Abstract
Barrett’s esophagus is a metaplasia of the distal esophagus that is the only recognized precursor of esophageal adenocarcinoma. Despite a characteristic histology, the pathogenesis of Barrett’s has remained obscure. A recent paper from the laboratories of Wa Xian and Frank McKeon presents evidence for a novel cell of origin of Barrett’s. Their work is based on studies of mice engineered to lack the squamous epithelial stem cell survival factor p63. These mice develop a metaplasia of the proximal stomach and esophagus that harbors substantial histological and molecular features of Barrett’s. The metaplasia appears to form from embryonic progenitor cells that normally persists post-natally only at the squamo-columnar junction. Moreover, in their model, the metaplasia is initiated not by mutation but by reduced competition between these cells and squamous epithelial cells.
Barrett’s esophagus is the only known precursor of esophageal adenocarcinoma, a major lethal cancer that has increased dramatically in incidence in recent decades.Citation1-Citation4 Barrett’s is readily recognized on endoscopy as a salmon coloration of the normally pale esophageal lining. It is confirmed by biopsy. Whereas the normal esophagus is lined by squamous epithelium (), Barrett’s exhibits a columnar epithelium with goblet cells, which are typical of the intestine. The cell of origin of Barrett’s has been a mystery, however. In an intriguing recent article in Cell, Wang et al. propose a novel explanation for the pathogenesis of Barrett’s esophagus.Citation5 They suggest that Barrett’s initiates from a unique cell of the embryo that persists at the esophago-gastric junction. Moreover, they postulate that the initial spread of this cell derives not from genetic alterations but from competitive interactions between cell lineages driven by opportunity.
Figure 1. Barrett’s esophagus and mouse models described in Wang et al. Top: human Barrett’s esophagus typically develops at the junction between the squamous epithelium of the esophagus (hatched) and the columnar epithelium of the stomach, in association with gastresophageal reflux disease (GERD). Bottom: in the mouse, the normal squamo-columnar junction is located within the proximal stomach (left). Wang et al. found that p63-null mice develop by birth a columnar metaplasia of the esophagus and proximal stomach that has molecular features of Barrett’s, including expression of Car4 and Krt7. Cells expressing these markers can be found at the squamo-columnar junction in normal adult mice. Expression of a toxin in the squamous epithelium of 3-week-old animals results in expansion of Krt7+ cells proximally into the squamous epithelium.
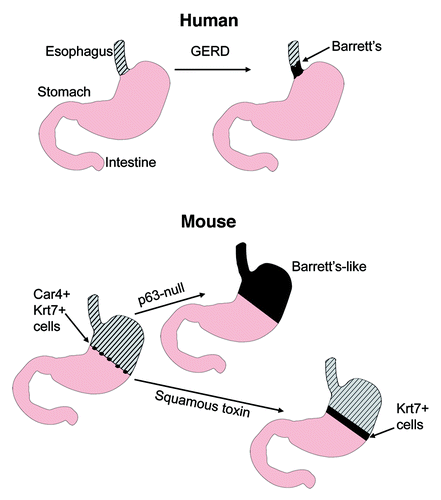
The paper is based primarily on studies in mice null for the p53 family member p63. p63 appears to be a survival factor for squamous epithelial progenitor cells. p63-null mice die post-natally with severe skin defects. Whereas the mouse upper gut is normally lined by squamous epithelium from the mouth to the mid-stomach, p63 null embryos instead show a well-developed columnar epithelium (). In their Cell paper, the authors present evidence that this epithelium is similar to human Barrett’s. The metaplasia in the p63-null mice demonstrates immunohistochemical staining for keratin 8, villin (Vil1), and anterior gradient 2 (Agr2). These are well known markers for Barrett’s esophagus.Citation6,Citation7 Human Barrett’s esophagus was shown to lack expression of p63 in previous studies.Citation8
Next, the authors compared gene expression profiles of p63-null metaplasia from different mice with those of distinct regions of the gastrointestinal tract of wild type and mutant animals, as well as Barrett’s esophagus. The profiles of p63-null metaplasia were more similar to distal stomach than normal proximal stomach or intestine but comprised a unique cluster. There was a strong correlation with genes differentially expressed in Barrett’s esophagus.Citation6,Citation7 Among the genes overexpressed in the p63-null metaplasia were mucin4 (Muc4), keratin 20 (Krt20), trefoil factor 2 (TFF2), and claudin 3 (Cldn3), as well as Agr2 and Vil1. These markers were validated by immunohistochemistry in human Barrett’s samples. Interestingly, in the p63-null metaplasia Caudal-type homeobox 2 (Cdx2), an intestinal transcription factor previously implicated in forming a Barrett’s-like phenotype, was not expressed.
The most robust marker of metaplasia was Car4. Car4+ epithelial cells were traced through embryonic development. They were expressed in a monolayer of cells in the proximal stomach on embryonic day 13 and 14. During development of wild type mice, p63+ cells increasingly extended among and under the Car4+ cells. Car4+ cells positioned on the basement membrane were highly proliferative, whereas Car+ cells that were undermined by p63+ cells showed reduced cell-cycle activity. Later in development, the undermined Car4+ cells detached from the epithelium and disappeared. In normal mice, only a discrete number of Car4+ cells remained, precisely at the squamo-columnar junction, with some cells expressing keratin (Krt) 7 (). The authors suggest that the Car4+ cells develop into a metaplasia in p63-null mice because of the absence of undermining by p63+ squamous epithelium.
A comparison of genes highly expressed at the squamo-columnar junction of wild type mice with those of the metaplasia in the p63-null mice and human Barrett’s revealed a consensus overlap of 87 genes, among them Spink4, Agr2, TFF1, TFF2, Krt8, Krt7, Krt18 and Vil1.
To test the hypothesis that the primitive junctional epithelial cells that persist post-natally can serve as origins of a Barrett’s like metaplasia, the authors generated mice in which diphtheria toxin A was conditionally expressed in basal cells of stratified epithelia in 3-week-old mice. A significant caveat to their model is that the Car4+ cells did not expand. However, the zone of Krt7+ cells did expand from the squamo-columnar junction into a more proximal band of Krt6+ cells ().
In summary, the work presented by the authors describes the evolution of a Barrett’s-like metaplasia in embryonic and adult mice from precursor cells associated by lineage. The authors suggest that, in this model and perhaps other precursors of malignancy, the earliest events depend not on genetic changes but rather on competition between cell lineages for access to the basal cell membrane.Citation9
Two distinct mechanisms for the development of Barrett’s metaplasia have been discussed in the literature. The first mechanism is direct conversation of differentiated cells, a process called transdifferentiation.Citation10 The second mechanism is the development from stem or progenitor cells. This work strongly supports a stem/progenitor cell model and argues against the need for transdifferentiation. In support of these mechanisms, four potential cellular origins of Barrett’s epithelium have been postulated: (1) differentiated squamous epithelial cells, (2) stem cells from esophageal submucosal glands, (3) stem or progenitor cells at the gastro-esophageal junction with unique or inherently plastic differentiation potential, or (4) bone marrow derived stem cells, in the context of inflammation.Citation11 This paper argues strongly for the third cell type, a gastro-esophageal progenitor cell.
Evidence has been building for some time that p63 is a pivotal protein in understanding the difference between normal esophageal epithelium and Barrett’s. p63 was shown to be mainly expressed in epithelial tissue and required for the development of stratified epithelia.Citation12 p63 shows homology to the p53 transcription factor in the DNA binding, transactivation and oligomerization domains. Targeted disruption of the murine p63 gene resulted in neonatal death.Citation12 Homozygotes were shown to have severe defects in forming and maintaining differentiation of tissues with stratified epithelium, such as the esophagus. p63 was shown to be highly expressed in epidermal stem cells, in particular. The lack of a stratified epidermis in p63 null mice suggests a role in maintenance of epidermal progenitor stem cells. Although p63 appears to mainly function during development, studies have shown evidence for p63’s role in maintaining stem cell populations of the epidermis.Citation13,Citation14 Further, p63 was found to influence conversion between a simple epithelium and stratified epithelial tissue.Citation15 Among neoplastic tissues, p63 expression was shown to be restricted to human squamous cell carcinomas; Barrett’s metaplasia and adenocarcinomas did not stain for p63.Citation8 Exposure of esophageal squamous epithelial cells to bile and acid led to the downregulation of p63, a possible explanation why p63 is lost in Barrett’s metaplasia.Citation16 However, the work discussed here argues for the origin of Barrett’s from a cell that is inherently p63-, rather than transdifferentiation of a p63+ cell.
What is to be made of the lack of Cdx2 in the p63-null metaplasia? Transdifferentiation of mature esophageal squamous epithelial cells to columnar epithelial cells was previously linked to overexpression of Cdx2 (reviewed by Burgess).Citation17 Cdx2 is a transcription factor of the caudal-related homeobox family that plays an important role in intestinal epithelial development and differentiation.Citation18-Citation20 Cdx2 expression is normally observed in epithelial cells of the human small and large intestine, but not in the esophagus. In contrast, several studies have shown that nondysplastic and dysplastic Barrett’s metaplasia and EAC often stained with anti-Cdx2 antibodies.Citation21,Citation22 Functionally, deletion of Cdx2 in the intestine was shown to result in squamous metaplasiaCitation23 and overexpression in the stomach results in intestinalization.Citation19 Although overexpression of Cdx2 in human keratinocytes did not induce intestinalization, treatment with 5-AzaC could induce markers of intestinalization in Cdx2 overexpressing cells. The authors of that study concluded that ectopic proliferation signals, alterations in epigenetic gene regulation and the inhibition of tumor suppressor mechanisms are required for Cdx2-mediated intestinalization of human esophageal keratinocytes in Barrett’s esophagus.Citation24 On the other hand, overexpression of Cdx2 has not been found in some Barrett’s studies.Citation6 In the current study, metaplastic tissue in p63 null mice did not show expression of Cdx2. The authors concluded that Cdx2 expression and transdifferentiation to an intestinal phenotype are probably not required for Barrett’s metaplasia. Nonetheless, a role for Cdx2 in Barrett’s is not excluded.
A major conclusion of this study is that certain precancerous lesions, such as Barrett’s, may initiate not from genetic alterations but from competitive interactions between cell lineages driven by opportunity. This is an interesting new concept in the pathogenesis of Barrett’s metaplasia. Moreover, the cell of origin of Barrett’s is proposed to be an obscure residual embryonic cell with no known role in the adult. Why these cells persist post-natally and whether they may play useful roles there, perhaps in response to esophageal damage, remain to be defined. Further studies of the role of these cells in Barrett’s are clearly warranted, in humans and mice. Study of more patients, using new markers of p63-null metaplasia identified by Wang et al. may be informative. In mice, it could be instructive to assess whether metaplasia of the proximal stomach induced by conditional, localized destruction of the squamous epithelium (e.g., by gavage), can be tolerated and predispose to neoplasia, with or without genetic lesions implicated in Barrett’s.
References
- Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst 2011; 103:1049 - 57; http://dx.doi.org/10.1093/jnci/djr203; PMID: 21680910
- Hvid-Jensen F, Pedersen L, Drewes AM, Sørensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med 2011; 365:1375 - 83; http://dx.doi.org/10.1056/NEJMoa1103042; PMID: 21995385
- Sharma P. Clinical practice. Barrett’s esophagus. N Engl J Med 2009; 361:2548 - 56; http://dx.doi.org/10.1056/NEJMcp0902173; PMID: 20032324
- Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer 2010; 10:87 - 101; http://dx.doi.org/10.1038/nrc2773; PMID: 20094044
- Wang X, Ouyang H, Yamamoto Y, Kumar PA, Wei TS, Dagher R, et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011; 145:1023 - 35; http://dx.doi.org/10.1016/j.cell.2011.05.026; PMID: 21703447
- Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, et al. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett’s esophagus. PLoS One 2008; 3:e3534; http://dx.doi.org/10.1371/journal.pone.0003534; PMID: 18953412
- Wang J, Qin R, Ma Y, Wu H, Peters H, Tyska M, et al. Differential gene expression in normal esophagus and Barrett’s esophagus. J Gastroenterol 2009; 44:897 - 911; http://dx.doi.org/10.1007/s00535-009-0082-2; PMID: 19468668
- Daniely Y, Liao G, Dixon D, Linnoila RI, Lori A, Randell SH, et al. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am J Physiol Cell Physiol 2004; 287:C171 - 81; http://dx.doi.org/10.1152/ajpcell.00226.2003; PMID: 15189821
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 2002; 70:537 - 46; http://dx.doi.org/10.1046/j.1432-0436.2002.700907.x
- Chen H, Fang Y, Tevebaugh W, Orlando RC, Shaheen NJ, Chen X. Molecular mechanisms of Barrett’s esophagus. Dig Dis Sci 2011; 56:3405 - 20; http://dx.doi.org/10.1007/s10620-011-1885-6; PMID: 21984436
- Dvorak K, Goldman A, Kong J, Lynch JP, Hutchinson L, Houghton JM, et al. Molecular mechanisms of Barrett’s esophagus and adenocarcinoma. Ann N Y Acad Sci 2011; 1232:381 - 91; http://dx.doi.org/10.1111/j.1749-6632.2011.06062.x; PMID: 21950830
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398:714 - 8; http://dx.doi.org/10.1038/19539; PMID: 10227294
- Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol 2010; 5:349 - 71; http://dx.doi.org/10.1146/annurev-pathol-121808-102117; PMID: 20078223
- Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 2007; 129:523 - 36; http://dx.doi.org/10.1016/j.cell.2007.02.045; PMID: 17482546
- Lefort K, Dotto GP. p63 and epithelial metaplasia: a gutsy choice. Cell 2011; 145:1003 - 5; http://dx.doi.org/10.1016/j.cell.2011.06.008; PMID: 21703442
- Roman S, Pétré A, Thépot A, Hautefeuille A, Scoazec JY, Mion F, et al. Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am J Physiol Gastrointest Liver Physiol 2007; 293:G45 - 53; http://dx.doi.org/10.1152/ajpgi.00583.2006; PMID: 17615180
- Burgess DJ. Disease mechanisms: Out with the old, in with the new. Nat Rev Cancer 2011; 11:536; http://dx.doi.org/10.1038/nrc3116; PMID: 21779007
- Beck F, Chawengsaksophak K, Waring P, Playford RJ, Furness JB. Reprogramming of intestinal differentiation and intercalary regeneration in Cdx2 mutant mice. Proc Natl Acad Sci U S A 1999; 96:7318 - 23; http://dx.doi.org/10.1073/pnas.96.13.7318; PMID: 10377412
- Silberg DG, Sullivan J, Kang E, Swain GP, Moffett J, Sund NJ, et al. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology 2002; 122:689 - 96; http://dx.doi.org/10.1053/gast.2002.31902; PMID: 11875002
- Silberg DG, Swain GP, Suh ER, Traber PG. Cdx1 and cdx2 expression during intestinal development. Gastroenterology 2000; 119:961 - 71; http://dx.doi.org/10.1053/gast.2000.18142; PMID: 11040183
- Souza RF, Krishnan K, Spechler SJ. Acid, bile, and CDX: the ABCs of making Barrett’s metaplasia. Am J Physiol Gastrointest Liver Physiol 2008; 295:G211 - 8; http://dx.doi.org/10.1152/ajpgi.90250.2008; PMID: 18556417
- Vallböhmer D, DeMeester SR, Peters JH, Oh DS, Kuramochi H, Shimizu D, et al. Cdx-2 expression in squamous and metaplastic columnar epithelia of the esophagus. Dis Esophagus 2006; 19:260 - 6; http://dx.doi.org/10.1111/j.1442-2050.2006.00586.x; PMID: 16866857
- Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell 2009; 16:588 - 99; http://dx.doi.org/10.1016/j.devcel.2009.02.010; PMID: 19386267
- Kong J, Nakagawa H, Isariyawongse BK, Funakoshi S, Silberg DG, Rustgi AK, et al. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis 2009; 30:122 - 30; http://dx.doi.org/10.1093/carcin/bgn227; PMID: 18845559