Abstract
Previously, we demonstrated potent antineoplastic activity of a distinctive histone deacetylase inhibitor (HDACI), AR42, against chemoresistant CP70 ovarian cancer cells in vitro and in vivo. Here, in follow-up to that work, we explored AR42 global mechanisms-of-action by examining drug-associated, genome-wide microRNA and mRNA expression profiles, which differed from those of the well-studied HDACI vorinostat. Expression of microRNA genes in negative correlation with their “target” coding gene (mRNA) transcripts, and transcription factor genes with expression positively correlated with coding genes having their cognate binding sites, were identified and subjected to gene ontology analyses. Those evaluations showed AR42 gene expression patterns to negatively correlate with Wnt signaling (> 18-fold induction of SFRP1), the epithelial-to-mesenchymal transition (40% decreased ATF1), and cell cycle progression (33-fold increased 14-3-3σ). By contrast, AR42 transcriptome alterations correlated positively with extrinsic (“death receptor”) apoptosis (> 2.3-fold upregulated DAPK) and favorable ovarian cancer histopathology and prognosis. Inhibition of Wnt signaling was experimentally validated by: (1) > 2.6-fold reduced Wnt reporter activity; and (2) 36% reduction in nuclear, activated β-catenin. Likely AR42 induction of multiple (type I or type II autophagic) cell death cascades was further supported by 57% decreased reliance upon reactive oxygen, increased mitochondrial membrane disruption, and caspase independence, as compared with vorinostat. Taken together, we demonstrate distinct antineoplastic pathway alterations, in aggressive ovarian cancer cells, following treatment with a promising HDACI, AR42. These combined computational and experimental approaches may also represent a straightforward means for mechanistic studies of other promising antineoplastics, and/or the identification of agents that may complement epigenetic therapies.
Introduction
Precise regulation of post-translational protein acetylation/deacetylation is vital to normal physiological homeostasis, includes essential biological processes such as transcription factor-to-DNA binding, chromatin structure (thus influencing gene expression), “chaperone”-mediated protein stabilization, and intracellular transport.Citation1,Citation2 Consequently, alterations in the balance of acetylation and deacetylation contributes to several pathologies, including cancer, metabolic disorders, neurodegenerative conditions, and inflammation.Citation1,Citation2
In the late 1990s, mechanistic studies of a number of leukemia cell-differentiating hydroxamic acids revealed these agents to enhance histone acetylation, via the inhibition of histone deacetylase enzymes.Citation3 Subsequent preclinical and clinical studies led to FDA approval of two such “histone deacetylase inhibitors” (HDACIs), suberoylanilide hydroxamic acid (SAHA, vorinostat) and the cyclic peptide romidepsin (depsipeptide), for cutaneous T-cell lymphoma.Citation4 Likewise, recent clinical trials of hematologic cancers demonstrated efficacy of other hydroxamate HDACIs, including belinostat, panobinostat, and SB939, while promising non-hydroxamate HDACIs include entinostat (formerly MS-275), apicidin, and an isoform-selective HDACI, mocetinostat (previously MGCD0103) (for more extensive HDACI review, see references Citation3 and Citation4). Preclinical studies by our group and others have similarly shown potential therapeutic efficacy of a “rationally designed” HDACI, AR42 (formerly OSU-HDAC42, Arno Therapeutics),Citation5 against multiple myeloma, adult T-cell lymphomas, and solid tumors of the ovary, liver, and prostate, both singly or combined with other agents.Citation6-Citation12
Various molecular mechanisms/processes have now been advanced to explain HDACI cancer cell-specific cytotoxicity (and non-toxicity toward normal cells), including upregulation of pro-apoptotic genes, downregulation of anti-apoptotic genes, abrogation of cell cycle checkpoints, and increased production of reactive oxygen species (concurrent with diminished anti-oxidative response).Citation2,Citation4 With some exceptions, however, many of those previously identified anticancer mechanisms were based on limited numbers of HDACI-altered genes, absent rigorous assessments of HDACI cumulative effects on signal pathways/ networks.Citation2,Citation4 In ovarian cancer specifically, clinical trials of single-agent HDACIs have shown marginal activity against high-grade serous (HGS) disease, but some efficacy against borderline cancer.Citation13,Citation14 Several preclinical studies of HGS ovarian cancer, however, have demonstrated potent antitumor activity of HDACIs when combined with conventional therapies, likely via resensitization of drug-resistant subclones (reviewed in referencesCitation14-Citation16). However, the precise mechanism(s) of HDACI-mediated chemosensitization, with regard to specific signal pathways/networks, remains somewhat speculative.Citation4,Citation15
In concert with aberrant protein acetylation, dysregulated expression of microRNAs (miRNAs) is also strongly linked to the progression of numerous solid tumors, including ovarian cancer.Citation14,Citation17-Citation21 Even so, relatively few reports have described HDACI effects on these non-coding, post-transcriptional gene product regulators. In chronic lymphocytic leukemia cells, the HDACI LAQ824 was shown to upregulate three silenced tumor suppressive miRNAs,Citation22 while HDACI treatment resulted in upregulation of 22, and downregulation of 10 miRNAs in breast cancer cells.Citation23 Similarly, numerous miRNAs were found dysregulated by vorinostat in colonCitation24 and lungCitation25 cancer cells (in concert with p53 gain-of-function), while LAQ824 altered the expression of over 60 miRNAs in breast cancer cells.Citation26 Another miRNA study, however, reported no HDACI effects on miRNA expression, using a panel of 91 cancer cell lines.Citation27 Consequently, in this follow-up to our previous study of AR42 chemosensitizing activity in vitro and in vivo,Citation6 we assessed the genome-wide effects of AR42 on both miRNA and protein-coding (mRNA) gene expression in drug-resistant ovarian cancer cells. Bioinformatic analyses of this data, combined with experimental validation, revealed AR42 to exhibit discrete (but likely cooperative) pleiotropic effects on multiple signal pathways that may jointly oppose progression of this lethal gynecological malignancy.
Results
HDAC inhibitor effects on microRNA and mRNA expression
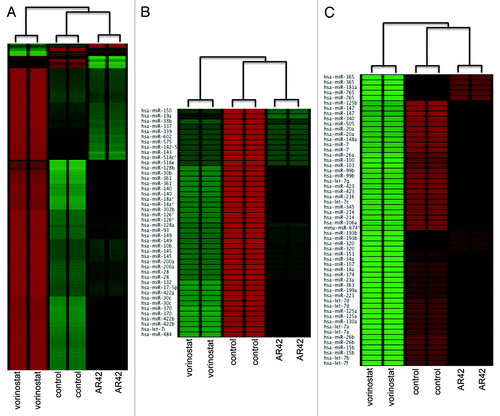
Previously, we demonstrated AR42 to differentially resensitize drug-resistant ovarian cancer cells and xenograft tumors to cisplatin, in parallel with the well-known HDACI vorinostat.Citation6 Based on that work, and other studies showing HDACI effects on microRNA (miRNA) expression,Citation22-Citation26 we further compared AR42 and vorinostat for differential effects on global miRNA and protein-coding gene (mRNA) expression levels. In accord with their similar structure/functional groups (hydroxamic acids, which act to chelate the essential HDAC cofactor Zn2+),Citation5 the two HDACIs exerted comparable expression effects on a large number of genes (). However, hierarchical clustering analysis also revealed distinct gene expression patterns (“clusters”) between duplicate arrays of untreated, AR42-, and vorinostat-treated CP70 cells (). Likewise, while AR42 and vorinostat similarly altered the expression of several miRNA genes, we also observed the two HDACI miRNA profiles to distinctly cluster, as compared with the untreated (DMSO vehicle) control ( and C). These discrete miRNA and gene expression clusters validated sample reproducibility (thus qualifying the data for additional analyses), thus justifying further examination of the genome-wide effects of AR42 on signal transduction pathways.Citation28,Citation29
Figure 1. Unsupervised cluster analysis of (A) coding-gene expression (Nimblegen 385K Gene Expression); (B) downregulation of mature; and (C) upregulation of mature microRNAs. Microarray data (124-feature custom array)Citation71 was subjected to quantile normalizationCitation72 and unsupervised clustering performed using EXPANDER.Citation29
Functional analyses of AR42 alterations in protein-coding and microRNA gene expression
To examine the global bases for the antineoplastic effects of AR42, we first identified transcription factor (TF) binding sites (TFBSs) localized within 4000 bp of the transcription start sites of AR42-differentially expressed genes (), using the webtool ECR (evolutionary conserved region) Browser.Citation30 We then examined the expression of TFs matching those TFBSs, followed by assay of TF expression changes with those of their TFBS-possessing “target” genes.Citation30,Citation31 The matched TF/target genes were then subjected to Gene Ontology (GO) analysis, using the web tool BiNGO,Citation32 revealing AR42-associated gene expression profiles to negatively correlate with the GO biological processes “epithelial-to-mesenchymal transition” (EMT, GO:0001837) and “canonical Wnt receptor signaling” (GO:0060070) (). Consistent with AR42 negative regulation of those tumor progression cascades, we also noted upregulation of genes repressive of the EMT (e.g., ONECUT2, WT1) and Wnt (e.g., SFRP1 ICAT, TAK1) pathways ( and , respectively). By contrast, the expression of AR42-altered TF/target gene transcription positively correlated with the GO terms “negative regulation of cell cycle processes” (GO:0010948) () and “induction of apoptosis by extracellular signals” (i.e., extrinsic apoptosis, GO:0008624) (). With regard to the latter two pathways, and in contrast with the Wnt and EMT cascades, AR42-upregulated agonists (14–3-3σ, p21, DAPK, and RIP), and downregulated antagonists (TopoII, BCL2) of cell cycle arrest and death receptor signaling ( and ). Overall, these four AR42-associated GO pathways concur with our previously observed AR42 antineoplastic effects on ovarian cancer cells, including G2/M cell cycle arrest (via repression of genes that stimulate that pathway), induction of epithelial morphology, and extensive apoptosis (following AR42 treatment in combination with the DNA-crosslinking agent cisplatin).Citation6
Table 1. Gene Ontology (GO) terms identified from gene expression changes following 48-h AR42 treatment
Table 2. Microarray AR42-induced expression changes in gene members of the GO term EPITHELIAL_TO_MESENCHYMAL_TRANSITION
Table 3. AR42-induced changes in members of the GO term CANONICAL_WNT_RECEPTOR_ SIGNALING_PATHWAY
Table 4. Selected AR42-altered genes in the GO term NEGATIVE_REGULATION_OF_CELL_CYCLE
Table 5. Selected AR42-altered genes in the GO term APOPTOTIC_PROCESS
Concordance of AR42 microRNA and mRNA expression profiles to pathway “gene expression” signatures
To further extend our functional analysis and provide greater mechanistic detail of AR42 antineoplastic activity, we examined drug-associated gene profiles for alignment to various published “gene expression signatures.” To first affirm that AR42 effects were indeed due to deacetylase inhibition, we showed AR42 gene alterations to positively correlate with an “HDAC inhibitor common gene set” expression signatureCitation33 (), including upregulation of CDNK1A (p21CIP1), a “benchmark” gene induced by all inhibitors of class I and class II HDACs.Citation2,Citation4
Figure 2. Comparison of AR42-associated gene expression changes for alignment with expression signatures for (A) genes commonly altered by 14 separate HDACIs;Citation33 (B) a set of 100 genes downregulated in cancer cells undergoing the epithelial-to-mesenchymal transition;Citation34 and (C) an 84-gene signature of upregulated genes in cancer cells possessing active signaling of the oncogenic mitogenic pathway Wnt (KEGG pathway hsa04310).Citation35
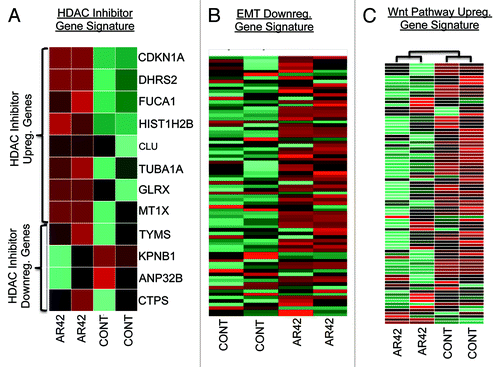
We also examined the AR42 transcriptome for expression of signature genes downregulated by EMT in breast cancer cells,Citation34 again supporting an AR42 inversely correlated expression profile (). Likewise, for Wnt signaling, we compared AR42 changes in gene expression to a publically available Wnt-upregulated gene signature (KEGG pathway hsa04310).Citation35 Similar to the inverse EMT profile, AR42 downregulated the majority of those (Wnt-upregulated) signature genes (), thus further supporting AR42 inhibition of Wnt signaling.
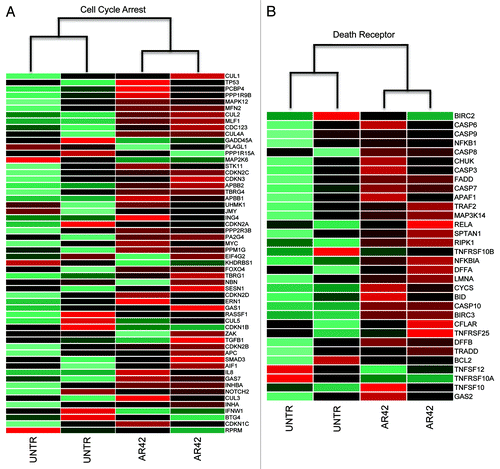
Similarly, we compared AR42 expression changes to other gene signatures of cell cycle arrest and death receptor-mediated apoptosis. Those evaluations revealed that in contrast to the EMT and Wnt pathways, AR42 transcriptional changes positively correlated with cell cycle arrest (KEGG pathway map04110) and death receptor apoptosis (MSigDB identifier M16635) gene expression signaturesCitation35 (, respectively). Within the cell cycle arrest signature, we noted upregulation of the cyclin-dependent kinase inhibitors CDKN1A (p21CIP1), CDKN1B (p15INK4B), and CDKN2D (p19INK4D), in addition to the p53 target gene and coactivator 14–3-3σ (). Interestingly, we also noted a slight (0.7-fold) downregulation of p53 (), which is overexpressed and dysfunctional in CP70 cells (likely contributing to chemotherapy resistance).Citation36,Citation37 These gene signature results concur with our prior observation of AR42-associated cell cycle arrest and epithelial differentiation, and previous studies demonstrating AR42 repression of oncogenic cascades, concurrent with derepression of tumor suppressive regulators, in various cancer types.Citation5-Citation11
Figure 3. Assessment of AR-altered expression patterns for signatures of (A) genes upregulated during cell cycle arrest (KEGG pathway map04110)Citation35 and (B) genes upregulated during “death receptor” (extrinsic) apoptosis (MSigDB identifier M16635).Citation35
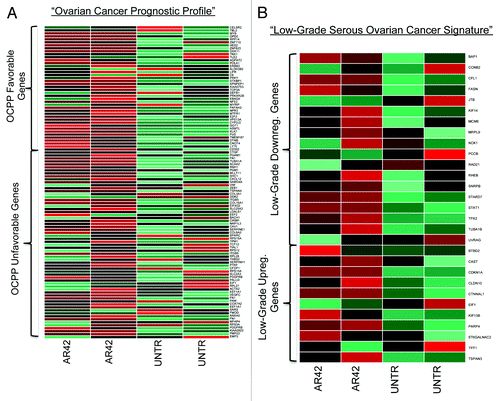
We also examined AR42 for disease-specific activity against ovarian cancer, by comparing AR42 expression patterns to two ovarian cancer clinically informative transcription signatures. Those analyses demonstrated AR42 expression changes to positively correlate with gene expression signatures for favorable prognosisCitation38 and low-grade serous (as compared with high-grade serous, HGS) diseaseCitation39 (). To further support specific antitumor activity against HGS malignancy, we also observed AR42 downregulation of various ovarian cancer oncogenic miRNAs, including miR-21 and miR-30 (),Citation18,Citation21 and upregulation of previously reported ovarian tumor-suppressive miRNAs, including those of the let-7 family and miRs 99, 100, and 125Citation17,Citation19,Citation21 (). Moreover, two other AR-42-upregulated miRNAs, miR-15 and miR-34 (a p53 target gene), were previously reported as epigenetically downregulated in ovarian cancer.Citation17,Citation19 Taken together, these gene set enrichment and ontology analyses support AR42 anti-ovarian cancer activity by circumvention of two aggressive tumor progression cascades, EMT and Wnt, coincident with upregulation of two growth-limiting cascades, cell cycle arrest and death receptor signaling.
Figure 4. Ovarian cancer-specific gene signature alterations by AR42 (A) with positive vs. negative ovarian cancer patient prognosis. Upper group represents genes whose expression correlates with favorable prognosis; lower group represents genes expressed in unfavorable prognosis (see reference Citation38). (B) AR42 alterations of genes correlated with low-grade ovarian adenocarcinoma histopathology (upper gene group are upregulated with low-grade pathology, while the lower gene group represent those downregulated with low-grade pathology (see reference Citation39).
Experimental assessment of AR42 effects on Wnt pathway signal transduction
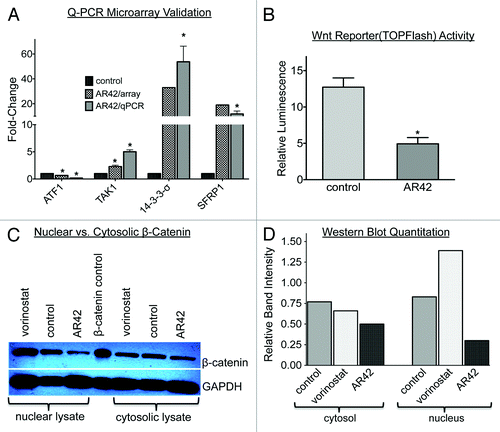
Based on our GO analyses (), we validated expression levels of selected genes from the various abovementioned pathways by quantitative, reverse transcription PCR (qRT-PCR), affirming upregulation of the Wnt antagonists SFRP1 and TAK1, the cell cycle inhibitor 14–3-3σ, and downregulation an EMT-activating TF gene, ATF1 (). To further assess the effects of AR42 on Wnt pathway signaling, we transfected CP70 cells with the Wnt luciferase reporter TOPFlash and its negative control, FOPFlash,Citation40 followed by 48-h treatment with vehicle or 1.0 µM AR42. As shown in , AR42 treatment resulted in > 2.6-fold decreased Wnt activity (luciferase activity). We also examined AR42 effects on the Wnt signal mediator β-catenin, which is dephosphorylated upon activation, followed by translocation to the nucleus.Citation40-Citation42 Using an antibody specific for dephosphorylated (and thus activated) β-catenin, we determined that AR42 caused a 57% decrease in nucleus-localized protein, and a 23% decrease in cytoplasmic protein ( and 5D). Taken together, these computational and experimental results provide additional support for Wnt signal blockade as a mechanism of AR42 disruption of ovarian cancer malignant phenotypes.
Figure 5. Experimental validation of AR42-decreased canonical Wnt signaling. (A) Quantitative PCR expression validation of selected microarray AR42-altered genes alter, including members of the Wnt signal pathway (TAK1, SFRP1), the epithelial-to-mesenchymal transition (ATF1), and cell cycle negative regulation (14-3-3σ). (*p < 0.05 vs. control). (B) Quantification of Wnt pathway signaling following 24 h, 1.0 µM AR42 treatment of CP70 cells transfected with a Wnt signal TOPFlash luciferase reporter.Citation40 Relative luminescence units (RLUs) were determined by normalizing the luminescence of TOPFlash-possessing cells to the total cellular protein from each lysate and to luminescence from mutant control vector (“FOPFlash”)-transfected cells.Citation70 (C) western blot analysis of cytoplasmic vs. nuclear extracts from CP70 ovarian cancer cells treated for 24 h with DMSO vehicle (“control,” lanes 2 and 6), 1.0 µM AR42 (lanes 3 and 7), or 1.0 µM vorinostat (lanes 1 and 5). (D) Quantification of western blot using ImageJ software (*p < 0.05, vs. control).
Experimental evaluation of AR42 induction of extrinsic (death receptor) apoptosis
In accord with our AR42 functional analyses described above, several other HDACIs have also been demonstrated to induce cell death via “death receptor” (extrinsic) apoptosis,Citation2,Citation15 a process that can occur in a mitochondria-independent manner (although “crosstalk” with mitochondrial apoptosis pathways may also take place).Citation43 Previously, we demonstrated that combined AR42/cisplatin treatment resulted in caspase activation in CP70 cells and xenografts.Citation6 While caspase induction associates with both the intrinsic (via intracellular signaling) and extrinsic apoptosis pathways,Citation43,Citation44 caspase-independent cell death has also been associated with mitochondrial disruption.Citation44,Citation45 Consequently, based on our gene ontology () and gene expression signature analyses ( and ), we assessed CP70 cell AR42 treatment in the presence or absence of the pan-caspase inhibitor z-VAD-fmk.Citation45 As shown in , caspase inhibition had little effect on AR42 dose-dependent growth inhibition. We then further investigated possible loss of mitochondrial membrane integrity and production of reactive oxygen species (ROS), two events that can also occur during extrinsic apoptosis and type II (autophagic) cell deathCitation43-Citation45 in 48-h AR42- and/or vorinostat-treated cells. With specific regard to ROS dependence, 48-h, HDACI-treated CP70 cells concurrently exposed to 15 mM of the antioxidant N-acetylcysteine (NAC) demonstrated a 37-fold increased 50% growth-inhibitory (GI50) dose for vorinostat (thus showing vorinostat as 37-fold less apoptotic in the absence of ROS). In contrast, NAC treatment increased the AR42 GI50 by only 21-fold (), suggesting AR42-induced cell death as less dependent on ROS production.
Figure 6. Experimental evaluation of the roles of caspase activity, reactive oxygen species (ROS), and mitochondrial disruptionCitation43,Citation44 in HDACI-mediated cell death. (A) Dose-dependent growth inhibition of CP70 cells treated for 48 h with increasing doses of AR42 in the continuous presence or absence of 20 µM pan-caspase inhibitor z-VAD-fmk. (B) HDACI dependence upon ROS, as determined by comparing 50% growth inhibitory doses (GI50 doses) of CP70 cells treated for 24 h with 1.0 µM vorinostat or 1.0 µM AR42 in the absence or presence of 15 mM antioxidant N-acetylcysteine (“NAC”). (C) Fluorescent micrographs of CP70 cells treated for 24 h with 1.0 µM AR42 (upper panel) or 1.0 µM vorinostat (lower panel) and exposed to the cell-permeable, mitochondrial inner membrane-accumulating fluorophore JC-1.Citation46 At normal membrane potentials, mitochondria-aggregated JC-1 fluoresces red, while loss of membrane potential results in JC-1 aggregation failure and monomer fluorescence emission of green.Citation46 (*p < 0.05, vs. control).
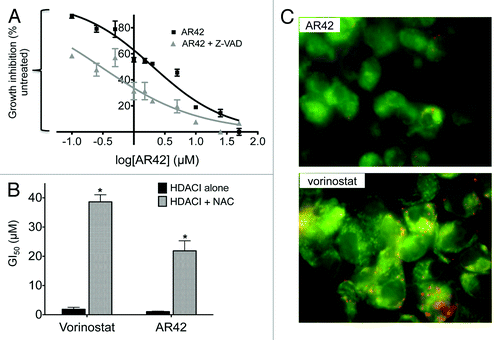
Caspase-independent cell death and ROS production may also associate with mitochondrial membrane permeation (and thus loss of mitochondrial membrane potential, ΔΨ).Citation43 To examine possible effects on mitochondrial integrity, we used a fluorescent ΔΨ reporter, JC-1,Citation46 following 48-h CP70 cell treatment with 1.0 µM AR42 or 1.0 µM vorinostat. At normal ΔΨ, JC-1 self-aggregates in the inner mitochondrial space, with aggregated JC-1 fluorescing red. By contrast, disrupted ΔΨ results in failed mitochondrial aggregation of the lipophilic dye, with the resulting (cytosolic) JC-1 monomers fluorescing green.Citation46 As shown in , following 48-h, 1.0 µM HDACI treatment, green fluorescence (i.e., monomeric and cytosolic JC-1) was predominant in AR42-treated cells (upper panel), while red fluorescence (mitochondria-aggregated JC-1) was more pronounced in vorinostat-treated cells (lower panel). These results suggest that at equal dose exposures, AR42 elicits greater loss of ΔΨ than vorinostat, in possible partial independence from ROS production and caspase activity.Citation43,Citation44
Discussion
In an earlier study, we examined growth suppression and drug-sensitization of chemoresistant ovarian cancer cells and mouse xenografts by a distinctive histone deacetylase inhibitor (HDACI), AR42.Citation6 While several reports have associated HDACI anticancer activity with limited numbers of altered genes,Citation2,Citation47,Citation48 here we endeavored to assess global mechanisms (i.e., pathways/ networks) of AR42 cancer cell-specific cytotoxicity, using a “systems biology” approach.Citation28,Citation29,Citation35 Moreover, with regard to the now well-established role of misexpressed (and frequently epigenetically altered) microRNAs (miRNAs) in ovarian tumorigenesis,Citation14,Citation17-Citation19,Citation21 we also considered dysregulation of these short, non-protein-coding, translation-regulatory oligonucleotides. Although a few studies have examined HDACI antineoplastic miRNA alterations,Citation22-Citation27 to our knowledge, the current work represents the first appraisal of monotherapeutic HDACI effects on miRNA expression (in association with altered signaling pathways) in ovarian cancer.
In earlier studies as monotherapies, HDACIs were found to induce only small numbers of genes, with greater gene re-expression requiring cotreatment with another class of epigenetic derepressive agents, DNA methyltransferase inhibitors (DNMTIs).Citation47,Citation48 Later reports, however, showed single-agent HDACIs to dysregulate (both induce and repress) considerably more genes.Citation2,Citation4,Citation8,Citation9 Interestingly, one recent report demonstrated DNMTIs to only marginally enhance HDACI antineoplastic activity in ovarian cancer cells and mouse xenografts.Citation49 The basis for recent demonstrations of greater HDACI gene dysregulation is somewhat uncertain, but could be related to previously used suboptimal or toxic doses, non-ideal durations of treatment, or secondary “downstream” gene regulatory events.Citation2 Even more impressively (but also controversially), several HDACIs have now been reported to reverse other (histone deacetylation-unrelated) repressive epigenetic modifications, including methylation of deoxycytosine, gain of gene-activating and/or loss of gene-repressive histone modifications (e.g., lysine methylation).Citation50-Citation52 Regardless of the extent of HDACI epigenomic reprogramming, however, it is widely held that these agents will be most beneficial when combined with conventional and/or targeted therapies, as demonstrated by our groupCitation6 and others.Citation4,Citation15,Citation16,Citation53,Citation54
In addition to chemosensitization, we previously demonstrated AR42 to differentially enhance protein acetylation and alter the expression of specific cell cycle-associated genes, in parallel with the HDACI vorinostat.Citation6 In the current work, comparisons of AR42- and vorinostat-altered microRNAomes and transcriptomes revealed distinct expression clusters (). This finding is consistent with earlier demonstrations of specific gene expression profiles for distinct HDACIs, possibly related to differences of inhibitor “fit” into the HDACI enzyme active site, HDAC-isoform preference(s), varying spatiotemporal HDAC enzyme location, and/or indirect (secondary, “downstream”) transcriptional effects.Citation2 However, while AR42 effects on miRNA expression were not previously studied in-depth, our finding of its distinctive transcriptome profile, as compared with that of vorinostat (), concurs with previously reported differences in gene expression by these two HDACIs.Citation6-Citation9
As vorinostat has now been extensively characterized and approved for human use for over five years,Citation2,Citation3 we directed our focus on characterizing global anticancer mechanisms of the “third generation” HDACI, AR42.Citation5 To further evaluate AR42 mechanisms, at the biological pathway/network signal level,Citation28,Citation29 we identified transcription factor (TF) genes whose expression correlated positively with that of their “target” genes (i.e., genes possessing specific binding sites for those TFs), followed by gene ontology (GO) assessmentCitation32 (). Those analyses showed that expression of AR42-altered TF target genes (mRNAs) anticorrelated with the biological processes epithelial-to-mesenchymal transition (EMT) ( and ), with upregulation of EMT-opposing TFs (GATA6 and WT1), and downregulation of EMT-promoting TF genes (SOX2 and ATF1) ( and ). While primary high-grade serous ovarian tumors actually gain epithelial attributes (i.e., undergo “MET”), late-stage disease phenotypes, including metastatic dissemination (“peritoneal seeding”), chemotherapy resistance (as exhibited by CP70 cells), and angiogenesis likely associate with restoration of the EMT program.Citation14,Citation55 Our current finding (possible EMT reversal) is consistent with our previous observation of AR42 induction of CP70 epithelial characteristics, based on cell morphology and the expression of multiple cytokeratins.Citation6 Additionally, while we did not examine Akt signaling in the current study, others have demonstrated that Akt signal inhibition (by AR42 and other agents) sensitized CP70 and other p53-dysfunctional, drug-resistant, ovarian cancer cells.Citation37,Citation56 As AR42 has also been demonstrated to downregulate Akt activity,Citation7-Citation9,Citation12 blockade of that oncogenic signal pathway might represent another HDACI mechanism of resensitization of chemoresistant ovarian cancer cells (an antineoplastic effect we previously observed).Citation6
Similar to EMT, gene set enrichment analysisCitation35 also demonstrated AR42 gene expression profiles to negatively associate with the EMT-dependent oncogenic signal cascade, Wnt (KEGG pathway hsa04310, ), while positively correlating with cell cycle antagonism (MSigDB_GO:0007050, ), and extrinsic (“death receptor”) apoptosis (MSigDB_M14971, ). We also note that in hepatocellular cancer cells, AR42 was found to potentiate type II, caspase-independent cell death via autophagy,Citation11 similar to vorinostat.Citation57 In the current work, we likewise saw AR42 downregulation of the anti-autophagy gene BCL2, and upregulation of two pro-autophagy genes, CHAF1A (p150) and BECN1 (Beclin1) (). However, we also observed downregulation of pro-autophagy (UVRAG, BNIP3), and upregulation of an anti-autophagy gene (AKT1S1) (), thus confounding firm conclusions regarding that type II cell death process.
With specific regard to ovarian cancer, we also found AR42-induced gene transcription patterns to correlate with gene signatures of favorable ovarian cancer prognosis () and low-grade disease ();Citation38,Citation39 AR42 also upregulated various ovarian cancer-repressed microRNAs, including miR-99, miR-100, and the miR-125 and let-7 familiesCitation18-Citation20,Citation58 (). Upregulation of miR-125 as a possible AR42 anti-ovarian cancer mechanism was also supported by a demonstration that miR-125 is repressed by epidermal growth factor signaling, and that miR-125 re-expression causes reversal of EMT.Citation20 Moreover, consistent with HDACI-mediated chromatin relaxation, we also observed AR42 to upregulate two miRNAs previously shown as epigenetically silenced in ovarian cancer, miRs 15a and 34 ().Citation17,Citation19
Experimental examination of Wnt pathway signaling (by a luciferase reporter assay) demonstrated Wnt activity to be 2.6-fold downregulated by AR42, consistent with other reports of HDACI inhibition of the Wnt cascade.Citation42,Citation59,Citation60 While overactive Wnt signaling often correlates with activating β-catenin mutations (and/or inactivating mutations of Wnt antagonists), many cancers (including ovarian) exhibit elevated Wnt signaling in the absence of Wnt pathway genetic alterations.Citation41,Citation42,Citation61 These findings suggest that epigenetic aberrations may play an analogous or even greater role in enhanced Wnt signaling. Indeed, numerous studies have now demonstrated chromatin-related silencing of the Wnt antagonists TAK1, SFRP1, ICAT, WIF1, and DKK1,Citation41,Citation42 genes we also identified as upregulated by AR42 ( and , and ).
EMT and Wnt also represent two interdependent pathways hypothesized to facilitate the genesis, proliferation, and tumor propagation characteristics of “cancer stem cells” (CSCs), neoplastic progenitors believed to govern (or possibly exacerbate) most or all malignant phenotypes.Citation41,Citation62 Additionally, the ovarian cancer cells used in this study, CP70, are highly aggressive, invasive, and chemoresistant,Citation36,Citation37,Citation56 three traits associated with cancer “stemness.”Citation14,Citation55,Citation62 CP70 cells also possess dysfunctional p53,Citation37 whose loss promotes self-renewal of normal and leukemia stem cells.Citation63,Citation64 Interestingly, our gene expression microarray results indicated a slight downregulation of p53 (0.7-fold, ), and upregulation of various p53 target genes including TP53I5 (6.7-fold), TP53I11 (4.3-fold), TP53I3 (1.3-fold), and TP53INP2 (1.3-fold) (). These findings would concur with previous demonstrations that HDACIs can downregulate abnormal p53 gene products, while also restoring “p53-like” pathways to cancer cells lacking functions (including apoptosis) of that tumor suppressor.Citation65-Citation67 Notably, we also observed AR42 upregulation of a p53 target microRNA gene, miR-34, whose re-expression, in ovarian and other cancers, strongly associates with activation of numerous p53 response pathways.Citation17,Citation68
Epigenetic modifications also regulate differentiation states in normal stem cells and adult tissues,Citation1,Citation16 and it has been shown that even highly aggressive cancer cells possess considerable phenotypic “plasticity,” associated with chromatin-remodeling ability.Citation41,Citation69 As HDACIs and DNA methyltransferase inhibitors (DNMTIs) are well-characterized differentiating agents, it is possible that these chromatin-relaxing agents could “reset” cancer progenitor cells to a differentiation-competent, drug-sensitive phenotype,Citation14,Citation16,Citation54,Citation62 thus decreasing the tumor subpopulation most responsible for perpetuating malignant phenotypes.Citation14,Citation41,Citation55,Citation62
Taken together, these combined computational and experimental approaches suggest that AR42 can upregulate tumor suppressor genes (including miRNAs) and antagonize specific tumor progression signal pathways, including two embryonic development cascades implicated in the genesis and maintenance of cancer stem cells. Overall, these findings suggest that AR42 (and perhaps other epigenetic modifiers), in possible combination with pathway-specific antagonists, might be clinically beneficial for the therapy of chemoresistant, high-grade serous ovarian cancer.
Materials and Methods
Reagents, cell culture, and HDAC inhibitor (HDACI) treatments
Cell culture reagents were obtained from Invitrogen Gibco/BRL. Except where specified, antibodies were purchased from KPL Laboratories. All other reagents were purchased from Sigma-Aldrich. For the specific study of advanced, chemoresistant ovarian cancer, we used the p53-dysfunctional CP70 ovarian cancer cell line previously shown resistant to platinum, adriamycin, and ionizing radiationCitation36,Citation37 (a kind gift from Dr. Thomas Hamilton, Fox Chase Cancer Center). The HDACIs AR42Citation5 and vorinostatCitation3 were kind gifts from Drs. Ching-Shih Chen and Samuel Kulp (College of Pharmacy, Ohio State University). For all HDAC inhibitor (HDACI) studies, CP70 cells (~70% confluency) were treated with vehicle (DMSO) control, 1.0 µM vorinostat, or 1.0 µM AR42 for 24–72 h, harvested from culture plates, centrifuged, and the pellets then further processed or stored at -80°C.
Mature miR and protein-coding (mRNA) gene expression
Total RNA was isolated from 24- or 48-h HDACI-treated or -untreated CP70 cells by suspension in TRIzol reagent (Invitrogen), as we have previously described.Citation70 Global analyses of mature miR expression were kindly performed by Dr. Michael Thomson (Vanderbilt University), using a 124-feature custom microarray.Citation71 For gene expression analyses, total RNA was qualified (Agilent Bioanalyzer 2100, Agilent Technologies), reverse transcribed to cDNA, labeled with Cy3-conjugated nonamers (Roche NimbleGen), and purified and hybridized to Human Gene Expression 385K Arrays, according to the manufacturer’s (Roche Nimblegen) protocol. Following washing, hybridized arrays were then scanned (single color green) using an Axon GenePix Professional 4200A Scanner (Molecular Devices).
Data processing and functional analyses of miR and mRNA expression
Scanned microarray images (GenePix files) were aligned using center fiducial controls, and the gridded files imported into NimbleScan analysis software (v2.5), followed by quantile normalization,Citation72 and gene calls made using Robust Multichip Averaging.Citation72 MicroRNA (miRNA) expression data (.xlsx format), provided by Dr. Thomson, was similarly processed using quantile normalization.Citation72 Unsupervised hierarchical clustering (a control for reproducibility and distinctiveness of the treatment groups) was then performed, using the integrated software platform Expander (EXPression ANalyzer and DisplayER, acgt.cs.tau.ac.il/expander, Tel Aviv University, Israel).Citation29
In this study, rather than restricting potential HDACI antineoplastic mechanisms to limited numbers of genes, we used a “systems biology” approachCitation28,Citation29 to delineate specific signal pathway alterations, and reveal possible transcriptional regulatory networks mediating the anti-ovarian cancer effects AR42. To that end, we compared genome-wide protein-coding gene (mRNA) and mature miRNA expression in AR42-treated vs. -untreated CP70 cells. Computationally, we first identified transcription factor (TF)-encoding genes transcribed in positive correlation with downstream “target” genes. Our rationale for the latter was that altered TF activity could result in pleitropic (and amplified) drug effects on specific pathways/processes.Citation28,Citation29 By this approach, using a signal intensity fold-change of 1.5, we identified TF genes whose expression positively correlated with AR42-altered genes having their cognate binding site motifs,Citation31 using the visualization tool ECR (evolutionary conserved region) Browser (ecrbrowser.dcode.org).Citation30 Those TF/target gene sets were then subjected to functional analysis, using the Web tool BiNGO v2.31.Citation32 Following normalization and microarray processing, we identified all genes 1.5-fold up- or downregulated by 48 h, 1.0 µM AR42 treatment. We then located the transcription start sites (TSSs) of those altered genes, as reported in research literature and publically available human genome databases.Citation30 Following identification of TSSs, we explored the presence of evolutionarily conserved transcription factor-binding sites (TFBSs) within 4 kb regions encompassing the TSSs, using the ECR browser (ecrbrowser.dcode.org).Citation30,Citation31 Genes encoding TFs having cognate TFBSs within misexpressed (protein-coding) genes were then identified (also by a fold-change cutoff of 1.5) and subjected to BiNGO functional analysisCitation32 to determine gene ontology (GO) biological process terms ().
Assessments of mitochondrial (intrinsic) or death receptor (extrinsic) apoptosis
To examine AR42 caspase dependence, CP70 cells were treated for 48-h with 1.0 µM AR42, in the absence or presence of 20 µM pan-caspase inhibitor z-VAD-fmk (Promega). To assess the role of reactive oxygen species in AR42-induced apoptosis, CP70 cells were similarly treated in the presence or absence of 15 mM of the antioxidant N-acetylcysteine. Separately, CP70 cells were grown on microscope coverslips, treated for 48 h with 1.0 µM vorinostat or 1.0 µM AR42, incubated for 20 min with 500 ng of the cationic mitochondrion-accumulating dye, JC-1 (Cayman Chemical Company), and photographed using a fluorescence microscope (Carl Zeiss) with filters for excitation/emission wavelengths of 485/530 nm (green) or 535/590 nm (red). Under normal mitochondrial transmembrane potential (ΔΨm), JC-1 self-aggregates within the mitochondrial inner membrane space, fluorescing red (multimeric JC-1). Loss of ΔΨm, however (as occurs during mitochondrial disruption), results in JC-1 mitochondrial aggregation failure, with JC-1 monomers remaining in the cytosol, fluorescing green.Citation46
Assessment of Wnt activity (β-catenin nuclear translocation)
For western blot assays, CP70 cells were treated with vehicle (DMSO), 1.0 μM AR42, or 1.0 µM vorinostat for 48 h, followed by isolation of total cytoplasmic or total nuclear protein, using a nuclear extraction kit (Active Motif). Fifteen micrograms of nuclear or cytoplasmic protein (with β-catenin positive control cell lysate, Lab Vision) were then electrophoresed through 8% SDS-PAGE, transferred to nylon membranes, blocked with 5% milk, and incubated overnight with diluted primary antibodies against activated (unphosphorylated) β-catenin (Lab Vision) and GAPDH (a gel-loading control, Millipore). Membranes were then washed and incubated with peroxidase-conjugated anti-rabbit or anti-mouse IgG secondary antibodies for 60 min, washed again, placed for 1 min in SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), and exposed to film (Kodak) for 2 min. Western blot band intensities were then determined by scanning and quantification using ImageJ Java-based imaging software (Research Services Branch, National Institute of Mental Health, rsb.info.nih.gov/ij).
Assessment of Wnt signal transactivity by luciferase reporter assay
For determination of Wnt signal activity at the pathway level, CP70 cells were grown in 6-well plates to 60–70% confluency. Using Lipofectamine 2000 (Invitrogen), cells were then transfected for 24 h with 1.0 μg of the luciferase reporter TOPFlash (a construct possessing four Wnt signal-transactivating TCF/LEF-binding sites ligated to the gene luc) or 1.0 µg of its negative control FOPflash (having four mutant luc–ligated TCF/LEF-binding sites).Citation40 Both TOPflash and FOPflash were purchased from Millpore. TOPFlash- or FOPFlash-transfected cells were then treated with DMSO vehicle or 1.0 μM AR42 or for 48 h, washed 2X with cold PBS, placed in 1X Reporter Lysis Buffer (Promega), and stored at -80°C. for at least 1 h. Following one freeze-thaw cycle, Luciferase Assay Reagent (Promega) was added to the AR42-treated or -untreated cell lysates, with luminescence intensity detected by a TD-20/20 luminometer (Turner Designs). Raw TOPFlash and FOPFlash luminescence values were then normalized for protein concentration, and the normalized TOPFlash:FOPFlash ratios then used to quantify Wnt reporter activity in untreated vs. AR42-treated CP70 cell lysates.Citation70
| Abbreviations: | ||
| BiNGO | = | Biological Networks Gene Ontology |
| DNMTI | = | DNA methyltransferase inhibitor |
| EMT | = | epithelial-to-mesenchymal transition |
| GO | = | gene ontology |
| HDACI | = | histone deacetylase inhibitor, NAC, N-acetylcysteine |
| ROS | = | reactive oxygen species |
| TF | = | transcription factor |
| TFBS | = | transcription factor-binding site |
Acknowledgments
The authors wish to thank Vasu Tumati and Michael Mand for technical assistance, and Dr. Meng Li and Rongye Lai for valuable informatics assistance. We also thank Dr. Michael Thomson (Vanderbilt University) for providing us microRNA microarray data for our samples. This work was supported by the National Cancer Institute awards CA113001 and CA085289 (to K.P.N.), the Walther Cancer Foundation (Indianapolis, IN) (to K.P.N.), the American Cancer Society (Institutional Research Grant 84-002-25, to C.B.) and The Ovar’coming Together Ovarian Cancer Foundation (Indianapolis, IN to C.B.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009; 10:32 - 42; http://dx.doi.org/10.1038/nrg2485; PMID: 19065135
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541 - 52; http://dx.doi.org/10.1038/sj.onc.1210620; PMID: 17694093
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25:84 - 90; http://dx.doi.org/10.1038/nbt1272; PMID: 17211407
- Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res 2009; 15:3947 - 57; http://dx.doi.org/10.1158/1078-0432.CCR-08-2787; PMID: 19509170
- Lu Q, Wang DS, Chen CS, Hu YD, Chen CS. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J Med Chem 2005; 48:5530 - 5; http://dx.doi.org/10.1021/jm0503749; PMID: 16107152
- Yang YT, Balch C, Kulp SK, Mand MR, Nephew KP, Chen CS. A rationally designed histone deacetylase inhibitor with distinct antitumor activity against ovarian cancer. Neoplasia 2009; 11:552 - 63, 3, 563; PMID: 19484144
- Sargeant AM, Rengel RC, Kulp SK, Klein RD, Clinton SK, Wang YC, et al. OSU-HDAC42, a histone deacetylase inhibitor, blocks prostate tumor progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res 2008; 68:3999 - 4009; http://dx.doi.org/10.1158/0008-5472.CAN-08-0203; PMID: 18483287
- Lu YS, Kashida Y, Kulp SK, Wang YC, Wang D, Hung JH, et al. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology 2007; 46:1119 - 30; http://dx.doi.org/10.1002/hep.21804; PMID: 17654699
- Bai LY, Omar HA, Chiu CF, Chi ZP, Hu JL, Weng JR. Antitumor effects of (S)-HDAC42, a phenylbutyrate-derived histone deacetylase inhibitor, in multiple myeloma cells. Cancer Chemother Pharmacol 2011; 68:489 - 96; http://dx.doi.org/10.1007/s00280-010-1501-z; PMID: 21072520
- Zimmerman B, Sargeant A, Landes K, Fernandez SA, Chen CS, Lairmore MD. Efficacy of novel histone deacetylase inhibitor, AR42, in a mouse model of, human T-lymphotropic virus type 1 adult T cell lymphoma. Leuk Res 2011; 35:1491 - 7; http://dx.doi.org/10.1016/j.leukres.2011.07.015; PMID: 21802726
- Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6:1057 - 65; http://dx.doi.org/10.4161/auto.6.8.13365; PMID: 20962572
- Jacob A, Oblinger J, Bush ML, Brendel V, Santarelli G, Chaudhury AR, et al. Preclinical validation of AR42, a novel histone deacetylase inhibitor, as treatment for vestibular schwannomas. Laryngoscope 2012; 122:174 - 89; http://dx.doi.org/10.1002/lary.22392; PMID: 22109824
- Takai N, Narahara H.. Histone deacetylase inhibitor therapy in epithelial ovarian cancer. J Oncol 2010; 2010:458431
- Balch C, Nephew KP. The role of chromatin, microRNAs, and tumor stem cells in ovarian cancer. Cancer Biomark 2010; 8:203 - 21; PMID: 22045354
- Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res 2009; 15:3970 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-08-2786; PMID: 19509171
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res 2010; 16:1119 - 28; http://dx.doi.org/10.1158/1078-0432.CCR-09-2642; PMID: 20145172
- Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res 2007; 67:8699 - 707; http://dx.doi.org/10.1158/0008-5472.CAN-07-1936; PMID: 17875710
- Zhang L, Volinia S, Bonome T, Calin GA, Greshock J, Yang N, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A 2008; 105:7004 - 9; http://dx.doi.org/10.1073/pnas.0801615105; PMID: 18458333
- Cowden Dahl KD, Dahl R, Kruichak JN, Hudson LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia 2009; 11:1208 - 15; PMID: 19881956
- Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, et al. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res 2008; 14:2690 - 5; http://dx.doi.org/10.1158/1078-0432.CCR-07-1731; PMID: 18451233
- Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood 2012; 119:1162 - 72; http://dx.doi.org/10.1182/blood-2011-05-351510; PMID: 22096249
- Rhodes LV, Nitschke AM, Segar HC, Martin EC, Driver JL, Elliott S, et al. The histone deacetylase inhibitor trichostatin A alters microRNA expression profiles in apoptosis-resistant breast cancer cells. Oncol Rep 2012; 27:10 - 6; PMID: 21971930
- Shin S, Lee EM, Cha HJ, Bae S, Jung JH, Lee SM, et al. MicroRNAs that respond to histone deacetylase inhibitor SAHA and p53 in HCT116 human colon carcinoma cells. Int J Oncol 2009; 35:1343 - 52; PMID: 19885557
- Lee EM, Shin S, Cha HJ, Yoon Y, Bae S, Jung JH, et al. Suberoylanilide hydroxamic acid (SAHA) changes microRNA expression profiles in A549 human non-small cell lung cancer cells. Int J Mol Med 2009; 24:45 - 50; PMID: 19513533
- Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 2006; 66:1277 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-05-3632; PMID: 16452179
- Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res 2006; 66:6097 - 104; http://dx.doi.org/10.1158/0008-5472.CAN-06-0537; PMID: 16778182
- Pe’er D, Hacohen N. Principles and strategies for developing network models in cancer. Cell 2011; 144:864 - 73; http://dx.doi.org/10.1016/j.cell.2011.03.001; PMID: 21414479
- Ulitsky I, Maron-Katz A, Shavit S, Sagir D, Linhart C, Elkon R, et al. Expander: from expression microarrays to networks and functions. Nat Protoc 2010; 5:303 - 22; http://dx.doi.org/10.1038/nprot.2009.230; PMID: 20134430
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res 2004; 32:Web Server issue W280-6; http://dx.doi.org/10.1093/nar/gkh355; PMID: 15215395
- Wang X, Xuan Z, Zhao X, Li Y, Zhang MQ. High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res 2009; 19:266 - 75; http://dx.doi.org/10.1101/gr.081638.108; PMID: 18997002
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005; 21:3448 - 9; http://dx.doi.org/10.1093/bioinformatics/bti551; PMID: 15972284
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther 2003; 2:151 - 63; PMID: 12589032
- Choi YL, Bocanegra M, Kwon MJ, Shin YK, Nam SJ, Yang JH, et al. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res 2010; 70:2296 - 306; http://dx.doi.org/10.1158/0008-5472.CAN-09-3141; PMID: 20215510
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545 - 50; http://dx.doi.org/10.1073/pnas.0506580102; PMID: 16199517
- Anderson K, Lawson KA, Simmons-Menchaca M, Sun L, Sanders BG, Kline K. Alpha-TEA plus cisplatin reduces human cisplatin-resistant ovarian cancer cell tumor burden and metastasis. Exp Biol Med (Maywood) 2004; 229:1169 - 76; PMID: 15564444
- Arafa SA, Zhu Q, Barakat BM, Wani G, Zhao Q, El-Mahdy MA, et al. Tangeretin sensitizes cisplatin-resistant human ovarian cancer cells through downregulation of phosphoinositide 3-kinase/Akt signaling pathway. Cancer Res 2009; 69:8910 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-09-1543; PMID: 19903849
- Spentzos D, Levine DA, Ramoni MF, Joseph M, Gu X, Boyd J, et al. Gene expression signature with independent prognostic significance in epithelial ovarian cancer. J Clin Oncol 2004; 22:4700 - 10; http://dx.doi.org/10.1200/JCO.2004.04.070; PMID: 15505275
- Meinhold-Heerlein I, Bauerschlag D, Hilpert F, Dimitrov P, Sapinoso LM, Orlowska-Volk M, et al. Molecular and prognostic distinction between serous ovarian carcinomas of varying grade and malignant potential. Oncogene 2005; 24:1053 - 65; http://dx.doi.org/10.1038/sj.onc.1208298; PMID: 15558012
- Warner DR, Greene RM, Pisano MM. Cross-talk between the TGFbeta and Wnt signaling pathways in murine embryonic maxillary mesenchymal cells. FEBS Lett 2005; 579:3539 - 46; http://dx.doi.org/10.1016/j.febslet.2005.05.024; PMID: 15955531
- Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011; 8:97 - 106; http://dx.doi.org/10.1038/nrclinonc.2010.196; PMID: 21151206
- Ying Y, Tao Q. Epigenetic disruption of the WNT/beta-catenin signaling pathway in human cancers. Epigenetics 2009; 4:307 - 12; http://dx.doi.org/10.4161/epi.4.5.9371; PMID: 19633433
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621 - 32; http://dx.doi.org/10.1038/nrm2952; PMID: 20683470
- Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene 2008; 27:6452 - 61; http://dx.doi.org/10.1038/onc.2008.311; PMID: 18955972
- Vandenabeele P, Vanden Berghe T, Festjens N.. Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006; 2006:pe44
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A 1991; 88:3671 - 5; http://dx.doi.org/10.1073/pnas.88.9.3671; PMID: 2023917
- Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999; 21:103 - 7; http://dx.doi.org/10.1038/5047; PMID: 9916800
- Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002; 31:141 - 9; http://dx.doi.org/10.1038/ng892; PMID: 11992124
- Chen MY, Liao WS, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011; 117:4424 - 38; http://dx.doi.org/10.1002/cncr.26073; PMID: 21491416
- Arzenani MK, Zade AE, Ming Y, Vijverberg SJ, Zhang Z, Khan Z, et al. Genomic DNA hypomethylation by histone deacetylase inhibition implicates DNMT1 nuclear dynamics. Mol Cell Biol 2011; 31:4119 - 28; http://dx.doi.org/10.1128/MCB.01304-10; PMID: 21791605
- Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008; 27:7274 - 84; http://dx.doi.org/10.1038/onc.2008.333; PMID: 18806826
- Huang PH, Chen CH, Chou CC, Sargeant AM, Kulp SK, Teng CM, et al. Histone deacetylase inhibitors stimulate histone H3 lysine 4 methylation in part via transcriptional repression of histone H3 lysine 4 demethylases. Mol Pharmacol 2011; 79:197 - 206; http://dx.doi.org/10.1124/mol.110.067702; PMID: 20959362
- Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, et al. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 2006; 5:2086 - 95; http://dx.doi.org/10.1158/1535-7163.MCT-06-0111; PMID: 16928830
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69 - 80; http://dx.doi.org/10.1016/j.cell.2010.02.027; PMID: 20371346
- Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. [Epub ahead of print] Oncogene 2011; http://dx.doi.org/10.1038/onc.2011.429; PMID: 21963846
- Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H, et al. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011; 13:1069 - 80; PMID: 22131882
- Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 2008; 216:495 - 504; http://dx.doi.org/10.1002/path.2434; PMID: 18850582
- Dahiya N, Sherman-Baust CA, Wang TL, Davidson B, Shih IeM, Zhang Y, et al. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One 2008; 3:e2436; http://dx.doi.org/10.1371/journal.pone.0002436; PMID: 18560586
- Galimberti S, Canestraro M, Maffei R, Marasca R, Guerrini F, Piaggi S, et al. Vorinostat interferes with Wnt and NF-kappaB pathways in the M-07e cell line. Leukemia 2009; 23:1935 - 8; http://dx.doi.org/10.1038/leu.2009.119; PMID: 19626048
- Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin SM. The class I HDAC inhibitor MGCD0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating Dickkopf-1 and non-canonical Wnt signaling. Oncotarget 2010; 1:596 - 605; PMID: 21317455
- Wiltse J. Mode of action: inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of beta-catenin--developmental effects of valproic acid. Crit Rev Toxicol 2005; 35:727 - 38; http://dx.doi.org/10.1080/10408440591007403; PMID: 16417040
- Tysnes BB. Tumor-initiating and -propagating cells: cells that we would like to identify and control. Neoplasia 2010; 12:506 - 15; PMID: 20651980
- Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev 2010; 24:1389 - 402; http://dx.doi.org/10.1101/gad.1940710; PMID: 20595231
- Krizhanovsky V, Lowe SW. Stem cells: The promises and perils of p53. Nature 2009; 460:1085 - 6; http://dx.doi.org/10.1038/4601085a; PMID: 19713919
- Condorelli F, Gnemmi I, Vallario A, Genazzani AA, Canonico PL. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol 2008; 153:657 - 68; http://dx.doi.org/10.1038/sj.bjp.0707608; PMID: 18059320
- Blagosklonny MV, Trostel S, Kayastha G, Demidenko ZN, Vassilev LT, Romanova LY, et al. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res 2005; 65:7386 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-04-3433; PMID: 16103091
- Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 2011; 18:1904 - 13; http://dx.doi.org/10.1038/cdd.2011.71; PMID: 21637290
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26:745 - 52; http://dx.doi.org/10.1016/j.molcel.2007.05.010; PMID: 17540599
- Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression?. Nat Clin Pract Oncol 2008; 5:280 - 90; http://dx.doi.org/10.1038/ncponc1089; PMID: 18349857
- Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 2006; 66:11954 - 66; http://dx.doi.org/10.1158/0008-5472.CAN-06-1666; PMID: 17178894
- Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods 2004; 1:47 - 53; http://dx.doi.org/10.1038/nmeth704; PMID: 15782152
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19:185 - 93; http://dx.doi.org/10.1093/bioinformatics/19.2.185; PMID: 12538238