Abstract
BRCA1 plays an important role in DNA damage and repair, homologous recombination, cell-cycle regulation and apoptosis. BRCA-mutated ovarian cancer often presents at an advanced stage, however, tend to have better response to platinum-based chemotherapy as compared with sporadic cases of epithelial ovarian cancer (EOC). In spite of this, most patients will develop a recurrence and eventually succumb to the disease. Preclinical studies are currently investigating natural compounds and their analogs for tumor-directed targets in ovarian cancer. The aim of this study is to investigate whether the STAT3 inhibitor HO-3867, a novel curcumin analog, has a therapeutic effect on BRCA1-mutated ovarian cancer. Our novel agent, HO-3867 and a commercial STAT3 inhibitor, STATTIC, significantly inhibited BRCA-mutated ovarian cancer cells in vitro in a dose- and time-dependent manner. BRCA-mutated ovarian cancer cells treated with HO-3867 exhibited a significant degree of apoptosis with elevated levels of cleaved caspase-3, caspase-7 and PARP. HO-3867 treatment induced more reactive oxygen species (ROS) in BRCA-mutated cells compared with wild-type cells, however, there was no increased ROS when benign ovarian surface epithelial cells were treated with HO-3867. BRCA1-mutated cancer cells had higher expression of Tyrosine-phosphorylated STAT3 (pTyr705) as compared with other STAT proteins. Furthermore, treatment of these cells with HO-3867 resulted in decreased expression of pTyr705 and its downstream targets cyclin D1, Bcl-2 and survivin. In addition, overexpression of STAT3 cDNA provided resistance to HO-3867-induced apoptosis. Our results show that HO-3867, a potent STAT3 inhibitor, may have a role as a biologically targeted agent for BRCA1-mutated cancers either as an adjunct to cytotoxic chemotherapy or as a single agent.
Introduction
Despite advances in treatment, epithelial ovarian cancer remains the most lethal of all gynecologic malignancies. In 2011, there were an estimated 22,000 new cases with approximately 15,500 deaths, making ovarian cancer the 5th most common cause of cancer-related death. Treatment for advanced ovarian cancer includes radical tumor cytoreductive surgery in combination with adjuvant chemotherapy. Unfortunately, the majority of ovarian cancers are diagnosed at an advanced stage and while there have been modest improvements in overall 5-y survival, from 30 to 50 percent, this trend does not apply to advanced–stage tumors which have only improved from 20 to 25 percent in the same timeframe.
Hereditary breast and ovarian cancer syndrome (HBOC) is attributed to germline mutations in the BRCA1 or BRCA2 tumor suppressor genes. BRCA mutations may account for as many as 90 percent of hereditary ovarian cancers and approximately 10 percent of all ovarian cancers.Citation1-Citation3 Women with BRCA1 mutations have a 40–50% cumulative lifetime risk of developing ovarian cancer (10–25% for BRCA2 mutations), a marked increase from the 1.3% lifetime risk in women who are not mutation carriers.Citation4-Citation8 Both BRCA proteins are involved in DNA repair via homologous recombination (HR) and thus cells with deficient BRCA are unable to repair DNA double strand breaks by HR. This can result in genomic instability and a predisposition to malignant transformation. Although patients with BRCA deficiency have similar histopathologic characteristics as sporadic ovarian cancers, the time-to-recurrence, response rates to chemotherapy and overall survival are significantly improved in advanced-stage hereditary ovarian cancer patients.Citation9,Citation10 It is theorized that the same DNA repair deficiency in hereditary tumors confers the improved survival rates to BRCA-deficient patients as these tumors are less likely to repair DNA cross-links caused by chemotherapeutic agents such as cisplatin.Citation11-Citation14
Curcumin has been used as a dietary spice and been consumed as a medicinal supplement for thousands of years. In 1949, curcumin was shown to be a biologically active compound with bactericidal attributes and more recently with anticancer properties. However, curcumin has poor bioavailability due to poor absorption, rapid metabolism and rapid elimination. Efforts have been made to improve curcumin’s bioavailability, for example, with the use of structural analogs.Citation15 Recently, we developed a novel class
of curcumin analogs, diarylidenyl piperidones (DAP), which were developed by incorporating a piperidone link to the β-diketone structure and fluorosubstitutions on the phenyl groups.Citation16-Citation18 The DAP compounds, in general, were more effective than curcumin in inhibiting the proliferation of a variety of cancer cell lines.Citation16 HO-3867, one of the DAP compounds with ortho-fluorinated phenyl groups, exhibited potent anticancer efficacy in vitro when tested using ovarian epithelial cancer cell lines as well as in vivo using a mouse model.Citation19 In this study, we investigated the effects of HO-3867 on the proliferation and viability of BRCA1-deficient ovarian cancer cells. Specifically, we evaluated the production of reactive oxygen species (ROS) and induction of apoptosis in BRCA1-mutated ovarian cancer cells. Our results show that the curcumin analog HO-3867 rapidly and selectively induces ROS and inhibition of STAT3, resulting in suppression of cell proliferation and induction of apoptosis.
Results
Effect of STAT3 inhibitor on BRCA1-mutated ovarian cancer cell proliferation
To determine the effect of our novel STAT3 inhibitor, HO-3867, on BRCA1-mutated ovarian cancer cell proliferation and viability, cells were treated with increasing doses of HO-3867 (1 μM, 5 μM, 10 μM and 20 μM) for 24 h. shows results of a clonogenic assay on cells treated with HO-3867 and depicts a dose-dependent decrease in the percentage of colonies formed. At 10 μM, a greater than 80% reduction in the number of colonies was observed. shows the effect of HO-3867 on the total number of cells after various time points of treatment with increasing doses of HO-3867. After 48 h incubation with 10 μM of HO-3867, there was a 9-fold decrease in the number of BRCA1-mutated cells when compared with controls. At 20 μM, 90 percent of cells were non-viable after 24 h incubation and no cells remained viable at 48 h. Next, control and treated cells grown on glass coverslips were stained with DAPI to confirm a decrease in proliferation. shows untreated cells with normal proliferative staining as compared with cells treated with HO-3867, which had markedly decreased staining. shows results of a MTT viability assay of four ovarian cancer cell lines, A2780, A2780R, SKOV3 and BRCA1- mutated cells. While cell viability is decreased in all four cell lines, the most marked reduction was in BRCA1 cells. Following this, BRCA cells were then treated with HO-3867, cisplatin, ABT (a commercial PARP inhibitor), curcumin, or STATTIC (a commercial STAT3 inhibitor). There was a significant decrease with all treatments, however, the most pronounced was with HO-3867 which appeared to be more effective than STATTIC, a known potent STAT3 inhibitor ().
Figure 1. HO-3867 inhibits BRCA1 ovarian cancer cell proliferation. (A) Clonogenic assay of BRCA1-mutated ovarian cancer cells treated with increasing concentrations of HO-3867 for 24 h (1, 5, 10 and 20 µM). There is a significant decrease in the percentage of cell colonies in a dose response progression (left panel). (B) After treatment with increasing concentrations of HO-3867 (1, 5, 10, 20 µM) at serially sequential times (12, 24, 48 h) cells were collected and counted. Cell counts were significantly decreased starting with the 5 µM concentration. After 24 h treatment with 20 µM there were no viable cells remaining. (C) Conformation of decreased proliferation was performed with immunohistochemistry staining with DAPI. This demonstrated that cells treated with 10 µM of HO-3867 showed a significant decrease in the number of proliferating cells. (D) MTT assay of four ovarian cancer cell lines (A2780, A2780R, SKOV3 and BRCA1-mutated cells) treated with HO-3867 or STATTIC. While all cell lines showed significant decreased viability, this was most observed in the BRCA1-mutated cells. (E) Of interest HO-3867 showed more of an effect than the potent STAT3 inhibitor STATTIC. E, MTT assay of BRCA1-mutated cells with different treatments [Curcumin, cisplatin (CP), PARP-inhibitor (ABT), STATTIC and HO-3867]. While all treatments decreased cell viability the most dramatic effect was seen in the HO-3867 treatment group.
![Figure 1. HO-3867 inhibits BRCA1 ovarian cancer cell proliferation. (A) Clonogenic assay of BRCA1-mutated ovarian cancer cells treated with increasing concentrations of HO-3867 for 24 h (1, 5, 10 and 20 µM). There is a significant decrease in the percentage of cell colonies in a dose response progression (left panel). (B) After treatment with increasing concentrations of HO-3867 (1, 5, 10, 20 µM) at serially sequential times (12, 24, 48 h) cells were collected and counted. Cell counts were significantly decreased starting with the 5 µM concentration. After 24 h treatment with 20 µM there were no viable cells remaining. (C) Conformation of decreased proliferation was performed with immunohistochemistry staining with DAPI. This demonstrated that cells treated with 10 µM of HO-3867 showed a significant decrease in the number of proliferating cells. (D) MTT assay of four ovarian cancer cell lines (A2780, A2780R, SKOV3 and BRCA1-mutated cells) treated with HO-3867 or STATTIC. While all cell lines showed significant decreased viability, this was most observed in the BRCA1-mutated cells. (E) Of interest HO-3867 showed more of an effect than the potent STAT3 inhibitor STATTIC. E, MTT assay of BRCA1-mutated cells with different treatments [Curcumin, cisplatin (CP), PARP-inhibitor (ABT), STATTIC and HO-3867]. While all treatments decreased cell viability the most dramatic effect was seen in the HO-3867 treatment group.](/cms/asset/e9cda24b-3a42-4bf7-b8f8-7de34c7bdcd5/kcbt_a_10920559_f0001.gif)
HO-3867 induces apoptosis in BRCA1-mutated ovarian cancer cells
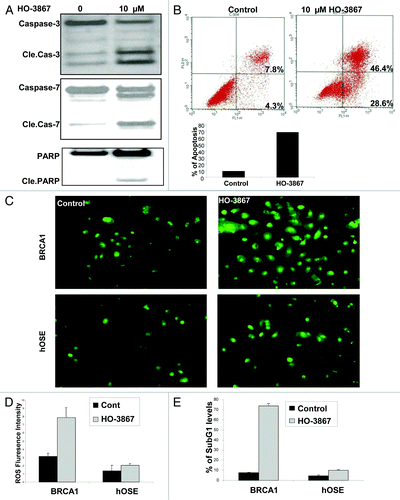
To determine whether HO-3867 not only decreases the proliferation of cells, but ultimately causes cell death, a series of experiments were performed to evaluate if HO-3867 induces apoptosis in BRCA1-mutated ovarian cancer cells. shows a western blot of control vs HO-3867-treated cells. This figure demonstrates that treatment results in cleavage of caspase 3, 7 and PARP, suggesting more cells are going through the apoptotic pathway. These results correlated with the results of annexin V flow cytometry seen in . Seventy-five percent of cells treated with HO-3867 were shown to be in either early or late phase apoptosis compared with 12% of untreated cells, respectively. We then wanted to show that HO-3867, while toxic to cancer cells, does relatively little harm to non-cancerous cells. shows the results of a ROS assay. For BRCA1-mutated cells there was an increase in the number of ROS correlating to DNA damage and eventual cell death compared with controls. However, in non-cancerous human ovarian surface epithelial (hOSE) cells, this phenomenon was not seen There was no difference in the amount of ROS staining seen between control and treatment groups in hOSE cells inferring that HO-3867, while damaging to cancer cells, may spare its harmful effects to non-cancerous cells. This was additionally confirmed using a subG1 analysis with flow cytometry (). Again, hOSE cells showed no difference in the percent of cells in the subG1 phase, however in BRCA1-mutated cells, there was a significant increase in the number of cells treated with HO-3867 that were unable to continue through the cell cycle.
Figure 2. Effect of HO-3867 on apoptosis in BRCA1 ovarian cancer cells. (A) Representative western blot showing a significant increase in cleaved caspase -3, -7 and cleaved PARP after treatment with HO-3867. (B) Flow cytometry with propidium iodide and annexin V. Treatment with HO-3867 showed a significant increase in the number of cells in apoptosis from controls (75.0% vs 12.1%, respectively). Graphical representation of flow cytometry results shown. (C and D) Measurement of intracellular ROS in BRCA1-mutated cancer cells and normal hOSE cells. At baseline there is an increased measure of ROS in BRCA1 cancer cells vs. controls. After treatment with 10uM HO-3867 for 24 h, a significant increase in the number of cells with ROS was detected in BRCA1 cells. This observation, however, was not seen in hOSE cells showing HO-3867 was selective against the BRCA1-mutated cancer cell line while sparing normal, non-cancerous cells. (E) Graphical results from a subG1 analysis after treatment with HO-3867 for 24 h. After treatment, a significant increase in the number of BRCA1 cells remained in the subG1 phase.
Expression of STAT proteins in BRCA1-mutated ovarian cancer cells
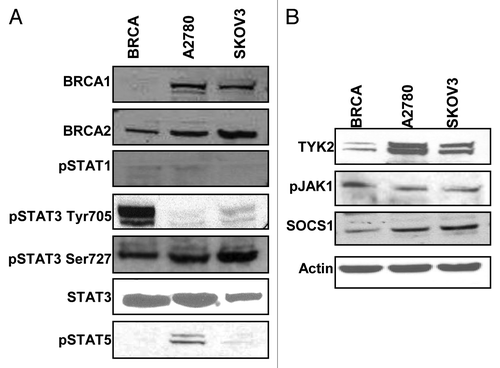
Previous studies have shown HO-3867 and other curcumin analogs exert their effects, at least in part, via inhibition of STAT proteins. In order to understand why HO-3867 had a greater effect on BRCA1-mutated cells as compared with non-mutated EOC cells western blot analysis of various STAT proteins was performed. As seen in , there was minimal expression of pSTAT1 and pSTAT5 among all three cell lines. There was, however, a markedly higher expression of pSTAT3 Tyr 705 (pTyr705) expression in BRCA1-mutated cells as compared with A2780 and SKOV3 cells. Total STAT3 and a second phosphorylation site, pSTAT3 Ser727 (pSer727), were observed to be similarly expressed in all three cell lines. The striking difference of pTyr705 expression in BRCA1-mutated cells compared with the other ovarian cancer cell lines gives a possible reason for the difference seen when treated with a potent STAT inhibitor. shows consistent expression of upstream proteins including TYK2, pJAK1 and JAK 2 in all cell lines. There was a decrease in the downstream negative regulator SOCS1, which may also play a role in treatment discrepancy.
Figure 3. Activation of the STAT pathway in BRCA1-mutated ovarian cancer cells. (A) BRCA1, A2780 and SKOV3 cells were cultured and collected for western blot for expression of proteins in the STAT family. There was minimal expression of STAT1 or STAT5 (A2780 cells minimally expressed STAT5). All cell lines uniformily expressed STAT3 and the activated form pSTAT3 Ser727, however, only BRCA1 cells showed expression of pSTAT3 Tyr705. (B) Representative western blots for protein expression in the STAT3 pathway. BRCA1 cells showed less expression of TYK2 and SOCS1. Both pJAK1 and JAK2 were consistently expressed in all lines.
HO-3867 inhibits STAT3 and downstream proteins
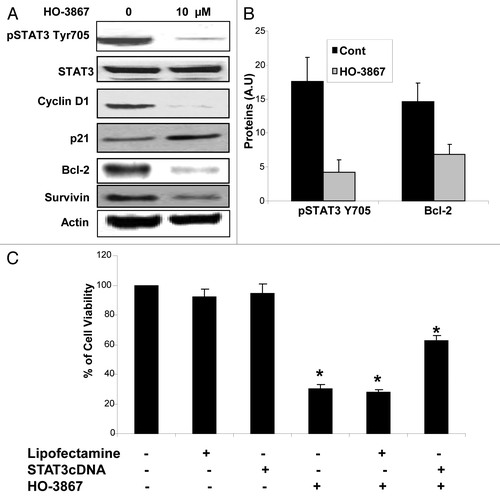
It was important to determine if HO-3867 alters protein expression of pTyr705 and cell-survival proteins downstream of the STAT pathway in BRCA1-mutated cells (). After treatment with 10 μM of HO-3867 for 24 h, a significant decrease in the expression of pTyr705 was observed. Effects on downstream proteins included decreased expression of cell proliferation and cell survival proteins (cyclin D1) and apoptotic proteins (Bcl-2, Survivin). There was also an increase in the cell cycle regulator protein p21. shows the quantitative depiction of average levels of expression between control and treated cells for pTyr705 and Bcl-2. In order to determine if decreased cell viability was truly from inhibition of STAT3, cells were transfected with STAT3 cDNA prior to treatment with HO-3867. Results seen in show that cells transfected with STAT3 cDNA had increased viability compared with non-transfected cells treated with HO-3867. This infers that BRCA 1-mutated cells transfected with STAT3 cDNA acquired some resistance to HO-3867.
Figure 4. Effect of HO-3867 on STAT pathway protein expression. (A) BRCA1 cells were treated with 10uM of HO3867 for 24 h and protein expression analyzed via western blot. There was a significant decrease in the expression of activated pSTAT3 Tyr705, the downstream prosurvival proteins pAKT and Cyclin D1, as well as anti-apoptotic proteins Bcl-2 and Survivin. Increased expression of the cell cycle regulator p21 was seen as well. (B) Graphical representation confirms decrease of pSTAT3705 and Bcl-2 expression after treatment with 10uM HO-3867 for 24 h. (C) STAT3 overexpression leads to HO-3867 resistance. BRCA1-mutated cells were transfected with STAT3 cDNA to see if overexpression of STAT3 could lead to resistance of HO-3867 on cell viability. Cells that were treated with 10uM of HO-3867 with only the lipofectamine vector showed identical loss in cell viability as cells treated with HO-3867 alone. However, HO-3867-treated cells that were transfected with STAT3 cDNA had a significant increase in viability from those cells that were not transfected. Although there was improved survival there was still a significant decrease in viability from control cells showing the potency of HO-3867 as a STAT inhibitor.
Effect of HO-3867 on BRCA1-mutated breast cancer cells
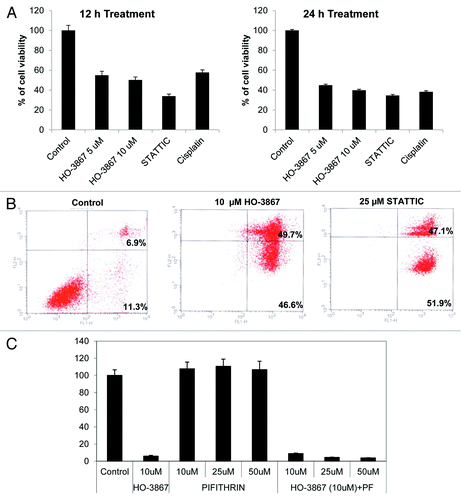
Finally, we looked at the effect HO-3867 had on other BRCA1-mutated cancer cells, namely SUM 149, a breast cancer cell line. SUM149 cells were treated for 12 and 24 h with 5 or 10 μM of HO-3867, 25 μM of STATTIC, or 10 μg/mL of cisplatin. shows the results of a MTT assay performed after treatment. A decrease in viability was seen in all treatments at both time points. While STATTIC showed the largest decrease in viability at 12 h, by 24 h, all treatments showed a 50–60% decrease in viability. In order to determine if HO-3867 had a similar effect on cell death in SUM 149 cells as with the ovarian cell population, an Annexin assay was performed. Treatment with either 10 μM of HO-3867 or 25 μM STATTIC for 24 h showed a significant increase in apoptosis vs. controls (> 95% vs. 18.2%) as seen in . Further, we determine whether or not the STAT3 inhibitor-induced apoptosis was dependent on p53, BRCA1 cancer cells were pretreated with pifithrin (pifithrin; Sigma) for 1 h and then treated with HO-3867 and STATTIC for 24 h. The cells were analyzed for cell viability by MTT assay. Pretreatment with pifithrin did not block HO-3867 and STATTIC-induced apoptosis (). These results indicate that the STAT3 inhibitor- induced apoptosis in the BRCA1 cells is not mediated by p53.
Figure 5. Effect of HO-3867 on viability and apoptosis in BRCA1-mutated breast cancer cells. (A) MTT assay of BRCA1-mutated breast cancer cell line (SUM149) treated with 5 or 10 μM HO-3867, 25 μM STATTIC or 10 μg/mL cisplatin for 12 or 24 h. Cells showed a significant decreased in viability as early as 12 h. By 24 h all treatments decreased viability by 50–60 percent. (B) Flow cytometry with propidium iodide and annexin V. Treatment with 10 μM HO-3867 and 25 μM STATTIC showed a significant increase in the number of cells in apoptosis from controls (96.3% and 99% vs 18.2%, respectively). Graphical representation of flow cytometry results shown. (C) Inhibition of p53 by pifithrin did not reverse the STAT3 inhibitor induced apoptosis. BRCA1 mutated ovarian cancer cells were pretreated with the pifithrin 10, 25 and 50 µM for 1 h and then treated with HO-3867 and STATTIC for 24 h. Cell viability was analyzed by MTT assay. STATTIC data not shown.
Discussion
Our novel anti-cancer agent HO-3867 has been shown to be a potent STAT3 inhibitor with anticancer properties. This is the first report showing HO-3867 and STATTIC, another potent STAT3 inhibitor, significantly suppresses BRCA1-mutated ovarian cancer cell proliferation and viability. This study furthermore demonstrates that HO-3867 selectively induces ROS and apoptosis in BRCA1-mutated human ovarian cancer cells in vitro. We have shown that pSTAT3 is highly expressed in BRCA1-mutated cancer cells compared with other STAT proteins. Our compound, HO-3867, inhibits BRCA1-mutated cancer cell proliferation and survival by targeting STAT3 pathways and altering the expression of pro-survival proteins cyclin D1, Bcl-2 and survivin.
Tumor cells typically exhibit increased levels of intracellular ROS, which in turn induce various gene mutations leading to metabolic malfunction and more ROS generation. Furthermore, ROS induces oxidative damage to proteins, lipids and other cellular components causing significant intracellular stress. Citation20,Citation21 In this study, a proposed therapeutic strategy against BRCA1-mutated ovarian cancer is to treat cancer cells with chemotherapeutic agents that have pro-oxidant properties, which increase the intracellular ROS generation immensely in cancer cells without harming normal cells. This hypothesis is supported by our recent findings that ROS-generating agents showed selective toxicity in tumor cells with increased ROS. Using EPR measurements, we have previously observed that HO-3867 underwent redox cycling to its corresponding nitroxide forms in vitro. We examined the role of the nitroxide moiety by comparing HO-3867 with its parent structure, H-4073. Cytotoxic effects of H-4073 and HO-3867 on A2780 cells were significantly higher compared with curcumin under similar conditions.Citation16 However, while H-4073 significantly reduced cell viability in hOSE cells, HO-3867 had no significant effect. Our results indicate that HO-3867 selectively drives ovarian cancer cells with a BRCA1-mutation to undergo apoptosis while sparing non-cancerous cells. Rational drug combinations that target matching specific and vital targets may kill cancer cells selectively.Citation22
In this study we also investigated the role of STAT3 in BRCA1-mutated ovarian cancer cells and the effect of HO-3867 on STAT3 activation and related proteins. STAT3 has a wide variety of biological functions, including acceleration of cell proliferation and activation of anti-apoptotic proteins such as Bcl-XL, Bcl-2, Mcl-1, c-Myc and Survivin. STAT3 is constitutively activated in a variety of tumor cell types including breast and ovarian cancer. Citation23-Citation26 The constitutively active STAT3 has been implicated in the induction of resistance to apoptosis, possibly through the expression of anti-apoptotic proteins and has recently become a proposed target for cancer therapy.Citation27,Citation28 This study is the first to show that, pTyr705 is highly expressed in BRCA1-mutated ovarian cancer cells compared with somatic ovarian cancer cells. In addition, we have shown that expression of pTyr705 is necessary for BRCA1 cancer cell proliferation and survival. Further studies will be required to fully understand the mechanistic role of STAT3 expression in BRCA1 ovarian cancer cells and its role in regulation of cell proliferation and survival.
Recently, STAT3 has attracted much attention as a pharmacologic target. Several reports have demonstrated that inhibition of STAT3 by flavonoids and synthetic compounds results in cancer cell apoptosis both in vitro and in vivo.Citation29,Citation30 HO-3867 inhibits STAT3 and STAT3-regulated anti-apoptotic genes such as Bcl-2 and Bcl-XL, contributing to decreased cancer cell survival via induction of apoptosis. Previous studies have shown that members of the Bcl-2 family play a key role in regulating caspase activation during apoptosisCitation31 and that Bcl-2 and Bcl-xL are negative regulators of caspase activation.Citation32,Citation33
We found that the levels of both Bcl-2 and Bcl-xL were decreased with a corresponding increase in cleaved caspase-3 and -7 in BRCA1-mutated cancer cells treated with HO-3867. In addition we confirmed that overexpression of STAT3 led to resistance to HO-3867, suggesting that STAT3 was a critical target of HO-3867 in BRCA1 ovarian cancer cells.
Results from this pilot study indicate that there is a role for STAT3 in BRCA1-mutated ovarian cancer cell survival and proliferation. A decrease in cell survival was also seen in BRCA1-mutated breast cancer cells, but the exact mechanism still needs to be elucidated. STAT3 inhibitors HO-3867 and STATTIC significantly suppress cancer cells through the induction of ROS and inhibition of STAT3. An additional benefit of HO-3867 is the selective induction of ROS in cancer cells without damage to non-cancerous cells, which provides a considerable advantage as a selective chemotherapeutic agent for BRCA1-mutation carriers. Future studies must be done in vivo with mice bearing BRCA1 mutations. Results from these studies will further validate our current findings and help decipher the role of STAT3 inhibitors at the clinical level.
Materials and Methods
Materials
HO-3867 was synthesized in the laboratory. Stock solutions of the compounds were freshly prepared in dimethylsulfoxide (DMSO). Cell-culture medium (RPMI 1640 and DMEM), fetal bovine serum (FBS), antibiotics, sodium pyruvate, trypsin, non-essential amino acids and phosphate-buffered saline (PBS) were purchased from Gibco. Polyvinylidene fluoride (PVDF) membrane and molecular-weight markers were obtained from Bio-Rad. Antibodies against caspase 3, cleaved caspase 3, caspase 7, cleaved caspase 7, PARP, cleaved PARP, pSTAT1, pSTAT3 Tyr705, pSTAT3 Ser727, pSTAT5, TYK, pJak1 and pAKT were purchased from Cell Signaling Technology. Antibodies specific for STAT3, JAK2, SOCS1, Actin, cyclin D1, p21, Bcl-xL and Bcl-2 were purchased from Santa Cruz Biotechnology. Enhanced chemiluminescence (ECL) reagents were obtained from Amersham Pharmacia Biotech (GE Healthcare). All other reagents, of analytical grade or higher, were purchased from Sigma-Aldrich.
Cell lines and cultures
A2780, A2780R, SKOV3, hOSE, SUM-149 and BRCA1-mutated human epithelial ovarian cancer cell lines were used in this study. The cells were grown in RPMI 1640, DMEM, F-12 Hams, or MEBM/RPMI medium supplemented with 5 or 10% FBS, 2% sodium pyruvate, 1% penicillin and 1% streptomycin with or without insulin. Cells were grown in a 75 mm flask to 70% confluence at 37°C in an atmosphere of 5% CO2 and 95% air. Cells were routinely trypsinized (0.05% trypsin/EDTA) and counted using an automated counter (NucleoCounter, New Brunswick Scientific).
Cell survival by clonogenic assay
Cell survival was assessed by clonogenic assay. Cells at ~80% confluence were trypsinized, rinsed, seeded onto 60-mm dishes (5 × 104 cells per dish), grown for 24 h at 37°C and treated afterward with increasing doses of HO-3867 (1 μM, 5 μM, 10 μM, 20 μM) for 24 h. Cells treated with equal amounts of DMSO alone served as controls. After treatment, the cells were washed twice with PBS, trypsinized, counted and plated in 60-mm dishes in triplicate and incubated for an additional 7 d. The colonies were then stained with crystal violet (in ethyl alcohol) and counted using an automated colony counter (ColCount, Oxford Optronix). Each experiment was repeated at least five times.
Cell proliferation
BRCA1-mutated ovarian cancer cells were cultured in MEBM/RPMI medium. They were seeded into 60mm culture dishes and cultured for 24 h. After 24 h the cells were treated with varying concentrations (1 μM, 5 μM, 10 μM, 20 μM) of HO-3867 and counted using a NucleoCounter (New Brunswick Scientific) at 12, 24, or 48 h of treatment.
Cell viability by MTT assay
Cell viability was determined by a colorimetric assay using MTT. In the mitochondria of living cells, yellow MTT undergoes a reductive conversion to formazan, giving a purple color. A2780, A2780R, SKOV3 and BRCA1-mutated ovarian cancer cells were grown to 80% confluence in 75 mm flasks, trypsinized, counted and seeded in 96-well plates with an average population of 7,000 cells/well. The cells were then incubated overnight and then treated with either STATTIC 25 μM or HO-3867 10 μM for 24 h. Cell viability was then calculated. Similarly, BRCA1-mutated cells were treated with curcumin 100 μM, cisplatin 10 μg/mL, ABT, STATTIC 25 μM or HO-3867 10 μM for 24 h and cell viability analyzed. The dose and time of incubation were determined from a set of preliminary experiments. All experiments were done using six replicates and repeated at least three times. Cell viability was expressed as a percentage of MTT viability of untreated cells.
Western blotting
A2780, SKOV3 and BRCA1-mutated ovarian cancer cells were incubated in their respective media. Cell lysates were prepared in non-denaturing lysis buffer (10 mM TRIS-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 0.3 mM phenylmethylsulfonyl fluoride, 0.2 mM sodium orthovanadate, 0.5% NP40, aprotinin (1 μg/ml) and leupetin. Cell lysates were centrifuged at 12,000 rpm for 20 min at 4°C and the supernatant was separated. The protein concentration in the lysates was determined using a Pierce detergent-compatible protein assay kit. For western blotting, 25 to 50 μg of protein lysate per sample was denatured in 2 × sample buffer and subjected to SDS-PAGE on a 10% or 12% tris-glycine gel. The separated proteins were transferred to a PVDF membrane and then blocked with 5% nonfat milk powder (w/v) in TBST (10 mM Tris, 100 mM NaCl, 0.1% Tween 20) for 1 h at room temperature or overnight at 4°C. The membranes were incubated with the primary antibodies described above. The bound antibodies were detected with horseradish peroxidase (HRP)-labeled sheep anti-mouse IgG or HRP-labeled donkey anti-rabbit IgG (Amersham) using an enhanced chemiluminescence detection system (ECL Advanced kit). Protein expressions were determined using Image Gauge version 3.45. The protocol was the same for cells treated with DMSO (control) or HO-3867 10 μM for 24 h. Equal volumes of DMSO (0.1% v/v) were present in both groups.
Apoptosis/flow cytometry
BRCA1-mutated ovarian cancer cells were treated with 10 μM of HO-3867 for 24 h. They were then trypsinized, washed in PBS and labeled with propidium iodide and Alexa Fluor 488 Annexin V using the Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Invitrogen). Apoptotic cells were measured by flow cytometry on a FACS Caliber (BD Bioscience).
Measurement of intracellular ROS
The ROS levels in cells treated with HO-3867 were determined using H2DCF-DA, a membrane-permeative fluorogenic probe. The acetate and acetoxymethyl ester groups of this probe are enzymatically cleaved inside living cells. The probe can then be oxidized by intracellular oxidants to give a product, DCF, which emits a strong, green fluorescence (λex = 504 nm; λem = 529 nm). The fluorescence intensity is proportional to the level of cellular oxidants. Cells, grown to 80% confluence on 6-mm glass coverslips, were treated with HO-3867 for 24 h followed by incubation with H2DCF-DA. The cells were further incubated in the dark for 20 min and washed with protein-free medium and then fluorescence images were immediately captured with a Nikon Eclipse TE2000-U camera system using excitation/emission at 495/520 nm. The captured images were then analyzed using MetaMorph image analysis software.
Sub G1 analysis
BRCA1-mutated and hOSE cells were treated with 10uM HO-3867 for 24 h. Cells were then trypsinized, collected by centrifugation, resuspended in PBS and fixed in 70% ethanol at -20°C overnight. After centrifugation, the cells were then washed in PBS and resuspended in potassium iodide (PI)-staining solution (PBS, PI, RNase) (Boehringer Mannheim Antisense bcl-xl 546 and Chemotherapy Co.). Specimens were incubated in the dark for 30 min at 37°C and then analyzed with the use of an EPICS Profile II flow cytometer (Coulter Corp.). All experiments were performed in triplicate.
Transfection of Wild-type STAT3 cDNA
The STAT3 overexpression experiments were performed using a wild-type STAT3 cDNA. The FLAG-tagged gene was transfected into BRCA1-mutated ovarian cancer cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. At 24 h after the transfection of the STAT3 gene, HO-3867 (10 μm) was added and incubated for 24 h. The cells were then subjected to a MTT viability assay.
Statistical analysis
Results were expressed as mean ± SE. Comparisons between groups were made by a Student’s t- test. The significance level was set at p ≤ 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was funded by the Ovarian Cancer Research Foundation (OCRF) grant and the Hungarian National Research fund OTKA K81123 (to K.H.)
Related Research Data
References
- Daly MB, Axilbund JE, Buys S, Crawford B, Farrell CD, Friedman S, et al, National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw 2010; 8:562 - 94; PMID: 20495085
- Fischer C, Engel C, Sutter C, Zachariae S, Schmutzler R, Meindl A, et al, on behalf of the German Consortium for Hereditary Breast and Ovarian Cancer. BRCA1/2 testing: uptake, phenocopies and strategies to improve detection rates in initially negative families. Clin Genet 2011; http://dx.doi.org/10.1111/j.1399-0004.2011.01788.x; PMID: 21919902
- Lane TF. BRCA1 and transcription. Cancer Biol Ther 2004; 3:528 - 33; http://dx.doi.org/10.4161/cbt.3.6.843; PMID: 15254397
- Schorge JO, Modesitt SC, Coleman RL, Cohn DE, Kauff ND, Duska LR, et al. SGO White Paper on ovarian cancer: etiology, screening and surveillance. Gynecol Oncol 2010; 119:7 - 17; http://dx.doi.org/10.1016/j.ygyno.2010.06.003; PMID: 20692025
- Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117 - 30; http://dx.doi.org/10.1086/375033; PMID: 12677558
- Chen S, Iversen ES, Friebel T, Finkelstein D, Weber BL, Eisen A, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006; 24:863 - 71; http://dx.doi.org/10.1200/JCO.2005.03.6772; PMID: 16484695
- Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol 2008; 26:20 - 5; http://dx.doi.org/10.1200/JCO.2007.11.6905; PMID: 18165636
- Skytte AB, Waldstrøm M, Rasmussen AA, Crüger D, Woodward ER, Kølvraa S. Identification of BRCA1-deficient ovarian cancers. Acta Obstet Gynecol Scand 2011; 90:593 - 9; http://dx.doi.org/10.1111/j.1600-0412.2011.01121.x; PMID: 21371001
- Boyd J, Sonoda Y, Federici MG, Bogomolniy F, Rhei E, Maresco DL, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA 2000; 283:2260 - 5; http://dx.doi.org/10.1001/jama.283.17.2260; PMID: 10807385
- Rubin SC, Benjamin I, Behbakht K, Takahashi H, Morgan MA, LiVolsi VA, et al. Clinical and pathological features of ovarian cancer in women with germ-line mutations of BRCA1. N Engl J Med 1996; 335:1413 - 6; http://dx.doi.org/10.1056/NEJM199611073351901; PMID: 8875917
- Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther 2006; 5:952 - 61; http://dx.doi.org/10.1158/1535-7163.MCT-05-0493; PMID: 16648566
- Banerjee S, Kaye S. PARP inhibitors in BRCA gene-mutated ovarian cancer and beyond. Curr Oncol Rep 2011; 13:442 - 9; http://dx.doi.org/10.1007/s11912-011-0193-9; PMID: 21913063
- Scully R, Xie A, Nagaraju G. Molecular functions of BRCA1 in the DNA damage response. Cancer Biol Ther 2004; 3:521 - 7; http://dx.doi.org/10.4161/cbt.3.6.842; PMID: 15280660
- Tassone P, Di Martino MT, Ventura M, Pietragalla A, Cucinotto I, Calimeri T, et al. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol Ther 2009; 8:648 - 53; http://dx.doi.org/10.4161/cbt.8.7.7968; PMID: 19333003
- Dayton A, Selvendiran K, Kuppusamy ML, Rivera BK, Meduru S, Kálai T, et al. Cellular uptake, retention and bioabsorption of HO-3867, a fluorinated curcumin analog with potential antitumor properties. Cancer Biol Ther 2010; 10:1027 - 32; http://dx.doi.org/10.4161/cbt.10.10.13250; PMID: 20798598
- Selvendiran K, Ahmed S, Dayton A, Kuppusamy ML, Tazi M, Bratasz A, et al. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: differential cytotoxicity in healthy and cancer cells. Free Radic Biol Med 2010; 48:1228 - 35; http://dx.doi.org/10.1016/j.freeradbiomed.2010.02.009; PMID: 20156552
- Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, et al. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem 2007; 282:28609 - 18; http://dx.doi.org/10.1074/jbc.M703796200; PMID: 17684018
- Selvendiran K, Ahmed S, Dayton A, Ravi Y, Kuppusamy ML, Bratasz A, et al. HO-3867, a synthetic compound, inhibits the migration and invasion of ovarian carcinoma cells through downregulation of fatty acid synthase and focal adhesion kinase. Mol Cancer Res 2010; 8:1188 - 97; http://dx.doi.org/10.1158/1541-7786.MCR-10-0201; PMID: 20713491
- Selvendiran K, Tong L, Bratasz A, Kuppusamy ML, Ahmed S, Ravi Y, et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol Cancer Ther 2010; 9:1169 - 79; http://dx.doi.org/10.1158/1535-7163.MCT-09-1207; PMID: 20442315
- Cook JA, Gius D, Wink DA, Krishna MC, Russo A, Mitchell JB. Oxidative stress, redox and the tumor microenvironment. Semin Radiat Oncol 2004; 14:259 - 66; http://dx.doi.org/10.1016/j.semradonc.2004.04.001; PMID: 15254869
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat Rev Drug Discov 2009; 8:579 - 91; http://dx.doi.org/10.1038/nrd2803; PMID: 19478820
- Blagosklonny MV. Prospective strategies to enforce selectively cell death in cancer cells. Oncogene 2004; 23:2967 - 75; http://dx.doi.org/10.1038/sj.onc.1207520; PMID: 15077157
- Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 2004; 4:97 - 105; http://dx.doi.org/10.1038/nrc1275; PMID: 14964307
- Huang M, Page C, Reynolds RK, Lin J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecol Oncol 2000; 79:67 - 73; http://dx.doi.org/10.1006/gyno.2000.5931; PMID: 11006034
- Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res 2006; 12:5055 - 63; http://dx.doi.org/10.1158/1078-0432.CCR-06-0861; PMID: 16951221
- Li L, Gao Y, Zhang LL, He DL. Concomitant activation of the JAK/STAT3 and ERK1/2 signaling is involved in leptin-mediated proliferation of renal cell carcinoma Caki-2 cells. Cancer Biol Ther 2008; 7:1787 - 92; http://dx.doi.org/10.4161/cbt.7.11.6837; PMID: 18787400
- Gest C, Mirshahi P, Li H, Pritchard LL, Joimel U, Blot E, et al. Ovarian cancer: Stat3, RhoA and IGF-IR as therapeutic targets. Cancer Lett 2011; PMID: 22120672
- Gao B, Shen X, Kunos G, Meng Q, Goldberg ID, Rosen EM, et al. Constitutive activation of JAK-STAT3 signaling by BRCA1 in human prostate cancer cells. FEBS Lett 2001; 488:179 - 84; http://dx.doi.org/10.1016/S0014-5793(00)02430-3; PMID: 11163768
- Selvendiran K, Koga H, Ueno T, Yoshida T, Maeyama M, Torimura T, et al. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: an implication for the antitumor potential of flavonoids. Cancer Res 2006; 66:4826 - 34; http://dx.doi.org/10.1158/0008-5472.CAN-05-4062; PMID: 16651438
- Selvendiran K, Ahmed S, Dayton A, Kuppusamy ML, Rivera BK, Kálai T, et al. HO-3867, a curcumin analog, sensitizes cisplatin-resistant ovarian carcinoma, leading to therapeutic synergy through STAT3 inhibition. Cancer Biol Ther 2011; 12:837 - 45; http://dx.doi.org/10.4161/cbt.12.9.17713; PMID: 21885917
- Huang Z. Bcl-2 family proteins as targets for anticancer drug design. Oncogene 2000; 19:6627 - 31; http://dx.doi.org/10.1038/sj.onc.1204087; PMID: 11426648
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 2003; 15:691 - 9; http://dx.doi.org/10.1016/j.ceb.2003.10.004; PMID: 14644193
- Su SJ, Chow NH, Kung ML, Hung TC, Chang KL. Effects of soy isoflavones on apoptosis induction and G2-M arrest in human hepatoma cells involvement of caspase-3 activation, Bcl-2 and Bcl-XL downregulation and Cdc2 kinase activity. Nutr Cancer 2003; 45:113 - 23; http://dx.doi.org/10.1207/S15327914NC4501_13; PMID: 12791511