinylated Ephs were added to ELISA plates and incubated for 1 h at 37°C, followed by washing. Neutravidin-HRP was added to plates for 1 h at 37°C, followed by washing. TMB substrate was then added to the plates and incubated for 5–10 min at room temperature. Reaction was stopped with equal volume of 0.2 M H2SO4. Plates were read on plate reader at 450 nM.
6C2 competition with EphrinB2 binding to EphB4 ELISA assay
96-well ELISA plates were coated with EphrinB2 overnight at 4°C, followed by wash and PBSTB (Invitrogen) blocking. Blocking was done for 1 h at room temperature, followed by washing. While plates were blocking, EphB4 was biotinylated and incubated with 6C2 or control Mabs. Biotinylation and incubation were performed at room temperature. EphB4-Mab combinations were added to ELISA plates and incubated for 1 h at 37°C, followed by washing. Neutravidin-HRP was added to plates for 1 h at 37°C, followed by washing. TMB substrate was then added to the plates and incubated for 5–10 min at room temperature. Reaction was stopped with equal volume of 0.2 M H2SO4. Plates were read on plate reader at 450 nM.
| Abbreviations: | ||
| Eph | = | erythropoietin-producing hepatoma amplified sequence |
| PP2A | = | protein phosphatase 2A |
| ERK | = | extracellular regulated MAP kinase |
| MEK | = | MAP kinase kinase or Erk kinase |
| EphB4 | = | ephrin type-B receptor 4 |
| HUVEC | = | human umbilical vein endothelial cells |
| FACS | = | fluorescence-activated cell sorting |
| siRNA | = | small interfering RNA |
| HNSCC | = | head and neck squamous cell carcinoma |
| c-Raf | = | v-raf-1 murine leukemia viral oncogene homolog 1 |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| VEGF | = | vascular endothelial growth factor |
| Ang-1 | = | angiopoietin 1 |
Acknowledgments
The authors wish to thank Mark Pearson, Simon Barry and David Blakey for their contributions to the development and characterization of clone 6C2 anti-EphB4 antibody.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008; 133:38 - 52; http://dx.doi.org/10.1016/j.cell.2008.03.011; PMID: 18394988
- Egea J, Klein R. Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell Biol 2007; 17:230 - 8; http://dx.doi.org/10.1016/j.tcb.2007.03.004; PMID: 17420126
- Himanen JP, Saha N, Nikolov DB. Cell-cell signaling via Eph receptors and ephrins. Curr Opin Cell Biol 2007; 19:534 - 42; http://dx.doi.org/10.1016/j.ceb.2007.08.004; PMID: 17928214
- Gerety SS, Wang HU, Chen ZF, Anderson DJ. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell 1999; 4:403 - 14; http://dx.doi.org/10.1016/S1097-2765(00)80342-1; PMID: 10518221
- Füller T, Korff T, Kilian A, Dandekar G, Augustin HG. Forward EphB4 signaling in endothelial cells controls cellular repulsion and segregation from ephrinB2 positive cells. J Cell Sci 2003; 116:2461 - 70; http://dx.doi.org/10.1242/jcs.00426; PMID: 12734395
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010; 10:165 - 80; http://dx.doi.org/10.1038/nrc2806; PMID: 20179713
- Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat Cell Biol 2006; 8:815 - 25; http://dx.doi.org/10.1038/ncb1438; PMID: 16862147
- Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, et al. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood 2010; 115:2412 - 9; http://dx.doi.org/10.1182/blood-2009-05-222208; PMID: 20061560
- Clevers H, Batlle E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res 2006; 66:2 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-05-3849; PMID: 16397205
- Cortina C, Palomo-Ponce S, Iglesias M, Fernández-Masip JL, Vivancos A, Whissell G, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet 2007; 39:1376 - 83; http://dx.doi.org/10.1038/ng.2007.11; PMID: 17906625
- Davalos V, Dopeso H, Castaño J, Wilson AJ, Vilardell F, Romero-Gimenez J, et al. EPHB4 and survival of colorectal cancer patients. Cancer Res 2006; 66:8943 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-4640; PMID: 16982731
- Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, et al. EphB receptor activity suppresses colorectal cancer progression. Nature 2005; 435:1126 - 30; http://dx.doi.org/10.1038/nature03626; PMID: 15973414
- Kumar SR, Masood R, Spannuth WA, Singh J, Scehnet J, Kleiber G, et al. The receptor tyrosine kinase EphB4 is overexpressed in ovarian cancer, provides survival signals and predicts poor outcome. Br J Cancer 2007; 96:1083 - 91; http://dx.doi.org/10.1038/sj.bjc.6603642; PMID: 17353927
- Kumar SR, Scehnet JS, Ley EJ, Singh J, Krasnoperov V, Liu R, et al. Preferential induction of EphB4 over EphB2 and its implication in colorectal cancer progression. Cancer Res 2009; 69:3736 - 45; http://dx.doi.org/10.1158/0008-5472.CAN-08-3232; PMID: 19366806
- Xia G, Kumar SR, Masood R, Zhu S, Reddy R, Krasnoperov V, et al. EphB4 expression and biological significance in prostate cancer. Cancer Res 2005; 65:4623 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-04-2667; PMID: 15930280
- Martiny-Baron G, Korff T, Schaffner F, Esser N, Eggstein S, Marmé D, et al. Inhibition of tumor growth and angiogenesis by soluble EphB4. Neoplasia 2004; 6:248 - 57; http://dx.doi.org/10.1593/neo.03457; PMID: 15153337
- Kertesz N, Krasnoperov V, Reddy R, Leshanski L, Kumar SR, Zozulya S, et al. The soluble extracellular domain of EphB4 (sEphB4) antagonizes EphB4-EphrinB2 interaction, modulates angiogenesis, and inhibits tumor growth. Blood 2006; 107:2330 - 8; http://dx.doi.org/10.1182/blood-2005-04-1655; PMID: 16322467
- Kim I, Ryu YS, Kwak HJ, Ahn SY, Oh JL, Yancopoulos GD, et al. EphB ligand, ephrinB2, suppresses the VEGF- and angiopoietin 1-induced Ras/mitogen-activated protein kinase pathway in venous endothelial cells. FASEB J 2002; 16:1126 - 8; http://dx.doi.org/10.1096/fj.01-0805fje; PMID: 12039842
- Elowe S, Holland SJ, Kulkarni S, Pawson T. Downregulation of the Ras-mitogen-activated protein kinase pathway by the EphB2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Mol Cell Biol 2001; 21:7429 - 41; http://dx.doi.org/10.1128/MCB.21.21.7429-7441.2001; PMID: 11585923
- Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, et al. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J Biol Chem 2005; 280:34008 - 18; http://dx.doi.org/10.1074/jbc.M502879200; PMID: 16051609
- Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol 2000; 2:62 - 9; http://dx.doi.org/10.1038/35000008; PMID: 10655584
- Noren NK, Pasquale EB. Paradoxes of the EphB4 receptor in cancer. Cancer Res 2007; 67:3994 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-07-0525; PMID: 17483308
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 2001; 353:417 - 39; http://dx.doi.org/10.1042/0264-6021:3530417; PMID: 11171037
- Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem 2005; 280:36029 - 36; http://dx.doi.org/10.1074/jbc.M506986200; PMID: 16129692
- Hu X, Wu X, Xu J, Zhou J, Han X, Guo J. Src kinase up-regulates the ERK cascade through inactivation of protein phosphatase 2A following cerebral ischemia. BMC Neurosci 2009; 10:74; http://dx.doi.org/10.1186/1471-2202-10-74; PMID: 19602257
- Adams DG, Coffee RL Jr., Zhang H, Pelech S, Strack S, Wadzinski BE. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J Biol Chem 2005; 280:42644 - 54; http://dx.doi.org/10.1074/jbc.M502464200; PMID: 16239230
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 2003; 13:1356 - 64; http://dx.doi.org/10.1016/S0960-9822(03)00535-9; PMID: 12932319
- Kumar SR, Singh J, Xia G, Krasnoperov V, Hassanieh L, Ley EJ, et al. Receptor tyrosine kinase EphB4 is a survival factor in breast cancer. Am J Pathol 2006; 169:279 - 93; http://dx.doi.org/10.2353/ajpath.2006.050889; PMID: 16816380