Abstract
Melanoma incidence and mortality rates continue to increase each year. Lack of clinically viable agents, drug combinations, effective targeted delivery approaches and success inhibiting targets in tumor tissue have made this disease one of the most difficult to treat, which makes prevention an important option for decreasing disease incidence and mortality rates. Inhibiting histone deacetylases (HDAC) is an approach currently being explored to more effectively treat melanoma but use for prevention has not been explored. In this study, novel selenium containing derivatives of the FDA approved HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) called 5-phenylcarbamoylpentyl selenocyanide (PCP-SeCN) and Bis{5-phenylcarbamoylpentyl} diselenide (B(PCP)-2Se) were created and efficacy tested for preventing early melanocytic lesion development in skin. Topical application of PCP-SeCN and B(PCP)-2Se inhibited melanocytic lesion development in laboratory-generated skin by up to 87% with negligible toxicological effect. Mechanistically, PCP-SeCN and B(PCP)-2Se inhibited HDAC activity and had new inhibitory properties by moderating Akt activity to induce cellular apoptosis as demonstrated by an increase in the sub-G0-G1 cell population, and cleaved caspase-3 as well as PARP levels. Furthermore, PCP-SeCN and B(PCP)-2Se inhibited cell proliferation by inhibiting cyclin D1 expression and increasing p21 levels. Thus, PCP-SeCN and B(PCP)-2Se are potential melanoma chemopreventive agents with enhanced efficacy compared with SAHA due to new PI3 kinase pathway inhibitory properties.
Introduction
Malignant melanoma incidence and mortality rates continue to increase in the US each year. It remains the most deadly form of skin cancer due to its high metastatic potential and rapid development of chemoresistance.Citation1,Citation2 Despite recent advances in the identification of vemurafenib (PLX-4032) and Yervoy (Ipilimumab) the prevalence and mortality rates continues to rise due to the development of drug resistance and associated side effects.Citation3,Citation4 About 10% of melanoma is inherited in families.Citation5,Citation6 Damage of epidermal melanocytes by UVB has been directly linked to the development of ~30% of non-inherited melanomas.Citation7,Citation8 The major chemopreventive agent to avoid UV damage and sun induced melanomas is sunscreen.Citation9 While most individuals in the US are aware of the dangers of sunburns and about sun protection, incidence and mortality rates for melanoma continue to rise.Citation5,Citation6 The cause of the remaining 60% of melanomas is yet to be determined making development of effective chemopreventive agents particularly important. Despite clinical trials testing a broad array of targeted and non-targeted therapeutic approaches, involving immunotherapy, radiotherapy, and chemotherapy, no effective long-term treatments have been identified for advanced-stage patients.Citation10,Citation11 Thus, the average survival of patients having advanced disease remains 6 to 10 mo.Citation12
Drugs inhibiting the activities of specific genes, signaling pathways or key processes promoting melanoma development are urgently needed, which can then be combined based on what is observed in a particular patient’s tumors for treating and preventing melanoma. The phosphodidylinositol 3-kinase (PI3K) pathway is a key signaling cascade playing a prominent role in melanoma development by relaying extra-cellular signals from cell surface to nucleus to regulate apoptosis.Citation13-Citation16 Epigenetic PTEN silencing in this pathway has been shown to play an important role in melanoma development.Citation17 PTEN is a unique 55 kDa dual specificity phosphatase, which dephosphorylates phosphoserine and phosphotyrosine residues in proteins as well as hydrolyzes the secondary messenger phosphatidylinositol 3,4,5-trisphosphate to regulate PI3K-Akt signaling in melanomas.Citation18 Loss of PTEN has been reported in 30 to 60% of non-inherited melanomas leading to increased PI3K activity measured as elevated levels of Akt3 activity.Citation19 Therefore, targeting epigenetic silencing to reduce Akt activity and that of other signaling cascades could be an important component of a therapeutic cocktail of drugs to treat or prevent melanoma.
Histone acetylation epigenetically regulates gene transcription by modulating DNA packaging to alter expression of proteins aiding cell proliferation and survival.Citation20 The enzymes regulating protein acetylation are histone acetyltransferases (HATs) and histone deacetylases (HDACs).Citation21 Mechanistically, histone acetylation relaxes chromatin to epigenetically promote gene transcription, whereas histone deacetylation induces chromatin condensation to epigenetically decrease gene transcription.Citation22,Citation23 Alterations in both HATs and HDACs contributes to aberrant gene expression, promoting cancer development.Citation24 Therefore, agents targeting these proteins could be an important part of a cocktail for treating or preventing melanomas.
Certain HDACs regulate cellular proliferation and differentiation to promote cancer development, but the function of others remains uncertain.Citation24,Citation25 Currently, 18 HDACs have been identified, including HDAC1 to 11 and SIRT (NAD-dependent deacetylase sirtuin; silent mating type information regulation) 1 to 7. SIRTs are members of a family of nicotinamide adenine dinucleotide (NAD)-dependent enzymes that regulate cell functions by deacetylating both histone and nonhistone targets and at least seven members of the protein family, termed “situins” or SIRTs have been identified in humans.Citation26 Current pharmacological HDAC inhibitors are not specific and therefore cause widespread histone acetylation to broadly induce expression of genes regulating differentiation, apoptosis, the cell cycle, redox pathways, DNA repair, cell migration, and angiogenesis.Citation22,Citation27-Citation30
The best-known HDAC inhibitor is SAHA (suberoylanilide hydroxamic acid), commercially known as Vorinostat or ZolinzaTM, which has been approved by the FDA for treating advanced cutaneous T cell lymphoma.Citation31-Citation34 SAHA had limited efficacy treating metastatic breast, bladder, prostate, colon, kidney, ovary or skin cancer, which has driven the search for chemical modifications to enhance its potency.Citation35 A variety of modifications have been made to SAHA to improve its cancer cell inhibitory efficacy but has resulted in compounds having poor pharmacokinetic properties and/or causing toxicity.Citation22 SAHA derivatives containing one or two selenium atoms have been developed and are called 5-phenylcarbamoylpentyl selenocyanide (PCP-SeCN) or Bis {5-phenylcarbamoylpentyl} diselenide (B(PCP)-2Se), respectively.Citation36
In this study, topical application selenium containing derivatives of SAHA having one or two selenium atoms were found to kill melanoma cells 2 to 4-fold more effectively than SAHA and decrease melanoma tumor development by up to 87% with negligible toxicity. Mechanistically, selenium derivatives of SAHA inhibited HDAC activity and had new selenium-mediated inhibitory properties that led to decreased PI3 kinase pathway activity to increase cellular apoptosis rates. Thus, PCP-SeCN and B(PCP)-2Se compounds has better efficacy for preventing melanoma than the parental compound.
Results
Selenium-containing derivatives of SAHA kill cancer cells more effectively than SAHA
Incorporating selenium into the chemical structure of therapeutic agents can make the drugs more effective melanoma cell killers.Citation37,Citation38 To determine whether incorporation of selenium into SAHA could increase the compound’s anticancer activity, a monomeric selenium containing derivative called PCP-SeCN and a dimeric version called B(PCP)-2Se were synthesized (). Names were based on IUPAC nomenclature.
Figure 1. Structures and activity of HDAC inhibitors. (A) Structures of HDAC inhibitors: SAHA was chemically modified by replacing amino hydroxyl (NH-OH) with free selenium cyanide (SeCN) to create PCP-SeCN. B(PCP)-2Se is a dimer of PCP-SeCN lacking the –CN functional group. (B) PCP-SeCN and B(PCP)-2Se inhibited melanocytic and melanoma cell viability in a time dependent manner. WM35 and UACC 903 cells were treated with increasing concentrations of SAHA, PCP-SeCN or B(PCP)-2Se for 24, 48 or 72 h and cell viability measured using MTS and IC50 values calculated using GraphPad Prism. Data represents average values of three independent experiments.
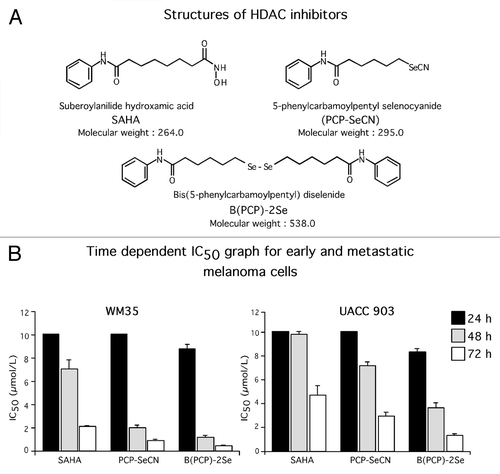
Melanoma cell killing efficacy of PCP-SeCN and B(PCP)-2Se was compared with SAHA following treatment of melanoma cell lines isolated from different stages of tumor progression (Table. 1). Therapeutic universality of the compounds for killing cancer cells was examined by treating pancreatic (MiaPaca-2), breast (MDA-MB-231), prostate (PC-3) or sarcoma (HT-1080) cell lines with the drugs and establishing the IC50 for each cell line (Table. 1). SAHA, PCP-SeCN and B(PCP)-2Se all decreased viability of cancer cells more efficiently than normal human melanocytes, fibroblasts or keratinocytes (). Melanoma cells derived from melanocytic lesion (radial and vertical growth phase) derived cell lines were 3–4 fold more sensitive than metastatic melanoma cells to the selenium derivatives, suggesting the compounds might be effective for melanoma prevention. Average IC50 of B(PCP)-2Se ranged from 0.3–1.4 µmol/L compared with normal melanocytes, fibroblasts and keratinocytes that was 17.05 µmol/L (). PCP-SeCN and B(PCP)-2Se further decreased viability of melanocytic and melanoma cell lines following 48 and 72 h treatment leading to a significant decrease in IC50 values compared with SAHA ( and ). No significant difference was observed in killing cultured melanoma cells between the monomeric selenium containing derivative (PCP-SeCN) and a dimeric version (B(PCP)-2Se) ().
Table 1. PCP-SeCN and B(PCP)-2Se more selectively inhibits cancer cell than SAHA
PCP-SeCN and B(PCP)-2Se retain HDAC inhibitory activity
To determine whether selenium containing SAHA derivatives retained HDAC inhibitory activity, HeLa nuclear extract, having high levels of HDAC activity, was incubated with 1.0 µmol/L of PCP-SeCN, B(PCP)-2Se or SAHA. Inhibitory activity order was found to be PCP-SeCN > B(PCP)-2Se > SAHA (). To establish whether HDAC expression correlated with sensitivity to PCP-SeCN or B(PCP)-2Se, level of HDAC expression was examined in primary melanoma or metastatic melanoma cell lines using an HDAC colorimetric activity assay kit. Higher levels of HDAC activity were observed in melanocytic lesion cell lines (WM35, WM3211, WM115 and WM278.1) compared with metastatic melanoma (A375M and UACC 903) cells, which had 2–3 fold lower levels (). HDAC protein expression also seemed to correlate with activity levels, suggesting that melanocytic cell lines having higher HDAC activity tended to be more sensitive to selenium containing SAHA derivatives than metastatic melanoma cell lines. This observation was used as the rationale to test whether these agents might be effective for preventing early melanocytic lesion development.
Figure 2. PCP-SeCN and B(PCP)-2Se inhibits HDAC activity more effectively than SAHA. (A) PCP-SeCN and B(PCP)-2Se retained HDAC inhibitory activity in HeLa cells. PCP-SeCN and B(PCP)-2Se effectively inhibited HDAC activity at concentrations as low as 1.0 µmol/L comparable to that occurring with SAHA. HDAC activity was expressed as percent inhibition compared with DMSO control. Data represented average of three independent duplicate experiments. (B) Expression and activity of HDAC was elevated in melanocytic lesion compared with metastatic melanoma cell lines. HDAC activity (µmol/L deacetylated histone / µg of nuclear protein / h) was measured in nuclear extracts of melanocytic or melanoma cell lines derived from radial (RGP) WM35, WM3211; vertical (VGP) WM115, WM278.1 and metastatic (MM) A375M; UACC 903 lesions. Compared with melanoma cells, higher HDAC activity was observed in radial and vertical growth phase cells derived from melanocytic lesions. HeLa nuclear extract served as positive control for the assay. Increased HDAC protein expression was observed in melanocytic lesion cell lines by western blotting. α-enolase served as a control for equal protein loading. Data constitute the averages of three independent experiments; bars ± SE (C) PCP-SeCN and B(PCP)-2Se inhibited endogenous HDAC activity in cells derived from primary melanomas. WM35 nuclear extract was incubated with 0.1, 0.5 and 1.0 µmol/L SAHA, PCP-SeCN or B(PCP)-2Se. A dose-dependent decrease in HDAC activity (µmol/L deacetylated histone / µg of nuclear protein / h) was observed with increasing inhibitor concentration. Data represents average of three independent experiments; bars ± SE (D) PCP-SeCN and B(PCP)-2Se increased levels of acetylated histone H3 and H4 in melanocytic lesion cells similar to that observed with SAHA. WM35 cells were treated with 0.25 to 2.0 µmol/L of SAHA, PCP-SeCN and B(PCP)-2Se for 72 h. Nuclear extracts were analyzed by western blotting to determine effect on expression of acetylated histone H3 and H4. A dose dependent increase in expression of acetylated histone H3 and H4 with increasing PCP-SeCN and B(PCP)-2Se concentration was observed, demonstrating HDAC inhibitory activity similar to that observed with SAHA. HDAC1 served as a control for equal protein loading.
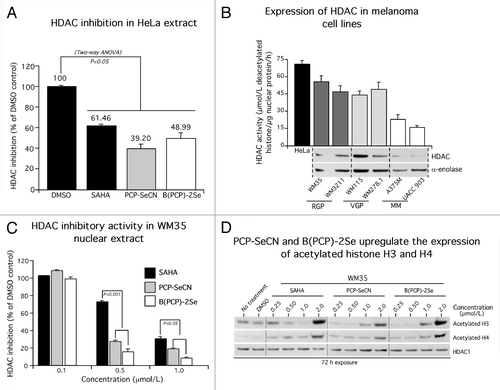
To measure the effect of PCP-SeCN and B(PCP)-2Se on cell lines having high HDAC activity, WM35 cells were treated and compared with SAHA. A dose dependent decrease in HDAC activity was observed with increasing concentration of selenium containing SAHA derivatives. SAHA inhibited HDAC activity 50–60% less than PCP-SeCN or B(PCP)-2Se (). PCP-SeCN and B(PCP)-2Se upregulated levels of acetylated histones in a manner similar to that occurring with SAHA, again confirming the HDAC inhibitory activity of the compounds ().
PCP-SeCN and B(PCP)-2Se inhibited melanoma cell proliferation and triggered apoptosis more effectively than SAHA
To unravel the mechanism by which selenium containing SAHA derivatives more effectively inhibited melanocytic and melanoma cell survival, cell proliferation and apoptosis following treatment were measured. PCP-SeCN and B(PCP)-2Se reduced cellular proliferation and induced apoptosis in a dose responsive manner (). Proliferation of WM35 cells was inhibited by ~80% at 1.25 µmol/L for PCP-SeCN and B(PCP)-2Se, while for UACC 903 cells, 1.25 µmol/L of PCP-SeCN or B(PCP)-2Se led to 25% and 60% inhibition, respectively (). Increased caspase-3/7 activity, an indicator of apoptosis, was observed consistently when WM35 or UACC 903 cells were exposed to PCP-SeCN and B(PCP)-2Se at concentration > 2.5 µmol/L compared with SAHA ().
Figure 3. PCP-SeCN and B(PCP)-2Se inhibited melanoma cell growth by reducing cellular proliferation, triggering apoptosis and elevating levels of sub-G0-G1 phase cells in the cell cycle. (A and B) Cell proliferation and apoptosis inhibited melanoma cell proliferation and triggered apoptosis more effectively than SAHA. WM35 and UACC 903 cells were treated with increasing concentrations of SAHA, PCP-SeCN and B(PCP)-2Se and cell proliferation and apoptosis rates measured after 72 h exposure using BrdU incorporation (A) and Apo-ONE homogenous caspase-3/7 assay kits (B). Data represents average of two to three independent experiments; bars ± SE (C) PCP-SeCN and B(PCP)-2Se increased the sub-G0-G1 population in melanoma cells. WM35 and UACC 903 cells were treated with increasing concentrations of SAHA, PCP-SeCN and B(PCP)-2Se for 72 h. Total cells (floating and adherent) were collected, stained with propidium iodide and analyzed using a FACScan analyzer. A 4–28 fold increase in sub-G0-G1 cells compared with vehicle DMSO treated cells was observed following treatment with PCP-SeCN or B(PCP)-2Se (C).
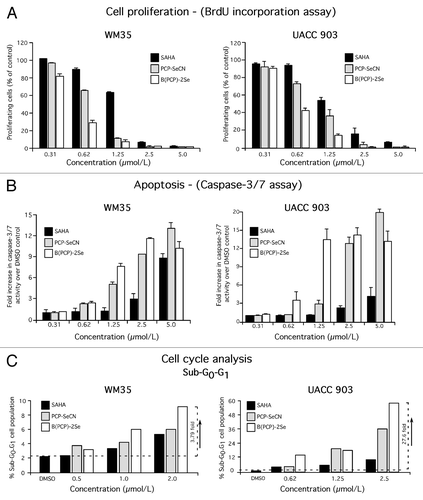
PCP-SeCN and B(PCP)-2Se increased the sub-G0-G1 population in melanoma cells
FACS analysis of WM35 or UACC 903 cells treated with selenium containing SAHA derivatives showed a consistent increase in the sub-G0-G1 population indicating a trend of elevated apoptosis compared with SAHA at concentrations > 1.0 µmol/L (). The sub-G0-G1 population in WM35 and UACC 903 cells increased more when treated with B(PCP)-2Se containing two selenium atoms than when exposed to PCP-SeCN, containing one. A 4–28 fold increase in sub-G0-G1 cells compared with vehicle DMSO treated cells was observed following cell treatment with B(PCP)-2Se ().
Compared with SAHA, PCP-SeCN and B(PCP)-2Se inhibited PI3K/Akt signaling in melanoma cells
The PI3 kinase pathway is a major signaling pathway promoting melanoma development by deregulating apoptosis to promote melanoma development; therefore, regulation of this pathway by these drugs was investigated.Citation14,Citation37 Compared with SAHA, PCP-SeCN and B(PCP)-2Se decreased PI3 kinase pathway activity in both WM35 and UACC 903 cells consistently with increasing the time and concentrations (). Inhibitory effects on this pathway occurred after 24 h of treatment and were most evident following 48 to 72 h exposure. Decreased pAkt and downstream pathway signaling inhibition was evident in both cell lines treated with PCP-SeCN and B(PCP)-2Se compared with SAHA or DMSO treated cells. Greatest inhibitory effect occurred following B(PCP)-2Se treatment, likely due to the presence of 2 selenium atoms. Downstream pPRAS40 (T246) levels also decreased significantly with increasing concentration, correlating with lower pAkt levels. In addition, decreased expression of cyclin D1 and increased p21, cleaved caspase-3 and PARP protein were observed with increasing time and concentrations ()
Figure 4. PCP-SeCN and B(PCP)-2Se but not SAHA inhibited the PI3K signaling pathway in melanoma cells to trigger apoptosis. WM35 (A) and UACC 903 (B) cells were treated with increasing concentrations of SAHA, PCP-SeCN and B(PCP)-2Se for 24–72 h and cell lysates analyzed to determine the expression as well as activity of Akt signaling proteins. PCP-SeCN and B(PCP)-2Se but not SAHA increased levels of cleaved caspase-3/7 and PARP levels mediated by decreased pAkt signaling resulting in reduced cyclin D1 and increased p21 levels. α-enolase served as a control for equal protein loading.

PCP-SeCN and B(PCP)-2Se reduced melanocytic lesion development in laboratory generated skin
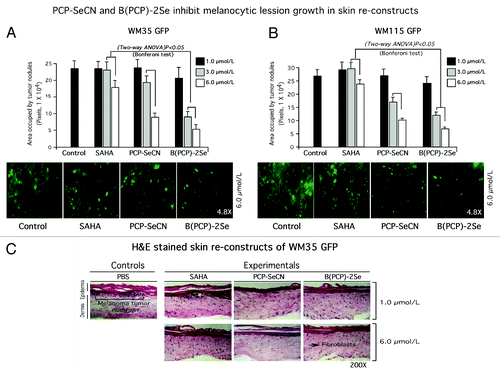
To establish the chemopreventive potential of PCP-SeCN or B(PCP)-2Se, laboratory generated skin reconstructs containing GFP expressing melanocytic lesion cell lines WM35 or WM115 cell lines were created and treated with the agents. This approach is an accepted strategy to measure the chemopreventive efficacy of topical agents.Citation16 Skins containing lesions were topically treated every day for one week and size as well as numbers of GFP cells derived from primary melanomas quantified using fluorescence microscopy. At 6 µmol/L of PCP-SeCN or B(PCP)-2Se, a 60–87% decrease in GFP tagged lesion development was observed compared with vehicle control or SAHA treated skins (). B(PCP)-2Se had the most dramatic effect leading to a consistent > 60% reduction in lesion development in the skin (p < 0.05, Two-way ANOVA). Examination of H&E stained skin to determine whether treatment conditions would damage skin cellular constituents, showed no discernable alteration in skin architecture, skin boundaries, structure or cell morphology following exposure to selenium-containing derivatives of SAHA compared with controls, indicating negligible toxicity ().
Figure 5. PCP-SeCN and B(PCP)-2Se inhibited melanocytic lesion development in laboratory generated skin reconstructs. (5A and 5B) Topical treatment of PCP-SeCN and B(PCP)-2Se inhibited melanocytic lesion development in laboratory-generated skin reconstructs. Skin reconstructs containing GFP expressing WM35 (A) or WM115 (B) melanoma cells were treated topically with 1, 3 and 6 µmol/L PCP-SeCN, B(PCP)-2Se or SAHA (in 200 µL PBS) daily for 8 d. Topical application of PCP-SeCN and B(PCP)-2Se, significantly decreased nodule development in the skin; p < 0.05, One-way ANOVA. No differences in tumor sizes were observed when comparing PBS vehicle control to SAHA treated skins. Magnification 4.8X. (C) Analysis of H and E stained sections showed no significant differences in cell or skin morphology compared with controls treated with PBS or SAHA. Untreated or PBS treated skin were compared with skin exposed to 1 or 6 µmol/L PCP-SeCN, B(PCP)-2Se or SAHA. Magnification 200X.
Discussion
Melanoma is the most invasive and deadly form of skin cancer with no clinically available chemopreventive agents to limit development of early melanocytic lesions into melanomas in skin.Citation39 Melanoma prevention programs, availability of UV protecting sunblocks, and surgical procedures for removing preinvasive lesions have all been employed to decrease the incidence and mortality rates of this disease.Citation40,Citation41 Despite some success, additional approaches are needed, which might involve preventing melanocytic lesions from developing or progressing past its earliest stages, using agents that could be added to creams, lotions, or sunblocks.Citation42 Addition of PCP-SeCN and B(PCP)-2Se to these topical agents could be one approach to prevent melanoma.
SAHA is approved by the FDA but is only marginally effective for treating solid tumors.Citation33 Therefore, a variety of derivatives of SAHA have been synthesized in an attempt to improve the cancer cell-killing efficacy of the drug for treatment and prevention purposes. A series of phosphorus, sulfur and thiol-based compounds have been created and HDAC inhibitory activity examined.Citation43,Citation44 Sulfur containing hydroxamate H40 in which the amidic bond was replaced with a sulfide, inhibited HDAC activity with enhanced efficacy and prevented development of prostate cancer.Citation43 Modified SAHA bearing a hydroxamic acid backbone was found to chelate zinc ions in the active site of HDAC in a bidentate fashion through its CO and OH groups, thereby reducing HDAC activity in a more effective manner.Citation45 This study builds on these prior reports by evaluating the efficacy of selenium-containing derivatives of SAHA, called PCP-SeCN and B(PCP)-2Se for preventing melanoma.Citation36 In this study, use of selenium for preventing cancers, substituting sulfur with selenium in SAHA shown to improve the efficacy of SAHA for preventing melanocytic lesion development. However, it is unclear whether this HDAC inhibitory activity is due to reversible binding of a Se species at the catalytic site on the enzyme, or an irreversible effect due to the redox modification of cysteine residues in the HDAC proteins.
Selenium can increase the potency of chemopreventive and therapeutic agents when this atom is substituted for sulfur in drugs, which generally acts by increasing rates of cellular apoptosis through inhibition of PI3 kinase signaling.Citation42,Citation46 Low selenium levels have also been reported in the serum of patients suffering from cancers including melanoma, which could be mitigated by the selenium released from these agents.Citation47,Citation48 Since incorporation of selenium in a drug can increase its efficacy for killing melanoma cells by inhibiting PI3 kinase pathway signaling, similar modifications were predicted to enhance the therapeutic potential of SAHA.Citation37,Citation38,Citation49,Citation50 Selenium containing PCP-SeCN and B(PCP)-2Se decreased pAkt (S473) and downstream pPRAS40 (T246) levels in a dose-dependent manner, leading to increased levels of apoptosis. Decreased cyclin D1 and increased p21 protein levels were also observed with selenium containing SAHA derivatives. Both PCP-SeCN and B(PCP)-2Se, containing 1 and 2 selenium atoms respectively, inhibited melanoma cell survival more effectively than SAHA and killed cancer cells selectively suggesting these agents might be good melanoma cell killers. These agents also inhibited the growth of cell lines derived from carcinomas of the pancreas, prostate, colon, breast and connective tissue, suggesting efficacy for targeting other cancer types.
Selenium-containing derivatives of SAHA inhibited HDAC activity in a manner similar to that of SAHA.Citation51 HDAC inhibitors TSA, SAHA and FK-228 have been reported to increase p21 levels in a variety of tumor cells.Citation52 Similarly, melanoma cells treated with PCP-SeCN and B(PCP)-2Se increased levels of p21 protein more significantly than observed following SAHA treatment. These compounds also decreased cyclin D1 levels, which resulted in suppression of cyclin dependent kinase activity. Thus, PCP-SeCN and B(PCP)-2Se can decrease expression of cyclin D1 and promote sub-G0-G1 arrest by upregulating p21 protein levels mediated though decreased Akt activity. p21 is a well-known inhibitor of cell cycle progression by reducing the cyclin/cdk2 complex and blocking DNA replication through binding to proliferating cell nuclear antigen.Citation52,Citation53
HDAC inhibitors such as LAQ824 (for leukemia), FK-228 (for cutaneous T cell lymphoma) and MS-275 (for melanoma) are currently being evaluated in clinical trials.Citation54-Citation58 Anti-tumor activity of orally administered HDAC inhibitors such as SAHA or MS-275 can be effective at inhibiting tumor development.Citation59,Citation60 Consistent with these reports, topical application of PCP-SeCN and B(PCP)-2Se inhibited growth of melanocytic lesions developing in laboratory-generated skin reconstructs, demonstrating the utility of these agents for preventing melanoma.
In conclusion, selenium containing derivatives of SAHA, called PCP-SeCN and B(PCP)-2Se have been shown to more effectively prevent melanoma development in skin that SAHA. Mechanistically, enhanced efficacy was due to novel Akt inhibitory properties mediated by selenium. Thus, selenium containing derivatives of SAHA have unique melanoma cell killing properties and can be used for preventing melanoma by acting as HDAC and PI3 kinase pathway inhibitors.
Materials and Methods
Cell lines and culture conditions
Human fibroblast FF2441 cells, melanocytic radial growth phase WM3211, metastatic melanoma cell lines A375M, SK-MEL-24, 1205 Lu and UACC 903, as well as cancer cell lines representing fibro sarcoma (HT-1080), prostate (PC-3), breast (MDA-MB-231) and pancreatic neoplasia (MiaPaca-2) were maintained in DMEM (Invitrogen) supplemented with 10% FBS (Hyclone) in a 5% CO2 atmosphere humidified 37°C incubator. Normal and GFP expressing melanocytic radial (WM35) and vertical (WM115, WM278.1) growth phase melanoma cell lines were maintained in Tu2% medium as described previously.Citation61 Normal human primary FOM103 melanocytes were cultured in melanoblast media: 1X MCDB 153 (Sigma), 2% FBS, 10% chelated FBS (Hyclone, Logan, UT), 100 nM ET3 (VWR International, Radnor, PA), 10 ng/mL SCF (R&D), 20 pM cholera toxin (Sigma), 4.5 ng/mL bFGF (Promega) and 2 mM L-Glutamine (Mediatech) as previously described.Citation62 Passage 2 to 5 human foreskin keratinocyte (HFK) cells were isolated and cultured in EpiLife E-medium as detailed previouslyCitation16
Cell viability, proliferation, apoptosis and cell cycle analysis
Viability and IC50 of normal human melanocytes, keratinocyte, fibroblast and melanoma cells following treatment with SAHA, PCP-SeCN or B(PCP)-2Se were measured using the MTS assay (Promega, Madison, WI).Citation37,Citation38 A total of 5 × 10Citation3 cells per well in 100 µL of media were grown in a 96-well plate for 48 or 72 h respectively for WM35, WM115, A375M, SK-MEL-24, 1205 Lu and UACC 903 or normal cell lines (FOM103, FF2441 and HFK) treated with either DMSO vehicle control or 0.62 to 10 µmol/L of SAHA, PCP-SeCN or B(PCP)-2Se for 24, 48 or 72 h. Cell viability compared with control treated cells was measured using the MTS assay.Citation37,Citation38 IC50 values for each compound in µmol/L for respective cell lines were measured from three independent experiments using GraphPad Prism version 4.01 (GraphPad Software).
Cellular proliferation and apoptosis
Rates were measured by seeding 5 × 10Citation3 cells in 96-well plates, followed by treatment for 72 h with each respective agent. Percentage proliferating and apoptotic cells were quantified using a colorimetric cell proliferation ELISA BrdU kit (Roche Diagnostics) and Apo-ONE Homogenous caspase-3/7 assay kit (Promega), respectively.Citation37,Citation38
Cell cycle analysis
Cell cycle analysis was undertaken by growing 1 × 10Citation6 WM35 and UACC 903 cell lines in 100-mm culture dishes followed by treatment with each respective agent for 72 h. Total floating and adherent cells were collected following trypsinization, and stained using 1 mL propidium iodide solution containing 100 µg/mL propidium iodide; (Sigma), 20 µg/mL Ribonuclease A (Roche Diagnostics) and 3 µg/mL Triton X-100 dissolved in 0.1% (W/V) sodium citrate for 30 min at 4°C. Stained cells were analyzed using the FACScan analyzer (Becton Dickinson) and data processed utilizing ModFit LT software (Verity Software House).Citation37,Citation38
HDAC activity assay
HDAC activity was measured on nuclear and cytosolic extracts using the colorimetric HDAC activity assay kit (BIOMOL) following the manufacturer's protocol. In brief, aliquots of nuclear extracts of melanoma cells or HeLa cell nuclear extract (supplied with the kit) were incubated with 1 mM Color de LysTM substrate at 37°C for 30–60 min in a total volume of 50 µL.Citation36 After incubation, 2 µmol/L TSA in a total volume of 50 µL of 1X developer was added to the sample and incubated at 37°C for 15 min. A series of deacetylated standard dilutions were prepared to establish a standard curve. Absorbance was measured at 405 nm using a SPECTRA max-M2 plate reader (Molecular Devices Corporation).
Western blot analysis
Cell lysates were harvested by addition of lysis buffers containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM sodium orthovanadate, 0.1 mM sodium molybdate, 1mM phenylmethylsulfonyl fluoride, 20 μg/mL aprotinin, and 5 μg/mL leupeptin. Whole cell lysates were centrifuged (≥ 10,000 X g) for 10 min at 4°C to remove cell debris. Protein concentrations were quantitated using the BCA assay from Pierce, and 30 μg of lysate loaded per lane onto NuPAGE Gels (Life Technologies). Following electrophoresis, samples were transferred to polyvinylidene difluoride membrane (Pall Corporation). Blots were probed with antibodies to total Akt, phospho-Akt (Ser473), phospho-PRAS40 (Thr246) caspase-3 and cleaved PARP from Cell Signaling Technology. Anti-acetyl histone H3 and H4 from Upstate Biotechnology. Total PRAS40 from Invitrogen; and, antibodies to cyclin D1, p21, α-enolase and secondary antibodies conjugated with horseradish peroxidase from Santa Cruz Biotechnology. Immunoblots were developed using the enhanced chemiluminescence (ECL) detection system (Thermo Fisher Scientific).
Generation of laboratory skin containing cells derived from primary melanomas
Reconstructed skin (average size measurements: length,14 mm; breadth, 21mm; height, 1mm) was created in a culture dish. Briefly, human FF2441 fibroblasts were trypsinized and resuspended in 10% reconstitution buffer, 10% DMEM (Mediatech), 2.4 µL/mL of 10 mol/L NaOH, and 80% collagen I on ice (Becton Dickinson) at a cell density of 3.75 × 10Citation5/mL. Mixture was then aliquoted into 6 or 12 well plates and incubated at 37°C for 3 h to form a dermal matrix. One mL aliquot of E-medium was added to each well containing a dermis and allowed to grow for 2 additional days. A mixture of normal human keratinocytes and GFP expressing melanoma cells WM35 GFP or WM115 GFP at a ratio of 1:10 were resuspended in 1 mL of E-medium and added on top of the dermal matrix to produce a keratinized layer containing non-invasive cells derived from primary or invasive melanomas. After 2 d, skin reconstructs were transferred onto wire grids in a tissue culture incubator to form a complete keratinized layer 7–8 d later. During this period, developing skin reconstructs were fed via diffusion from E-medium (replaced every other day) below the wire grids.Citation16
Topical drug treatment of skin reconstructs
Skin reconstructs containing nodules of WM35 GFP or WM115 GFP having similar number and size were grouped into vehicle PBS control (200 µL), SAHA, PCP-SeCN or B(PCP)-2Se (1, 3 and 6 µmol/L) treatment groups (n = 3 skin reconstructs in each group). They were exposed to agents every day for 8 consecutive days at which time, a Nikon SMZ 1500 fluorescent microscope (Nikon Instruments) was used to measure the number and area of melanoma nodules using IP Lab software after photographically recording and analyzing of images (BD Biosciences). Average area occupied by nodules for each treatment group were measured and plotted against each drug concentration.Citation16
Histology and morphological characterization of artificial skin reconstructs
Cellular morphology and architecture of skin reconstructs prior to, and at the end of each treatment regime were analyzed following fixation with 10% paraformaldehyde (Electron Microscopy Science). Skin reconstructs were trimmed into strips, frozen in O.C.T compound and sectioned. Formalin fixed paraffin embedded sections were stained with H&E to examine skin architecture, whereas frozen sections were used to photograph location and structure of GFP expressing nodules.Citation16
Statistical analysis
Statistical analysis was performed using Prism 4.0 GraphPad Software. One-way or Two-way Analysis Of Variance (ANOVA) was used for groupwise comparisons, followed by the Tukey’s or Bonferroni’s post hoc tests. For comparison between two groups, the t test was used. Results represent at least two to three independent experiments and are shown as averages ± SEM. Results with a p value less than 0.05 (95% CI) were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Arati Sharma, Omer Kuzu, Keen Chung and Kenneth Huang for technical assistance.
NIH CA-127892–01A, NIH NCI contract (NO2-CB-56603), The Foreman Foundation for Melanoma Research and Melanoma Research Foundation.
Note
PCP-SeCN (5-phenylcarbamoylpentyl selenocyanide) and B(PCP)-2Se (Bis {5-phenylcarbamoylpentyl} diselenide) are also referred to as SelSA-2 (containing one selenium atom) and SelSA-1 (containing 2 selenium atoms) respectively in the published reports by Desai et al.Citation36 Since names were misleading regarding the number of selenium atoms, this manuscript uses names derived from the established IUPAC nomenclature.
Related Research Data
References
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature 2007; 445:851 - 7; http://dx.doi.org/10.1038/nature05661; PMID: 17314971
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225 - 49; http://dx.doi.org/10.3322/caac.20006; PMID: 19474385
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711 - 23; http://dx.doi.org/10.1056/NEJMoa1003466; PMID: 20525992
- Jemal A, Devesa SS, Hartge P, Tucker MA. Recent trends in cutaneous melanoma incidence among whites in the United States. J Natl Cancer Inst 2001; 93:678 - 83; http://dx.doi.org/10.1093/jnci/93.9.678; PMID: 11333289
- Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52:23 - 47; http://dx.doi.org/10.3322/canjclin.52.1.23; PMID: 11814064
- Ley RD, Reeve VE. Chemoprevention of ultraviolet radiation-induced skin cancer. Environ Health Perspect 1997; 105:Suppl 4 981 - 4; PMID: 9255591
- Stratton SP, Dorr RT, Alberts DS. The state-of-the-art in chemoprevention of skin cancer. Eur J Cancer 2000; 36:1292 - 7; http://dx.doi.org/10.1016/S0959-8049(00)00108-8; PMID: 10882869
- Einspahr JG, Bowden GT, Alberts DS. Skin cancer chemoprevention: strategies to save our skin. Recent Results Cancer Res 2003; 163:151 - 64, discussion 264-6; http://dx.doi.org/10.1007/978-3-642-55647-0_14; PMID: 12903851
- Helmbach H, Rossmann E, Kern MA, Schadendorf D. Drug-resistance in human melanoma. Int J Cancer 2001; 93:617 - 22; http://dx.doi.org/10.1002/ijc.1378; PMID: 11477569
- Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bardia A, et al, Melanoma Study Group of the Mayo Clinic Cancer Center. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc 2007; 82:364 - 80; PMID: 17352373
- DeLuca AM, Srinivas A, Alani RM. BRAF kinase in melanoma development and progression. Expert Rev Mol Med 2008; 10:e6; http://dx.doi.org/10.1017/S1462399408000604; PMID: 18279546
- Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res 2007; 67:3626 - 36; http://dx.doi.org/10.1158/0008-5472.CAN-06-4234; PMID: 17440074
- Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res 2004; 64:7002 - 10; http://dx.doi.org/10.1158/0008-5472.CAN-04-1399; PMID: 15466193
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 54; http://dx.doi.org/10.1038/nature00766; PMID: 12068308
- Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, et al. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res 2008; 68:7638 - 49; http://dx.doi.org/10.1158/0008-5472.CAN-07-6614; PMID: 18794153
- Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res 2006; 66:6546 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-06-0384; PMID: 16818626
- Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res 2009; 22:400 - 19; http://dx.doi.org/10.1111/j.1755-148X.2009.00585.x; PMID: 19493313
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res 2003; 63:2881 - 90; PMID: 12782594
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737 - 49; http://dx.doi.org/10.1042/BJ20021321; PMID: 12429021
- Wiech NL, Fisher JF, Helquist P, Wiest O. Inhibition of histone deacetylases: a pharmacological approach to the treatment of non-cancer disorders. Curr Top Med Chem 2009; 9:257 - 71; http://dx.doi.org/10.2174/156802609788085241; PMID: 19355990
- Marson CM. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med Chem 2009; 9:661 - 92; PMID: 19601748
- Buchwald M, Krämer OH, Heinzel T. HDACi--targets beyond chromatin. Cancer Lett 2009; 280:160 - 7; http://dx.doi.org/10.1016/j.canlet.2009.02.028; PMID: 19342155
- Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol 2008; 615:261 - 98; http://dx.doi.org/10.1007/978-1-4020-6554-5_13; PMID: 18437899
- Martínez-Iglesias O, Ruiz-Llorente L, Sánchez-Martínez R, García L, Zambrano A, Aranda A. Histone deacetylase inhibitors: mechanism of action and therapeutic use in cancer. Clin Transl Oncol 2008; 10:395 - 8; http://dx.doi.org/10.1007/s12094-008-0221-x; PMID: 18628067
- Han SH. Potential role of sirtuin as a therapeutic target for neurodegenerative diseases. J Clin Neurol 2009; 5:120 - 5; http://dx.doi.org/10.3988/jcn.2009.5.3.120; PMID: 19826562
- Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, Horinouchi S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother Pharmacol 2001; 48:Suppl 1 S20 - 6; http://dx.doi.org/10.1007/s002800100300; PMID: 11587361
- Wang C, Fu M, Mani S, Wadler S, Senderowicz AM, Pestell RG. Histone acetylation and the cell-cycle in cancer. Front Biosci 2001; 6:D610 - 29; http://dx.doi.org/10.2741/1wang1; PMID: 11282573
- Fang JY. Histone deacetylase inhibitors, anticancerous mechanism and therapy for gastrointestinal cancers. J Gastroenterol Hepatol 2005; 20:988 - 94; http://dx.doi.org/10.1111/j.1440-1746.2005.03807.x; PMID: 15955204
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5:769 - 84; http://dx.doi.org/10.1038/nrd2133; PMID: 16955068
- Kavanaugh SM, White LA, Kolesar JM. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am J Health Syst Pharm 2010; 67:793 - 7; http://dx.doi.org/10.2146/ajhp090247; PMID: 20479100
- Mitsiades CS, Hayden PJ, Anderson KC, Richardson PG. From the bench to the bedside: emerging new treatments in multiple myeloma. Best Pract Res Clin Haematol 2007; 20:797 - 816; http://dx.doi.org/10.1016/j.beha.2007.09.008; PMID: 18070720
- Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene 2007; 26:1351 - 6; http://dx.doi.org/10.1038/sj.onc.1210204; PMID: 17322921
- Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors--development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol 2005; 2:150 - 7; http://dx.doi.org/10.1038/ncponc0106; PMID: 16264908
- Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 2009; 15:3958 - 69; http://dx.doi.org/10.1158/1078-0432.CCR-08-2785; PMID: 19509172
- Desai D, Salli U, Vrana KE, Amin S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg Med Chem Lett 2010; 20:2044 - 7; http://dx.doi.org/10.1016/j.bmcl.2009.07.068; PMID: 20167479
- Sharma A, Sharma AK, Madhunapantula SV, Desai D, Huh SJ, Mosca P, et al. Targeting Akt3 signaling in malignant melanoma using isoselenocyanates. Clin Cancer Res 2009; 15:1674 - 85; http://dx.doi.org/10.1158/1078-0432.CCR-08-2214; PMID: 19208796
- Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol Cancer Ther 2008; 7:1297 - 308; http://dx.doi.org/10.1158/1535-7163.MCT-07-2267; PMID: 18483317
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene 2003; 22:3138 - 51; http://dx.doi.org/10.1038/sj.onc.1206454; PMID: 12789290
- Berwick M, Erdei E, Hay J. Melanoma epidemiology and public health. [viii.] Dermatol Clin 2009; 27:205 - 14, viii; http://dx.doi.org/10.1016/j.det.2008.12.002; PMID: 19254665
- Greinert R. Skin cancer: new markers for better prevention. Pathobiology 2009; 76:64 - 81; http://dx.doi.org/10.1159/000201675; PMID: 19367127
- Nguyen N, Sharma A, Nguyen N, Sharma AK, Desai D, Huh SJ, et al. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev Res (Phila) 2011; 4:248 - 58; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0106; PMID: 21097713
- Gu W, Nusinzon I, Smith RD Jr., Horvath CM, Silverman RB. Carbonyl- and sulfur-containing analogs of suberoylanilide hydroxamic acid: Potent inhibition of histone deacetylases. Bioorg Med Chem 2006; 14:3320 - 9; http://dx.doi.org/10.1016/j.bmc.2005.12.047; PMID: 16434199
- Kapustin GV, Fejér G, Gronlund JL, McCafferty DG, Seto E, Etzkorn FA. Phosphorus-based SAHA analogues as histone deacetylase inhibitors. Org Lett 2003; 5:3053 - 6; http://dx.doi.org/10.1021/ol035056n; PMID: 12916979
- Mai A, Massa S, Rotili D, Cerbara I, Valente S, Pezzi R, et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev 2005; 25:261 - 309; http://dx.doi.org/10.1002/med.20024; PMID: 15717297
- Chung CY, Madhunapantula SV, Desai D, Amin S, Robertson GP. Melanoma prevention using topical PBISe. Cancer Prev Res (Phila) 2011; 4:935 - 48; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0202; PMID: 21367959
- Dennert G, Zwahlen M, Brinkman M, Vinceti M, Zeegers MP, Horneber M. Selenium for preventing cancer. Cochrane Database Syst Rev: CD005195.
- Jensen JD, Wing GJ, Dellavalle RP. Nutrition and melanoma prevention. Clin Dermatol 2010; 28:644 - 9; http://dx.doi.org/10.1016/j.clindermatol.2010.03.026; PMID: 21034988
- Chen CS, Weng SC, Tseng PH, Lin HP, Chen CS. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem 2005; 280:38879 - 87; http://dx.doi.org/10.1074/jbc.M505733200; PMID: 16186112
- Bali P, Pranpat M, Swaby R, Fiskus W, Yamaguchi H, Balasis M, et al. Activity of suberoylanilide hydroxamic Acid against human breast cancer cells with amplification of her-2. Clin Cancer Res 2005; 11:6382 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-05-0344; PMID: 16144943
- Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett 2009; 280:192 - 200; http://dx.doi.org/10.1016/j.canlet.2009.03.013; PMID: 19345475
- Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol 2007; 39:1367 - 74; http://dx.doi.org/10.1016/j.biocel.2007.03.001; PMID: 17412634
- Madhunapantula SV, Hengst J, Gowda R, Fox TE, Yun JK, Robertson GP. Targeting sphingosine kinase-1 to inhibit melanoma. Pigment Cell Melanoma Res 2012; 25:259 - 74; http://dx.doi.org/10.1111/j.1755-148X.2012.00970.x; PMID: 22236408
- Piekarz RL, Sackett DL, Bates SE. Histone deacetylase inhibitors and demethylating agents: clinical development of histone deacetylase inhibitors for cancer therapy. Cancer J 2007; 13:30 - 9; http://dx.doi.org/10.1097/PPO.0b013e31803c73cc; PMID: 17464244
- Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs 2007; 16:659 - 78; http://dx.doi.org/10.1517/13543784.16.5.659; PMID: 17461739
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27:5459 - 68; http://dx.doi.org/10.1200/JCO.2009.22.1291; PMID: 19826124
- Cang S, Ma Y, Liu D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J Hematol Oncol 2009; 2:22; http://dx.doi.org/10.1186/1756-8722-2-22; PMID: 19486511
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem 2009; 107:600 - 8; http://dx.doi.org/10.1002/jcb.22185; PMID: 19459166
- Kato Y, Yoshimura K, Shin T, Verheul H, Hammers H, Sanni TB, et al. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin Cancer Res 2007; 13:4538 - 46; http://dx.doi.org/10.1158/1078-0432.CCR-07-0014; PMID: 17671140
- He R, Chen Y, Chen Y, Ougolkov AV, Zhang JS, Savoy DN, et al. Synthesis and biological evaluation of triazol-4-ylphenyl-bearing histone deacetylase inhibitors as anticancer agents. J Med Chem 2010; 53:1347 - 56; http://dx.doi.org/10.1021/jm901667k; PMID: 20055418
- Quong RY, Bickford ST, Ing YL, Terman B, Herlyn M, Lassam NJ. Protein kinases in normal and transformed melanocytes. Melanoma Res 1994; 4:313 - 9; http://dx.doi.org/10.1097/00008390-199410000-00008; PMID: 7858416
- Satyamoorthy K, DeJesus E, Linnenbach AJ, Kraj B, Kornreich DL, Rendle S, et al. Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res 1997; 7:Suppl 2 S35 - 42; http://dx.doi.org/10.1097/00008390-199708001-00007; PMID: 9578415
- Karelia N, Desai D, Hengst JA, Amin S, Rudrabhatla SV, Yun J. Selenium-containing analogs of SAHA induce cytotoxicity in lung cancer cells. Bioorg Med Chem Lett 2010; 20:6816 - 9; http://dx.doi.org/10.1016/j.bmcl.2010.08.113; PMID: 20855208