Abstract
The small GTPase Rac1 regulates many cellular processes, including cytoskeletal reorganization, cell migration, proliferation, and survival. Additionally, Rac1 plays a major role in activating NF-κB-mediated transcription. Both Rac1 and NF-κB regulate many properties of the malignant phenotype, including anchorage-independent proliferation and survival, metastasis, and angiogenesis. Despite these findings, the roles of Rac1and NF-κB in non-small cell lung carcinoma, a leading cause of cancer deaths, have not been thoroughly investigated. Here, we compared the effects of Rac1 siRNA to that of the Rac1 inhibitor NSC23766 on multiple features of the NSCLC malignant phenotype, including NF-κB activity. We show that the siRNA-mediated silencing of Rac1 in lung cancer cells results in decreased cell proliferation and migration. The decrease in proliferation was observed in both anchorage-dependent and anchorage-independent assays. Furthermore, cells with decreased Rac1 expression have a slowed progression through the G1 phase of the cell cycle. These effects induced by Rac1 siRNA correlated with a decrease in NF-κB transcriptional activity. Additionally, inhibition of NF-κB signaling with BAY 11–7082 inhibited proliferation; indicating that the loss of cell proliferation and migration induced by the silencing of Rac1 expression may be attributed in part to loss of NF-κB activity. Interestingly, treatment with the Rac1 inhibitor NSC23766 strongly inhibits cell proliferation, cell cycle progression, and NF-κB activity in lung cancer cells, to an even greater extent than the inhibition induced by Rac1 siRNA. These findings indicate that Rac1 plays an important role in lung cancer cell proliferation and migration, most likely through its ability to promote NF-κB activity, and highlight Rac1 pathways as therapeutic targets for the treatment of lung cancer.
Introduction
Rac1 is a member of the Rho family of small GTPases and participates in numerous pathways inducing cytoskeleton reorganization, gene transcription, cell proliferation, and survival.Citation1,Citation2 The importance of Rac1 in neoplastic transformation is indicated by the finding that Rac1 is necessary for K-Ras-induced transformation of fibroblasts as well as the development of K-Ras-induced lung cancer in mice.Citation3-Citation5 Rac1 is overexpressed in breast, colon, and lung carcinomas,Citation6-Citation9 and increased expression of Rac1 is correlated with poor prognosis in non-small cell lung cancer (NSCLC).Citation10 These and other findings highlight the roles of Rac1 in neoplastic transformation, tumor progression, and metastasis.Citation1,Citation2
Nuclear factor-kappaB (NF-κB) is also implicated in cancer development and progression.Citation11-Citation13 NF-κB is a ubiquitously expressed transcription factor which regulates over 200 different genes involved in numerous pathways including inflammation, apoptosis/survival, cell cycle progression, and migration.Citation14-Citation16 NF-κB can be regulated by Rac1 through multiple mechanisms,Citation17,Citation18 indicating that the Rac1-dependent regulation of NF-κB likely plays a role in cancer. Interestingly, many cancers have been shown to rely on constitutive NF-κB signaling, which is now referred to as NF-κB addiction.Citation19
Although some studies have implicated the role of Rac1 and other small GTPases in NSCLC,Citation6,Citation20-Citation22 the ability of Rac1 to regulate NF-κB activity in NSCLC has not been previously investigated. In this study, we investigated the ability of Rac1 to regulate NF-κB activity and promote cell migration and proliferation in NSCLC cell lines. To study the role of Rac1 in NSCLC, we took advantage of two methods of Rac1 inhibition; Rac1-specific siRNA, and Rac1 inhibition with the Rac1-specific inhibitor NSC23766. Rac1-specific siRNA inhibits expression of Rac1, whereas NSC23766 inhibits the interaction of Rac1 with specific guanine nucleotide exchange factors (GEFs). Specifically, NSC23766 was shown to decrease Rac1 activation by inhibition of the Rac1 GEFs Trio and Tiam1.Citation23 We also utilized the well-known NF-κB inhibitor BAY 11–7082. BAY 11–7082 is characterized as an irreversible IκB kinase (IKK) inhibitor and has been shown to inhibit TNFα induced NF-κB activity.Citation24,Citation25
We found that silencing of Rac1 expression decreases NSCLC migration and proliferation, as indicated by slowed progression through G1 phase of the cell cycle and reduced proliferation in both anchorage-dependent and -independent conditions. Additionally, we found that silencing of Rac1 in NSCLC decreases the basal transcriptional activity of NF-κB, without detectably diminishing the ability of NF-κB to be activated by TNFα. Interestingly, the Rac1 inhibitor NSC23766, and the NF-κB inhibitor BAY 11-7082, produced a greater inhibitory effect on NSCLC proliferation and inhibited both basal and TNFα-induced NF-κB activity. These results highlight the differences between using Rac1 siRNA and NSC23766 as methods of Rac1 inhibition. These findings also identify Rac1 as a major regulator of proliferation and migration in NSCLC, and implicate NF-κB as a mediator of these processes.
Results
Silencing of Rac1 reduces cell proliferation in NSCLC cell lines
To investigate the role of Rac1 in NSCLC cell lines, we utilized siRNAs, which specifically target Rac1 and reduce its expression. Rac1 siRNAs #1 and #2 diminish the expression of Rac1 in two different NSCLC cell lines (NCI-H1703 and A549) while having no effect on GAPDH protein levels. The non-targeting (NT) siRNA had no significant effect on Rac1 or GAPDH protein expression ( and ). Similar responses were induced by siRNAs in two other NSCLC cell lines, NCI-H23 and NCI-H1650 (results not shown).
Figure 1. Cell proliferation is significantly decreased by silencing Rac1 expression in NSCLC cell lines. (A) To assess the silencing of Rac1 expression in NSCLC cell lines, NCI-H1703 and A549 cells were either subjected to mock transfection without siRNA (lane 1) or transfected with nontargeting (NT) siRNA (lane 2) or the indicated Rac1 siRNAs (lanes 3 and 4). Cell lysates were prepared and subjected to ECL-Western blotting using antibodies to GAPDH and Rac1. Results are representative of three independent experiments. (B) Quantitative densitometry was conducted to determine the protein levels of Rac1 in the ECL-Western blots described in A. The values are normalized to densitometric values obtained from ECL-Western blots of cells subjected to NT siRNA transfection. Results are the mean ± SE from three independent experiments. (C) Cell proliferation was assayed by measuring [3H]thymidine uptake 72 h after transfecting the cells with 25 nM of the indicated siRNAs. The values are normalized to [3H]thymidine uptake by cells subjected to NT siRNA transfection. Results are the mean ± SE from three independent experiments conducted with six replicates for each treatment in each experiment. (D) Cell proliferation rate was assayed by measuring [3H]thymidine uptake at the indicated time points 72 h after transfecting the cells with 25 nM of the indicated siRNAs. Results are the mean ± SE from three independent experiments conducted with four replicates for each treatment, in each experiment. Symbols above a column indicate a statistical comparison between the indicated sample and the control sample of cells transfected with NT siRNA (*, p < 0.01)
![Figure 1. Cell proliferation is significantly decreased by silencing Rac1 expression in NSCLC cell lines. (A) To assess the silencing of Rac1 expression in NSCLC cell lines, NCI-H1703 and A549 cells were either subjected to mock transfection without siRNA (lane 1) or transfected with nontargeting (NT) siRNA (lane 2) or the indicated Rac1 siRNAs (lanes 3 and 4). Cell lysates were prepared and subjected to ECL-Western blotting using antibodies to GAPDH and Rac1. Results are representative of three independent experiments. (B) Quantitative densitometry was conducted to determine the protein levels of Rac1 in the ECL-Western blots described in A. The values are normalized to densitometric values obtained from ECL-Western blots of cells subjected to NT siRNA transfection. Results are the mean ± SE from three independent experiments. (C) Cell proliferation was assayed by measuring [3H]thymidine uptake 72 h after transfecting the cells with 25 nM of the indicated siRNAs. The values are normalized to [3H]thymidine uptake by cells subjected to NT siRNA transfection. Results are the mean ± SE from three independent experiments conducted with six replicates for each treatment in each experiment. (D) Cell proliferation rate was assayed by measuring [3H]thymidine uptake at the indicated time points 72 h after transfecting the cells with 25 nM of the indicated siRNAs. Results are the mean ± SE from three independent experiments conducted with four replicates for each treatment, in each experiment. Symbols above a column indicate a statistical comparison between the indicated sample and the control sample of cells transfected with NT siRNA (*, p < 0.01)](/cms/asset/26e15e73-0aaa-4625-8b5f-0933099ddcd5/kcbt_a_10920082_f0001.gif)
To assess the role of Rac1 in NSCLC cell proliferation, we first tested the ability of Rac1 to regulate anchorage-dependent proliferation by measuring [3H]thymidine uptake by NSCLC cells proliferating in tissue culture plates. In this anchorage-dependent environment, silencing of Rac1 expression significantly decreased cell proliferation by approximately 60% in the NCI-H1703 and A549 cell lines (), and the NCI-H23 and NCI-H1650 cell lines (data not shown). To further confirm the ability of Rac1 siRNA to decrease NSCLC proliferation, we also tested the rate of proliferation by measuring [3H]thymidine uptake over time. Indeed, Rac1 siRNA also decreased the rate of proliferation of NCI-H1703 cells ().
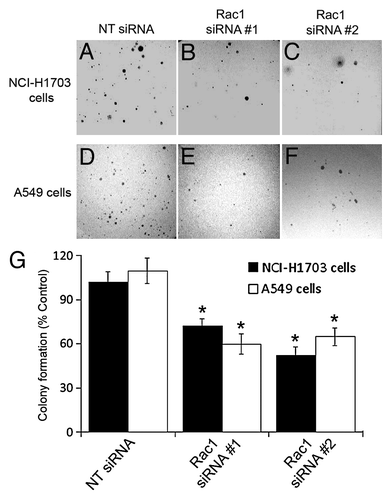
Since anchorage-independent proliferation is a hallmark of transformed cells, we next examined the ability of Rac1 to promote proliferation in an anchorage-independent environment. The cells were treated with Rac1 siRNA, re-plated in soft agar, and allowed to form colonies for five weeks. In all cases, silencing of Rac1 reduced the ability of the NSCLC cell lines to proliferate in soft agar (). Together, these data indicate that silencing of Rac1 in NSCLC inhibits both anchorage-dependent and -independent proliferation.
Figure 2. Silencing of Rac1 expression in NSCLC cells diminishes colony formation in soft agar. NCI-H1703 cells (A–C and G) and A549 cells (D–F and G) were transfected with the indicated siRNAs and an equal number of live cells were plated 24 h later in soft agar. Digital images of the cells were collected 5 weeks later (A–F), and the numbers of visible colonies in the agar were counted (G). The results in G are the mean ± SE from three independent experiments conducted with triplicate samples. Symbols above a column indicate a statistical comparison between the indicated sample and the control sample of cells transfected with NT siRNA (*p < 0.05).
Silencing of Rac1 slows cell cycle progression through the G1 phase of the cell cycle in NSCLC cell lines
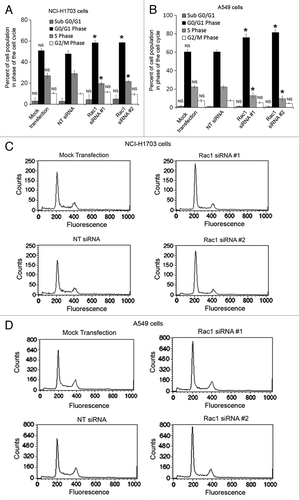
The decrease in proliferation observed in both assays lead us to question whether the siRNA-treated NSCLC cell lines were actually slowing in their proliferation or undergoing apoptosis. To address whether Rac1 siRNA could affect cell cycle progression or induce apoptosis, we conducted cell cycle analysis of the NCI-H1703 and A549 cells using flow cytometry. The cell cycle analysis revealed that Rac1 siRNA slowed cell cycle progression through the G1 phase of the cell cycle and reduced entrance into S phase (). This observation is consistent with previous reports that Rac1 is necessary for progression through the G1/S cell cycle checkpoint.Citation26,Citation27 Interestingly, this analysis also revealed that few cells were undergoing apoptosis or necrosis due to Rac1 siRNA treatment, as indicated by the absence of sub-G1 peaks in the cell cycle histograms (). This finding indicates that Rac1 siRNA slows cell cycle progression rather than inhibiting cell survival.
Figure 3. Silencing of Rac1 in NSCLC cells slows progression through the G1 phase of the cell cycle. Changes in cell cycle progression were determined by transfecting NCI-H1703 cells (A) or A549 (B) cells without siRNA (mock transfection) or with the indicated siRNAs and staining the cells with propidium iodide 72 h post-transfection, followed by flow cytometry analysis. Results are the mean ± SE from three independent experiments. Representative histograms of cell cycle analysis conducted with either NCI-H1703 (C) cells or A549 (D) cells treated with the indicated siRNAs. Symbols above a column indicate a statistical comparison of progression through each phase of the cell cycle by the indicated cells vs. the control cells transfected with NT siRNA (NS, not significant; *p < 0.05).
Silencing of Rac1 inhibits migration in NCI-H1703 cells
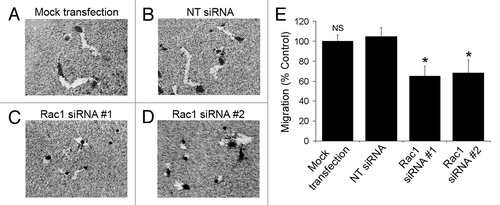
We next tested the ability of Rac1 siRNA-treated NSCLC cells to migrate on collagen-coated plates. The migration was assessed by measuring the area of the trails left by individual cells as they migrated over collagen-coated colloidal gold. Consistent with the known critical role of Rac1 for cell migration,Citation1 silencing of Rac1 in NCI-H1703 cells significantly reduced their ability to migrate ().
Figure 4. Silencing of Rac1 expression slows the migration of individual cells. (A–D) NCI-H1703 cells were transfected without siRNA (mock transfection) or with the indicated siRNAs, incubated for 48 h, and equal numbers of live cells were placed on colloidal gold coated with collagen Type I. Digital images of the cells were collected 24 h later. (E) The areas of the migration trails were measured after the cells migrated for 24 h, and the values were normalized to migration by the mock-transfected cells. Results are the mean ± SE from three independent experiments. Symbols above a column indicate a statistical comparison of migration by the indicated cells compared with control cells transfected with NT siRNA (NS, not significant; *p < 0.05).
Inhibition of Rac1 with NSC23766 reduces proliferation and slows progression through the G1 phase of the cell cycle
In support of our Rac1 siRNA experiments, we also tested the effects of the well-known Rac1 inhibitor NSC23766. First, to assess the effectiveness of NSC23766 on the inhibition of Rac1 and not Rho activity in NSCLC, we performed active Rac and Rho pull-down assays in NCI-H1703 cells treated with or without NSC23766. As expected, NSC23766 effectively decreased Rac1 activation, but did not decrease Rho activation (). We next tested the effect of NSC23766 treatment on NCI-H1703 cell proliferation. Treatment with NSC23766 caused significant dose-dependent loss of cell proliferation () and also decreased the rate of proliferation (). The reduced proliferation induced by NSC23766 was also supported by a slowed progression through the G1 phase of the cell cycle ().
Figure 5. Pharmacological inhibition of Rac1 decreases proliferation and slows progression through the G1 phase of the cell cycle. (A) The effects of NSC23766 on Rac and Rho activity were measured by active Rac or Rho pull-down. Quantitative densitometry was conducted to determine the ratio of active GTPase to total GTPase detected in the immunoblots. The values are normalized to densitometric values obtained from ECL-Western blots of cells not treated with NSC23766. Results are the mean ± SE from three independent experiments. (B) Proliferation of NCI-H1703 cells was assayed by measuring [3H]thymidine uptake after a 24 h incubation with the indicated concentrations of the Rac inhibitor NSC23766. The values are normalized to [3H]thymidine uptake by cells treated without the drug. Results are the mean ± SE from three independent experiments conducted with six replicates for each treatment in each experiment. (C) The rate of proliferation was assessed by measuring [3H]thymidine uptake at the indicated times after incubation with NSC23766 for 24 h. (D) Changes in cell cycle progression were determined by incubation of NCI-H1703 cells with the Rac inhibitor NSC23766 (100 μg/ml) for 24, followed by flow cytometry analysis. (E) Histograms of cell cycle analysis conducted with NCI-H1703 cells treated with or without the Rac1 inhibitor NSC23766. Results are the mean ± SE from three independent experiments. Symbols above a column indicate a statistical comparison of cells treated with NSC23766 and control cells treated without NSC23766 (NS, not significant; *p < 0.01).
![Figure 5. Pharmacological inhibition of Rac1 decreases proliferation and slows progression through the G1 phase of the cell cycle. (A) The effects of NSC23766 on Rac and Rho activity were measured by active Rac or Rho pull-down. Quantitative densitometry was conducted to determine the ratio of active GTPase to total GTPase detected in the immunoblots. The values are normalized to densitometric values obtained from ECL-Western blots of cells not treated with NSC23766. Results are the mean ± SE from three independent experiments. (B) Proliferation of NCI-H1703 cells was assayed by measuring [3H]thymidine uptake after a 24 h incubation with the indicated concentrations of the Rac inhibitor NSC23766. The values are normalized to [3H]thymidine uptake by cells treated without the drug. Results are the mean ± SE from three independent experiments conducted with six replicates for each treatment in each experiment. (C) The rate of proliferation was assessed by measuring [3H]thymidine uptake at the indicated times after incubation with NSC23766 for 24 h. (D) Changes in cell cycle progression were determined by incubation of NCI-H1703 cells with the Rac inhibitor NSC23766 (100 μg/ml) for 24, followed by flow cytometry analysis. (E) Histograms of cell cycle analysis conducted with NCI-H1703 cells treated with or without the Rac1 inhibitor NSC23766. Results are the mean ± SE from three independent experiments. Symbols above a column indicate a statistical comparison of cells treated with NSC23766 and control cells treated without NSC23766 (NS, not significant; *p < 0.01).](/cms/asset/e18f411c-381a-47f8-880f-f99ecf66e3d1/kcbt_a_10920082_f0005.gif)
Silencing or inhibiting Rac1 reduces NF-κB-mediated transcription in NSCLC cells
Our results indicate that the inhibition of Rac1 in NSCLC has multiple adverse effects on the cells’ ability to both proliferate and migrate. Since Rac1 is known to be able to activate NF-κB, which in turn promotes many pathways leading to cell proliferation and migration,Citation18,Citation28,Citation29 we tested the status of NF-κB activation in cells treated with Rac1 siRNA. We conducted a reporter gene assay using an NF-κB promoter driving luciferase gene expression. Silencing of Rac1 in NCI-H1703 cells led to a decrease in NF-κB promoter activity in the absence of TNFα, indicating that Rac1 is needed for basal NF-κB activity in NSCLC (). Treatment with TNFα induced a ~1.5-fold increase in NF-κB activity in the control cells transfected with non-targeting siRNA. Similarly, TNFα induced a ~1.7-fold increase in NF-κB activity in cells treated with Rac1 siRNA (), indicating that TNFα-induced NF-κB activation is still functional in cells treated with Rac1 siRNA. These results indicate that treating cells with Rac1 siRNA diminishes basal NF-κB activity more than it diminishes the TNFα-induced activation of NF-κB in NSCLC cells. We also found that a dose-dependent treatment with NSC23766 significantly diminished basal NF-κB activity (), similar to the effects of Rac1 siRNA. However, unlike the effects of Rac1 siRNA, we found that treatment with higher concentrations of NSC23766 also inhibited TNFα-stimulated NF-κB activity ().
Figure 6. Inhibition of Rac1 reduces NF-κB transcriptional activity and treatment with BAY 11-7082 inhibits NF-κB activity and proliferation. (A) To examine NF-κB activity, NCI-H1703 cells were transfected without siRNA (mock transfection) or with the indicated siRNAs, cultured for 48 h, and then co-transfected with the pNifty-Luc NF-κB reporter plasmid and a β-galactosidase plasmid. After 24 h, the cells were incubated in the absence or presence of TNF-α (100 ng/ml) for 3 h, and luciferase activity was measured. Symbols above a column indicate a statistical comparison of the indicated treatment groups compared with control cells transfected with NT siRNA. (NS, not significant; *p < 0.05). (B and C) Cells were transfected with pNifty-Luc NF-κB reporter plasmid and a β-galactosidase plasmid and then incubated with the indicated concentrations of NSC23766 for 24 h or BAY 11-7082 for 12 h. Cells were then incubated in the absence or presence of TNF-α (100 ng/ml) for 3 h, and luciferase activity was measured. All NF-κB transcriptional activity results are the mean ± SE from three independent experiments normalized to β-galactosidase. (NS, not significant; *p < 0.01). (D) Cells were incubated with the indicated concentrations of BAY 11-7082 for 12 h and proliferation measured by [3H]thymidine uptake at the indicated time points. Symbols above a column indicate a statistical comparison of the indicated treatment groups compared with control cells transfected with NT siRNA or treated without drug (NS, not significant; *p < 0.001).
![Figure 6. Inhibition of Rac1 reduces NF-κB transcriptional activity and treatment with BAY 11-7082 inhibits NF-κB activity and proliferation. (A) To examine NF-κB activity, NCI-H1703 cells were transfected without siRNA (mock transfection) or with the indicated siRNAs, cultured for 48 h, and then co-transfected with the pNifty-Luc NF-κB reporter plasmid and a β-galactosidase plasmid. After 24 h, the cells were incubated in the absence or presence of TNF-α (100 ng/ml) for 3 h, and luciferase activity was measured. Symbols above a column indicate a statistical comparison of the indicated treatment groups compared with control cells transfected with NT siRNA. (NS, not significant; *p < 0.05). (B and C) Cells were transfected with pNifty-Luc NF-κB reporter plasmid and a β-galactosidase plasmid and then incubated with the indicated concentrations of NSC23766 for 24 h or BAY 11-7082 for 12 h. Cells were then incubated in the absence or presence of TNF-α (100 ng/ml) for 3 h, and luciferase activity was measured. All NF-κB transcriptional activity results are the mean ± SE from three independent experiments normalized to β-galactosidase. (NS, not significant; *p < 0.01). (D) Cells were incubated with the indicated concentrations of BAY 11-7082 for 12 h and proliferation measured by [3H]thymidine uptake at the indicated time points. Symbols above a column indicate a statistical comparison of the indicated treatment groups compared with control cells transfected with NT siRNA or treated without drug (NS, not significant; *p < 0.001).](/cms/asset/15463b73-ad6d-4504-8a9e-f01108bb924d/kcbt_a_10920082_f0006.gif)
Inhibition of NF-κB-mediated transcription with BAY 11-7082 reduces proliferation in NCI-H1703 cells
To further investigate the role of NF-κB in NSCLC proliferation, we utilized the NF-κB pathway inhibitor BAY 11-7082. As expected, BAY 11-7082 was able to effectively inhibit both basal and TNFα stimulated NF-κB luciferase activity in a dose dependent manner (). We also found that treatment with varying concentrations of BAY 11-7082 strongly inhibits the rate of proliferation in NCI-H1703 cells (). All together, these results implicate NF-κB in the Rac1-dependent cell proliferation of NSCLC cells.
Discussion
This study demonstrates that Rac1 promotes processes that are associated with the malignant phenotype in NSCLC cells, including cell proliferation and migration. These findings are consistent with previous reports indicating the importance of Rac1 in cancer.Citation1,Citation2 We found that inhibiting Rac1 either by the use of Rac1 siRNAs or NSC23766 significantly reduces NF-κB activity in NSCLC. Additionally, pharmacological inhibition of the NF-κB signaling pathway with BAY 11–7082 also inhibited NSCLC proliferation, similar to that of Rac1 inhibition. NF-κB is known to promote features of the malignant phenotype, including anti-apoptotic signaling in NSCLC cells.Citation30 Previous studies indicate that Rac1 promotes NF-κB activity in other cell types, resulting in enhanced cell proliferation, migration, and survival,Citation12,Citation13 but the ability of Rac1 to regulate NF-κB in NSCLC has not been previously characterized. Our finding that Rac1 promotes NF-κB activity in NSCLC cells may account in part for our finding that Rac1 promotes both cell proliferation and migration in NSCLC cells.
Rac1 may promote NSCLC cell migration through both NF-κB-dependent and -independent pathways. It is well documented that Rac1 can promote migration through its regulation of actin nucleation, branching, and cytoskeletal organization.Citation1 Indeed, we show that Rac1-specifc siRNA reduced single cell migration of H1703 cells. Additionally, Rac1 can promote migration by activating NF-κB, which in turn can induce transcription of various proteins necessary for cell migration.Citation14 Thus, multiple mechanisms undoubtedly contribute to the ability of Rac1 to regulate NSCLC cell migration, a critical process that contributes to metastasisCitation31.
Rac1 can regulate cell proliferation through many different pathways involving JNK, p38, and NF-κB.Citation1,Citation27,Citation32,Citation33 Much of the ability of Rac1 to affect proliferation has been attributed to its ability to regulate progression through the G1/S cell cycle check point.Citation26,Citation27 Consistent with this observation, we found that NSCLC cells arrest in G1 phase after treatment with Rac1 siRNA or NSC23766. There have been reports that Rac1 also promotes progression through the G2 phase of the cell cycle. This response was first reported by Moore et al., who demonstrated that Rat 2 fibroblasts transfected with dominant negative (DN) Rac1 accumulate in G2/M.Citation34 More recently, Michaelson et al. hypothesized that Rac1 is important for cell cycle progression through the G2 phase of the cell cycle, due to their observation that Rac1 accumulates in the nucleus of cells in the G2 phase.Citation35 Interestingly, we did not observe a significant change in the progression of NSCLC cells through the G2 phase when Rac1 activity was suppressed by siRNA or NSC32766.
There are several possible reasons why we observed that suppressing Rac1 activity with siRNA or NSC32677 diminishes progression though the G1 phase rather than the G2 phase of the cell cycle. First, our assays were conducted using asynchronous populations, and it is possible that any changes in G2 phase progression were not detectable in our assays. Second, previous studies did not examine NSCLC cells and, therefore, differences observed between these previous studies and our study utilizing NSCLC cells may be due to cell type-specific responses. Finally, and perhaps most importantly, previous studies reporting that Rac1 regulates G2 progression utilized DN-Rac1 to suppress Rac1 activity,Citation34 rather than Rac1 siRNA or NSC23766. Cell cycle progression might be affected in a unique way by the use of DN-Rac1. DN-Rac1 competitively binds GEFs that are normally bound by endogenous Rac1. This competitive inhibition diminishes the interaction of Rac1 GEFs with endogenous Rac1 and with any other endogenous small GTPases that normally interact with these Rac1 GEFs. There are several Rac1 GEFs that can bind other small GTPases in addition to Rac1. For example, VAV2 has been shown to activate Rac1, Cdc42, and RhoA,Citation36 whereas DOCK6 has been shown to activate Rac1 and Cdc42.Citation37,Citation38 Thus, the interaction of DN-Rac1 with GEFs such as VAV2 and DOCK6 might suppress the activities of RhoA and Cdc42, in addition to Rac1. Therefore, G2 arrest induced by DN-Rac1 might be due to the inhibition of pathways mediated by multiple small GTPases in addition to Rac1.
Since we found that Rac1 can affect the proliferation of NSCLC cells in an anchorage-dependent environment, we investigated whether Rac1 can also affect proliferation in an anchorage-independent environment, a hallmark of cancer. We found that silencing of Rac1 in NCI-H1703 cells significantly reduces colony formation in an anchorage-independent environment. This assay only measured the number of colonies and not the size of each colony. Therefore, it is possible that silencing of Rac1 was affecting cell survival in soft agar, rather than cell proliferation. Interestingly, NF-κB is also known to regulate the ability of cells to grow in anchorage-independent environmentsCitation39 and has been reported to promote cell metastasis, a process which requires the ability of cells to survive in an anchorage-independent environment.Citation40-Citation43 The ability of Rac1 to regulate both anchorage-dependent and -independent proliferation in NSCLC may be only partly explained by the ability of Rac1 to regulate NF-κB activity in NSCLC. Rac1 has been shown to regulate proliferation by many mechanisms in addition to NF-κB activity.Citation1,Citation2 Therefore, further investigation will be necessary to elucidate the many possible pathways by which Rac1 can regulate proliferation through both NF-κB-dependent and –independent mechanisms in NSCLC.
We found that NSCLC cell survival was not detectably altered by treating the cells with NSC23766 or Rac1 siRNA, as indicated by the lack of sub-G1 signals in our cell cycle analysis. This result might be viewed as surprising, since these treatments decreased NF-κB activity, and NF-κB is classically known as a pro-survival factor that can induce the transcription of anti-apoptotic genes.Citation30 Several reasons could account for the lack of effect of NSC23766 or Rac1 siRNA on NSCLC cell survival. It is probable that NSCLC cells have many mechanisms which suppress apoptosis, and therefore the reduced activity of NF-κB in cells treated with NSC23766 or Rac1 siRNA might not have been sufficient to induce apoptosis. Additionally, the role of NF-κB in anti-apoptotic signaling in NSCLC cells is believed to arise mainly from NF-κB suppressing apoptosis induced by stressors such as irradiation and chemotherapy.Citation30 The absence of these stressors in our assays may account for our finding that loss of NF-κB activity in the treated cells did not detectably alter their survival. However, further experimentation is necessary to fully define possible Rac1-mediated changes in apoptosis.
We observed that many of the responses induced by Rac1 siRNA were also induced by treating the cells with NSC23766. However, some unique responses were induced by NSC23766. Most notably, we found that NSC23766, at higher concentrations, decreases both basal NF-κB activity and TNFα-induced NF-κB activation. In contrast, Rac1 siRNA decreases basal NF-κB activity but does not significantly inhibit TNFα-induced NF-κB activation. Additionally, NSCLC cell proliferation was inhibited more effectively by treating the cells with NSC23766 than by Rac1 siRNA.
The different effects of Rac1 siRNA and NSC23766 on proliferation and NF-κB activity may be explained by their different modes of action. Rac1 siRNA reduces the amount of available Rac1 for signaling but does not completely eliminate its expression (). Therefore, there is still a small amount of active Rac1 available for signaling in siRNA-treated cells. In contrast, NSC23766 is a chemical inhibitor which can inhibit Rac1 activity by directly binding to Rac1, thereby inhibiting the ability of Rac1 to interact with Rac1-specific GEFs, such as Tiam1 and Trio.Citation23 This pharmacological inactivation has the potential to inhibit nearly all of the Rac1 present in the cell and, therefore, might be expected to produce a more potent effect as compared with Rac1 siRNA treatment. In support of this hypothesis, only at higher concentrations did NSC23766 inhibit both basal and TNFα-mediated NF-κB luciferase activity. In addition, although NSC23766 was previously shown to inhibit Rac1 activation by Tiam1 and Trio, NSC23766 did not inhibit Rac1 activation by Vav1, another Rac1-specific GEF.Citation23 Therefore, NSC23766 does not inhibit Rac1 activation by all Rac1-specific GEFs and thus some Rac1 signaling pathways may still be fully functional. This is yet another way in which it may be possible to observe differences in the phenotypic outcome when using Rac1-specifc siRNA, NSC23766, or DN-Rac1to inhibit Rac1 function.
The observation that treatments with Rac1 siRNA, DN-Rac1, or NSC23766 produce different phenotypic outcomes raises some interesting questions regarding the mechanisms by which Rac1 mediates its effects in cells, as well as the clinical implications of targeting Rac1 in the treatment of lung cancer. Our study and others have clearly shown the importance of Rac1 and other small GTPases in cancer, which makes them attractive targets in the treatment of cancer. However, the direct targeting of small GTPases with small molecule inhibitors has been difficult. Additionally, although the Rac1 inhibitor NSC23766 works well in tissue culture, the IC50 of NSC23766 is potentially too high for clinical use.Citation44 Optimistically, one might hope that modifications to NSC23766 could be made to improve its ability to be used therapeutically. Indeed, attempts to improve the efficacy of NSC23766 have been made with modest success.Citation44 More recently, another Rac1 inhibitor (EHT 1864) has been developed which inhibits guanine nucleotide association, thus rendering Rac1 inactive.Citation45-Citation47
The results of this study demonstrate the important roles of Rac1 in NSCLC cell proliferation and migration and compare the effects of Rac1-specific siRNA to that of Rac1 inhibition by NSC23766. One possible mechanism contributing to these multiple roles of Rac1 may be through the Rac1-dependent regulation of NF-κB transcriptional activity in NSCLC. Our studies highlight the importance of Rac1 in NSCLC and supports the idea that Rac1, and Rac1-related pathways, may be viable therapeutic targets in the treatment of lung cancer.
Materials and Methods
Cell culture, siRNA transfection, and NSC23766 or BAY 11-7082 treatments
The NSCLC cell lines NCI-H1703, NCI-H23, NCI-H1650 and A549 were obtained from the American Type Tissue Collection (Bethesda) and cultured in complete NSCLC medium (RPMI 1640, 10% heat-inactivated fetal bovine serum, and antibiotics). All siRNA duplexes were purchased from Dharmacon. Two different Rac1-specific siRNA sequences were tested and named Rac1 siRNA #1 (5′ - UUUACCUACAGCUCCGUCUUU - 3′) and Rac1 siRNA #2 (5′ - UACAGCACCAAUCUCCUUAUU - 3′). Non-targeting (NT) siRNA (5′ - UGGUUUACAUGUUUUCUGA - 3′), which does not target any known human genes, was also tested. The cells were transfected with 25 nM siRNA using Dharmafect-3 transfection reagent (Dharmacon T-2003-03) according to the manufacturer’s instructions, and incubated for 72 h. In some assays, cells were treated with the Rac1 specific inhibitor NSC23766 from EMD Biosciences (Cat #553502) for 24 h or the IKK inhibitor BAY 11–7082 from Sigma Aldrich (Cat #B5556) for 12 h.
Enhanced chemiluminescence (ECL) western blotting
Equal numbers of transfected cells were boiled in 2X Laemmli sample buffer and subjected to SDS-PAGE. The proteins were transferred to PVDF and immunoblotted using an antibody to Rac1 (BD Transduction Labs 610651) or GAPDH (Santa Cruz Biotechnology sc-32233). Bound antibodies were visualized using HRP-linked anti-mouse IgG (GE Healthcare NA931) and ECL reagents (Perkin Elmer Life Sciences NEL105001EA).
[3H]Thymidine uptake
Cells were transfected with siRNA in 96-well microtiter plates and then cultured for 72 h in complete NSCLC medium, treated with NSC23766 for 24 h, or treated with BAY 11-7082 for 12 h. Cells were incubated with [3H]thymidine for 3 h or for the indicated time points after the treatment with siRNA or pharmacological inhibitor. The cells were collected on filters using an automatic cell harvester (Skatron) and radioactivity on the filters was measured by β-scintillation counting, as previously described.Citation48
Cell cycle analysis
Treated cells were fixed in 50% ethanol, incubated with RNase cocktail consisting of 500 U/ml RNase A and 20,000 U/ml RNase T1 (Ambion 2286) for 30 min, and stained with propidium iodide (100 μg/ml). The cells were examined using flow cytometry and the results were analyzed using the ModFit program.
Cell migration
The migration of individual cells was examined by plating transfected cells at a density of 3 x 103 cells/600 μl complete medium in wells of a 24-well plate coated with collagen Type 1 (10 μg/ml in PBS) and colloidal gold, as previously described.Citation49 Digital images of the cells were collected after 24 h. The areas of the migration trails and cell bodies were analyzed using the NIH Image-J software program. Cells that were in contact with other cells, or whose migration trails crossed those of other cells, were not included in the analysis.
Clonogenic assay
Cells were transfected with the indicated siRNA and incubated for 24 h. The cells were then re-plated in soft agar at a final concentration of 2.2 x 103 live cells/well and cultured for 5 weeks. Digital images of the wells were obtained and colony formation was assessed by counting the number of detectable colonies.
Luciferase reporter gene assay
Treated cells were co-transfected with the pNifty-Luc NF-κB-Luciferase reporter cDNA and a β-galactosidase cDNA using Lipofectamine 2000 (Invitrogen, 11668-019) following the manufacturer’s guidelines, and cultured for 24 h. Transfected cells were then incubated in the absence or presence of TNF-α (100 ng/ml) for 3 h. Luciferin (0.15 mg/ml) was then added to each well, followed by measurement of luminescence using a FLUOstar Omega (BMG Labtech) plate reader. Cells were washed with PBS and incubated with β-galactosidase assay reagent (Pierce 75705) for 30 min. Absorbance at 405 nm was measured using a colorimetric plate reader. NF-κB activity was defined as the luminescence normalized to the optical density of the colorimetric detection of β-galactosidase.
Rac and Rho Activation Assays
Cells were treated with or without NSC23766 for 24 h and activities were assessed by active GTPase pull-down using Cytoskeleton Rac or Rho activation kits (Cat #s BK035, BK036) according to manufacturer’s protocols. Lysates were subjected to SDS-PAGE, transferred to a PVDF membrane, and western blotting performed with Rac1 (BD Transduction Labs 610651) or RhoA (Cytoskeleton ARH03) antibodies.
| Abbreviations: | ||
| DN | = | dominant negative |
| GEF | = | guanine nucleotide exchange factor |
| GTP | = | guanosine-5′-triphosphate |
| IKK | = | IkappaB Kinase |
| NF-κB | = | nuclear factor-kappaB |
| NSCLC | = | non-small cell lung carcinoma |
| siRNA | = | small interfering ribonucleic acid |
| TNFα | = | tumor necrosis factor alpha |
Acknowledgments
This work was supported by funding from NCI R01 CA136799 and Advancing a Healthier Wisconsin.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci 2009; 66:370 - 4; http://dx.doi.org/10.1007/s00018-008-8552-x; PMID: 19151919
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008; 582:2093 - 101; http://dx.doi.org/10.1016/j.febslet.2008.04.039; PMID: 18460342
- Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, Sweet-Cordero A, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res 2007; 67:8089 - 94; http://dx.doi.org/10.1158/0008-5472.CAN-07-2300; PMID: 17804720
- Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol 1997; 17:1324 - 35; PMID: 9032259
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature 1995; 374:457 - 9; http://dx.doi.org/10.1038/374457a0; PMID: 7700355
- Liu Y, Wang Y, Zhang Y, Miao Y, Zhao Y, Zhang PX, et al. Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer 2009; 63:375 - 82; http://dx.doi.org/10.1016/j.lungcan.2008.12.012; PMID: 19162367
- Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, et al. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000; 19:3013 - 20; http://dx.doi.org/10.1038/sj.onc.1203621; PMID: 10871853
- Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer 1999; 81:682 - 7; http://dx.doi.org/10.1002/(SICI)1097-0215(19990531)81:5<682::AID-IJC2>3.0.CO;2-B; PMID: 10328216
- Jordan P, Brazåo R, Boavida MG, Gespach C, Chastre E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999; 18:6835 - 9; http://dx.doi.org/10.1038/sj.onc.1203233; PMID: 10597294
- Yuan K, Qian C, Zheng R. Prognostic significance of immunohistochemical Rac1 expression in survival in early operable non-small cell lung cancer. Med Sci Monit 2009; 15:BR313 - 9; PMID: 19865048
- Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 2009; 8:33 - 40; http://dx.doi.org/10.1038/nrd2781; PMID: 19116625
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006; 441:431 - 6; http://dx.doi.org/10.1038/nature04870; PMID: 16724054
- Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 2006; 25:6817 - 30; http://dx.doi.org/10.1038/sj.onc.1209942; PMID: 17072330
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 2004; 18:2195 - 224; http://dx.doi.org/10.1101/gad.1228704; PMID: 15371334
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell 2002; 109:Suppl S81 - 96; http://dx.doi.org/10.1016/S0092-8674(02)00703-1; PMID: 11983155
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol 2002; 3:221 - 7; http://dx.doi.org/10.1038/ni0302-221; PMID: 11875461
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol 2000; 1:533 - 40; http://dx.doi.org/10.1038/82797; PMID: 11101877
- Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol 1996; 16:7115 - 21; PMID: 8943367
- Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 2011; 30:1615 - 30; http://dx.doi.org/10.1038/onc.2010.566; PMID: 21170083
- Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene 2009; 28:3597 - 607; http://dx.doi.org/10.1038/onc.2009.217; PMID: 19617897
- Chen QY, Xu LQ, Jiao DM, Yao QH, Wang YY, Hu HZ, et al. Silencing of Rac1 modifies lung cancer cell migration, invasion and actin cytoskeleton rearrangements and enhances chemosensitivity to antitumor drugs. Int J Mol Med 2011; 28:769 - 76; PMID: 21837360
- Jayachandran G, Sazaki J, Nishizaki M, Xu K, Girard L, Minna JD, et al. Fragile histidine triad-mediated tumor suppression of lung cancer by targeting multiple components of the Ras/Rho GTPase molecular switch. Cancer Res 2007; 67:10379 - 88; http://dx.doi.org/10.1158/0008-5472.CAN-07-0677; PMID: 17974981
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 2004; 101:7618 - 23; http://dx.doi.org/10.1073/pnas.0307512101; PMID: 15128949
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, et al. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem 1997; 272:21096 - 103; http://dx.doi.org/10.1074/jbc.272.34.21096; PMID: 9261113
- Gasparian AV, Guryanova OA, Chebotaev DV, Shishkin AA, Yemelyanov AY, Budunova IV. Targeting transcription factor NFkappaB: comparative analysis of proteasome and IKK inhibitors. Cell Cycle 2009; 8:1559 - 66; http://dx.doi.org/10.4161/cc.8.10.8415; PMID: 19372735
- Page K, Li J, Hodge JA, Liu PT, Vanden Hoek TL, Becker LB, et al. Characterization of a Rac1 signaling pathway to cyclin D(1) expression in airway smooth muscle cells. J Biol Chem 1999; 274:22065 - 71; http://dx.doi.org/10.1074/jbc.274.31.22065; PMID: 10419534
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 1995; 269:1270 - 2; http://dx.doi.org/10.1126/science.7652575; PMID: 7652575
- Miyano K, Ueno N, Takeya R, Sumimoto H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J Biol Chem 2006; 281:21857 - 68; http://dx.doi.org/10.1074/jbc.M513665200; PMID: 16762923
- Moldovan L, Mythreye K, Goldschmidt-Clermont PJ, Satterwhite LL. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc Res 2006; 71:236 - 46; http://dx.doi.org/10.1016/j.cardiores.2006.05.003; PMID: 16782079
- Denlinger CE, Rundall BK, Jones DR. Modulation of antiapoptotic cell signaling pathways in non-small cell lung cancer: the role of NF-kappaB. Semin Thorac Cardiovasc Surg 2004; 16:28 - 39; http://dx.doi.org/10.1053/j.semtcvs.2003.12.004; PMID: 15366685
- Sun D, Xu D, Zhang B. Rac signaling in tumorigenesis and as target for anticancer drug development. Drug Resist Updat 2006; 9:274 - 87; http://dx.doi.org/10.1016/j.drup.2006.12.001; PMID: 17234445
- Chiariello M, Marinissen MJ, Gutkind JS. Regulation of c-myc expression by PDGF through Rho GTPases. Nat Cell Biol 2001; 3:580 - 6; http://dx.doi.org/10.1038/35078555; PMID: 11389443
- Aznar S, Lacal JC. Rho signals to cell growth and apoptosis. Cancer Lett 2001; 165:1 - 10; http://dx.doi.org/10.1016/S0304-3835(01)00412-8; PMID: 11248412
- Moore KA, Sethi R, Doanes AM, Johnson TM, Pracyk JB, Kirby M, et al. Rac1 is required for cell proliferation and G2/M progression. Biochem J 1997; 326:17 - 20; PMID: 9337845
- Michaelson D, Abidi W, Guardavaccaro D, Zhou M, Ahearn I, Pagano M, et al. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 2008; 181:485 - 96; http://dx.doi.org/10.1083/jcb.200801047; PMID: 18443222
- Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, et al. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem 2000; 275:10141 - 9; http://dx.doi.org/10.1074/jbc.275.14.10141; PMID: 10744696
- Miyamoto Y, Yamauchi J, Sanbe A, Tanoue A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp Cell Res 2007; 313:791 - 804; http://dx.doi.org/10.1016/j.yexcr.2006.11.017; PMID: 17196961
- Kulkarni K, Yang J, Zhang Z, Barford D. Multiple factors confer specific Cdc42 and Rac protein activation by dedicator of cytokinesis (DOCK) nucleotide exchange factors. J Biol Chem 2011; 286:25341 - 51; http://dx.doi.org/10.1074/jbc.M111.236455; PMID: 21613211
- Jiang Y, Cui L, Yie TA, Rom WN, Cheng H, Tchou-Wong KM. Inhibition of anchorage-independent growth and lung metastasis of A549 lung carcinoma cells by IkappaBbeta. Oncogene 2001; 20:2254 - 63; http://dx.doi.org/10.1038/sj.onc.1204293; PMID: 11402320
- Wu K, Bonavida B. The activated NF-kappaB-Snail-RKIP circuitry in cancer regulates both the metastatic cascade and resistance to apoptosis by cytotoxic drugs. Crit Rev Immunol 2009; 29:241 - 54; PMID: 19538137
- Royuela M, Rodríguez-Berriguete G, Fraile B, Paniagua R. TNF-alpha/IL-1/NF-kappaB transduction pathway in human cancer prostate. Histol Histopathol 2008; 23:1279 - 90; PMID: 18712680
- Brown M, Cohen J, Arun P, Chen Z, Van Waes C. NF-kappaB in carcinoma therapy and prevention. Expert Opin Ther Targets 2008; 12:1109 - 22; http://dx.doi.org/10.1517/14728222.12.9.1109; PMID: 18694378
- Holcomb B, Yip-Schneider M, Schmidt CM. The role of nuclear factor kappaB in pancreatic cancer and the clinical applications of targeted therapy. Pancreas 2008; 36:225 - 35; http://dx.doi.org/10.1097/MPA.0b013e31815b3207; PMID: 18362834
- Hernández E, De La Mota-Peynado A, Dharmawardhane S, Vlaar CP. Novel inhibitors of Rac1 in metastatic breast cancer. P R Health Sci J 2010; 29:348 - 56; PMID: 21261173
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem 2007; 282:35666 - 78; http://dx.doi.org/10.1074/jbc.M703571200; PMID: 17932039
- Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol 2008; 439:111 - 29; http://dx.doi.org/10.1016/S0076-6879(07)00409-0; PMID: 18374160
- Désiré L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem 2005; 280:37516 - 25; http://dx.doi.org/10.1074/jbc.M507913200; PMID: 16150730
- Strassheim D, Shafer SH, Phelps SH, Williams CL. Small cell lung carcinoma exhibits greater phospholipase C-beta1 expression and edelfosine resistance compared with non-small cell lung carcinoma. Cancer Res 2000; 60:2730 - 6; PMID: 10825148
- Varker KA, Williams CL. Involvement of the muscarinic acetylcholine receptor in inhibition of cell migration. Biochem Pharmacol 2002; 63:597 - 605; http://dx.doi.org/10.1016/S0006-2952(01)00901-7; PMID: 11992627