Abstract
Stress treatment generally causes the post-translational modification and accumulation of the p53 protein, although the role of these aspects has not been always understood in relation to this protein’s tumor suppressor activity. We analyzed these attributes of p53 in eight different breast cancer cell lines, with either wild-type or mutant p53 protein, in response to oxidative stress. We found that the wild-type p53 protein from MCF-7 and ZR-75-1 cells binds with different affinity to 12 gene sequences covering several pathways regulated by p53. Treatment of MCF-7 cells with H2O2 caused an increase in this binding affinity while this same treatment of ZR-75-1 cells caused the p53 protein to lose binding affinity to several genes. The mutant p53 proteins from all cell lines had minimal to weak binding to these sequences even after treatment with H2O2. The p53 protein from the ZR-75-1 cells and three cell lines with mutant p53 showed serine 15 phosphorylated protein, but we found no correlation between that modification and the levels or localization of this protein although DNA binding affinity of wild-type protein might be affected by this modification. From this and other work, it appears that the mutation status of the TP53 gene alone cannot predict the activity of this tumor suppressor since cell lines with the same genetic information do not show the same properties of this protein.
Introduction
p53 has been called the ‘Guardian of the Genome’Citation1 owing to its contribution in maintaining the integrity of cells mainly by acting as a transcriptional regulator of a wide variety of target genes. The genes controlled by p53 perform essential functions in cell cycle regulation, apoptosis, DNA repair, cell proliferation, and angiogenesis.Citation2 There are low levels of p53 in normal cells and tissues, but these levels increase following a variety of treatments, primarily due to the lack of degradation of p53 mediated by Mdm-2.Citation3 The control of p53-regulated genes is also facilitated by post-translational modifications and the subcellular localization of the tumor suppressor.Citation4,Citation5 Regulation of these genes serves to activate checkpoints of the cell cycle to prevent inheritance of genomic instability and related anomalies in daughter cells. The stabilization and activation of wild-type p53 in response to stress have been associated with post-translational modifications on key residues of the tumor suppressor protein. DNA damage has been shown to induce phosphorylation of nearly 20 serine and threonine residues in the p53 protein, some of which are known to affect p53 accumulation, tetramerization and sequence specific DNA binding.Citation6 Acetylation of lysine residues in the DNA binding and C-terminal domains of p53 is known to stabilize the protein and augment sequence specific DNA binding following DNA damage.Citation7,Citation8
Cancer cells bypass the cell cycle checkpoints and evade the normal regulation by tumor suppressors. The TP53 gene is mutated in 50% of human cancers, and over 90% of these mutations occur in the DNA binding domain, potentially altering the ability of this protein to bind DNA.Citation8 A number of investigations have been done to study the correlation between p53 alteration and clinical outcome in breast cancer.Citation10 Mutations in the TP53 gene are found in only 20% of human primary breast cancers, but these mutations are linked with aggressive variants of this disease.Citation11 Mutant p53 may exert its effect in a couple of different ways. The mutant protein may aggregate with wild-type protein in cells containing both forms of the tumor suppressor preventing this protein from acting as a transcription factor.Citation12 Alternatively, the mutant p53 may actually provide a gain-of-function by regulating new genes or altering the regulation of genes normally controlled by the wild-type protein. This latter aspect may be reflected in differential DNA binding by mutant p53 proteins isolated from cancer cells. There is already evidence for this from our previous workCitation13,Citation14 as well as that of other.Citation15 Some mutant forms of p53 can be post-translationally modified which may result in accumulation of mutant p53 in the nucleus where it can act as oncogene.Citation16 Increasing evidence suggests that post-translational modification of mutant p53 contributes to tumorigenesis.Citation17,Citation18
Since 80% of the breast tumors express wild-type p53 protein, other mechanisms for compromising wild-type protein functions have been suggested. These include cytoplasmic retention of p53, inactivation of co-activators of wild-type p53 or inactivation of downstream targets of p53. Cytoplasmic sequestration of wild-type p53 in undifferentiated neuroblastoma, colon carcinoma and breast cancer cells have been observed.Citation19,Citation20 BRCA1 associates with the C-terminus of wild-type p53 protein to mediate transcription from p53 regulated genes whereas BRCA1 mutations in tumors compromise this co-activator function.Citation21 Studies have shown that σ, one of the direct targets of p53 that mediates G2 checkpoint maintenance, undergoes inappropriate silencing in several primary breast carcinomas.Citation22 Post-translational regulation of p53 may also be altered.
To understand the importance of p53 in breast cancer, we investigated the DNA binding affinity and post-translational modification of this tumor suppressor in several breast cancer cell lines and tried to correlate that to the mutation status of the TP53 gene and the sub-cellular localization of the protein. The effect of oxidative stress on alterations in p53 levels and localization was also investigated in 5 of the cell lines. We found that mutant p53 proteins accumulated in breast cancer cell lines, predominately in the nucleus but showed minimal binding to all of the DNA sequences tested. Wild-type p53 in 2 breast cancer cell lines bound to different sequences with unique specificities. When one specific post-translational modification was tested, we found that serine 15 phosphorylation may correlate with changes in wild-type p53 DNA binding, but may not affect the location and accumulation of this tumor suppressor.
Materials and Methods
Cell culture and treatment
All the breast cancer cell lines selected for this study () were obtained from American Type Culture Collection (ATCC) (HTB-22, HCC2157, HCC70, HCC1395, SK-BR-3, MDA-MB-468) or from Dr. Gokul Das (T-47D and ZR-75–1) (Roswell Park Cancer Institute, Buffalo, NY). The cells were maintained in Dulbecco’s Minimal Essential Medium (ATCC, 30–2002) with appropriate supplements of 10% fetal bovine serum (Thermofisher Scientific, 30910.02), penicillin/streptomycin (Lonza, 17–602) and insulin (Sigma, I6634) at 37°C in the presence of 5% CO2.
Table 1. Breast cancer cell lines being used for this study
Nuclear and cytoplasmic extract preparation
The cells were grown to 80–90% confluency and cytoplasmic and nuclear proteins were harvested following a protocol by Jagleská and colleaguesCitation23 from ‘untreated’ cells or cells treated with 0.2 mM freshly prepared H2O2 (J.T Baker Inc., 2186–01) for three hours. Total protein concentration for both the nuclear and cytoplasmic fractions was determined using the bicinchoninic protein assay procedure (Sigma, B9643) using BSA as a standard. The p53 concentration for both fractions was determined using the Pantropic p53 ELISA Kit from Calbiochem-EMD (QIA107) or the Roche p53 ELISA kit (Roche Diagnostics, 11828789001).
Streptavidin magnetic bead assay
This assay was performed as described previouslyCitation13 utilizing 50pg (as estimated from ELISA) of wild-type or mutant p53 from the nuclear extract from the cell lines and 20 pmoles of the biotinylated p53 gene regulatory sequences (custom synthesis by Integrated DNA Technologies) (). Following each binding reaction, the ‘unbound’ and ‘bound’ fractions were obtained using streptavidin magnetic beads (Roche, 11641778001), separated by SDS-PAGE (precast gels from Bio-Rad, 456–1033) along with a pre-bound sample and then transferred onto a PVDF (Millipore, IPVH000–10) membrane for western blot analysis.
Table 2. Gene sequences used to analyze p53 DNA binding categorized according to regulatory pathways
Western blots
The PVDF membranes following transfer were blocked overnight with 5% BSA in 1xPBST (10mM Phosphate pH-8, 0.9% NaCl, 1% Tween 20). The membrane was subsequently probed with primary antibody at room temperature using either DO-7 for p53 (Calbiochem-EMD, OP140) (at a dilution of 1:2000 for 1.5 h) or Phosphodetect Ser 15 for ser15 phosphorylated p53 (Calbiochem-EMD, PC386) (at a dilution of 1:1000 for 2.5 h). This was followed by washes and secondary antibody (1:10,000 diluted), either anti-mouse horseradish peroxidase (HRP) (for DO-7) (Cell Signaling, 7076S) or anti-rabbit HRP (for Phosphodetect Ser 15) (Cell Signaling, 7074) and incubation of the membrane at room temperature for 50 min. The signal was detected using a chemiluminiscent substrate (Supersignal West Pico, Pierce, 34080) with a chemiluminescent detector camera system (Bio-Rad, Chemidoc XRS system). The signal intensities were quantitated with the Quantity One software (Bio-Rad 70–9600-Q1–465PC). To determine the percentage of p53 bound to a particular gene regulatory sequence, the intensities in the ‘bound’ and ‘unbound’ fractions were added, and used to determine the percentage in each fraction. The level of change in serine 15 phosphorylated p53 was quantitated in the same way and compared with the density of the total p53 detected on a blot using the DO-7 antibody.
Electrophoretic mobility shift assay
An aliquot of nuclear p53 (50pg, as estimated from ELISA) was allowed to react with 20 pmoles of biotinylated double stranded DNA sequence (custom synthesis by Integrated DNA Technologies) as described.Citation13 The reaction mixture was separated on a 5% TBE (Tris borate EDTA) polyacrylamide gel (BioRad; 456–5013) and transferred to a nylon membrane. The membrane was subsequently blocked in 5% BSA in 1X PBST. The membrane was incubated with alkaline phosphatase conjugated streptavidin at a dilution of 1:10,000 (Cell Signaling; 7055). The membrane was subsequently developed colorimetrically with the BCIP (5-bromo-4-chloro-3-indolyl-phosphate)/NBT (nitro blue tetrazolium) substrate (Kirkegaard and Perry Laboratories, Inc., 50–81–18).
Results
DNA binding of wild-type p53 From MCF-7 and ZR-75–1 cells
The tumor suppressor function of the p53 protein is exerted by its sequence specific DNA binding activity. Thus, we analyzed the affinity of the wild-type p53 protein from 2 different breast cancer cell lines to 12 p53 gene regulatory sequences utilizing a streptavidin magnetic bead assay (see for genes and sequences used).Citation13 The wild-type p53 from untreated MCF-7 cells bound with moderate affinity (~30–60% of bound p53) to the regulatory sequences from the fas, p21, cyclin G, pcna and survivin genes, with weak affinity to the regulatory sequences from genes such as bax, cdc25C, noxa and puma and with minimal affinity to sequences from genes like kai1, mdm2 and mdr1 ( and ; Table S1). The wild-type p53 protein from the nuclear extract of ZR-75–1 cells had a different sequence specificity as it bound to the regulatory sequences from the pcna, cyclin G, p21, survivin, cdc25C, bax and noxa genes with moderate affinity, from the fas gene with high affinity (greater than 60% of p53 bound) and to the regulatory sequence from the puma and mdm-2 genes with weak or minimal affinity ( and ; Table S1). The p53 from nuclear extracts of stressed MCF-7 cells (treated with H2O2) displayed a significant increase in binding affinity to some of the gene regulatory sequences used in this study such as those from the bax, cdc25C, fas, mdm-2, noxa, p21 and survivin genes ( and ; also noted in Ray et al.Citation14). In contrast, wild-type p53 from H2O2 treated ZR-75–1 cells displayed a decrease in binding affinity to some of the gene regulatory sequences such as from the p21, survivin and cdc25C genes ( and ; Table S1).
Figure 1. Streptavidin DNA binding assay with wild-type p53 from two cell lines. Aliquots of nuclear extracts from either MCF-7 cells (A) or ZR-75–1 cells (B and C) with or without treatment with 0.2 mM H2O2 for 3 h (H2O2) containing 50 pg of the p53 protein were reacted with 20 pmoles of biotinylated gene regulatory sequences (marked at the bottom of each panel) and DNA binding performed as described in Materials and Methods. The unbound (U) and bound (B) fractions from each reaction along with a pre-bound control (P) were loaded on an SDS-PAGE, transferred to a PVDF membrane and probed with DO-7 to determine the percent p53 bound to each sequence.
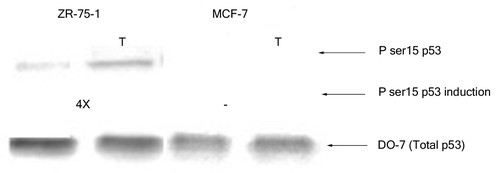
Table 3. Percent of wild-type p53 bound to p53 gene regulatory sequences
Change in serine 15 phosphorylation of wild-type p53
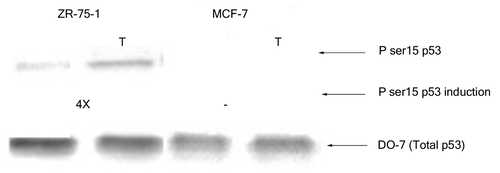
Next, we monitored the phosphorylation at serine 15 of wild-type p53 in extracts from the above mentioned breast cancer cell lines prior to and after treatment with H2O2. The p53 from nuclear extracts of untreated MCF-7 cells did not display any significant phosphorylation at serine 15 although that from ZR-75–1 cells had minimal modification at this particular residue (). Oxidative stress (3 h H2O2 treatment) did not induce any apparent change in the serine 15 phosphorylation of p53 from MCF-7 cells even when the cells were treated for up to 8 h (data not shown). On the other hand, nuclear p53 from ZR-75–1 cells displayed an increase (4X) in phosphorylation of the serine 15 residue upon exposure to H2O2 for 3 h ().
Figure 2. Effect of H2O2 stress on phosphorylation of serine 15 of wild-type p53. Nuclear extracts containing 100 pg p53 protein were loaded from each breast cancer cell line and probed with PhosphodetectTM to detect serine 15 phosphorylated wild-type p53, and subsequently with DO-7 to detect the total amount of wild-type p53 for normalization. The values shown for the increase in serine 15 phosphorylation following oxidative stress were calculated based on the relative intensities of the phospho-serine 15 band normalized for the total p53. The significance of the difference in phosphorylated serine 15 between treated and untreated for the ZR-75–1 cells was analyzed using two-tailed t-test from three independent runs of the experiment at p < 0.05.
DNA-binding and phosphorylation of mutant p53 from breast cancer cells
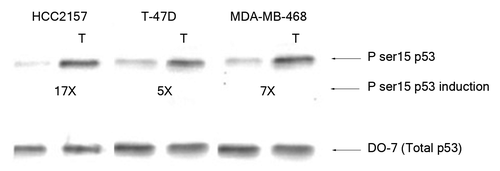
Sequence specific p53 DNA binding was also analyzed in nuclear extracts from breast cancer cells expressing different p53 hot spot mutants (). The mutant p53 proteins from breast cancer cells showed either a weak binding affinity toward selected gene regulatory sequences (the p53 from T-47D cells to the regulatory sequences from the pcna and fas genes) or minimal affinity to the other sequences tested here (; ; Table S1). The mutant p53 from the breast cancer cells HCC2157 or MDA-MB-468 cells showed no alteration in binding affinity for the gene regulatory sequences following exposure to oxidative stress (Table S1). Moreover, binding by the p53 mutant from T-47D cells to fas and pcna became negligible upon exposure to stress. The DNA binding of the p53 protein from the cytoplasmic fractions from the MCF-7, HCC2157, T-47D and MDA-MB-468 cell lines was tested with the gene regulatory sequences from the bax, noxa, p21 and survivin genes, but no binding was observed (data not shown). DNA binding was also analyzed by electrophoretic mobility shift assay and a similar difference in binding affinity of wild-type and mutant p53 to particular gene regulatory sequences was noted (Fig. S1). We observed a basal level of serine 15 phosphorylated mutant p53 in the HCC2157, T-47D and MDA-MB-468 breast cancer cells (). Oxidative stress in cells encoding mutant p53 was observed to cause a significant increase (~17X) in the serine 15 modification on p53 from the nuclear extracts from HCC2157 cells, and a more modest increase (5–7 times) in the modification of p53 from T-47D and MDA-MB-468 cells ().
Figure 3. Streptavidin DNA binding assay with mutant p53 from HCC2157 cell line. Nuclear extracts from HCC2157 containing 50 pg of the p53 protein were reacted with 20 pmoles of biotinylated gene regulatory sequence (marked at the bottom of each panel) and DNA binding was performed as described in Materials and Methods. The unbound (U) and bound (B) fractions from each reaction along with a pre-bound control (P) were loaded on an SDS-PAGE, transferred to a PVDF membrane and probed with DO-7 to determine the percent p53 bound to each sequence.
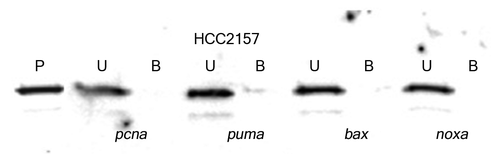
Figure 4. Effect of H2O2 stress on phosphorylation of serine 15 of mutant p53. Nuclear extracts containing 100 pg p53 protein were loaded from the HCC2157, T-47D and MDA-MB-468 breast cancer cells and probed with PhosphodetectTM to detect serine 15 phosphorylated mutant p53, and subsequently with DO-7 to detect the total amount of mutant p53 for normalization. The values shown for the increase in serine 15 phosphorylation following oxidative stress were calculated based on the relative intensities of the phospho-serine 15 band normalized for the total p53. The significance of the difference in phosphorylated serine 15 between treated and untreated for the cells was analyzed using two-tailed t-test from three independent runs of the experiment at p < 0.05.
Level and localization of the p53 protein in the breast cancer cell lines
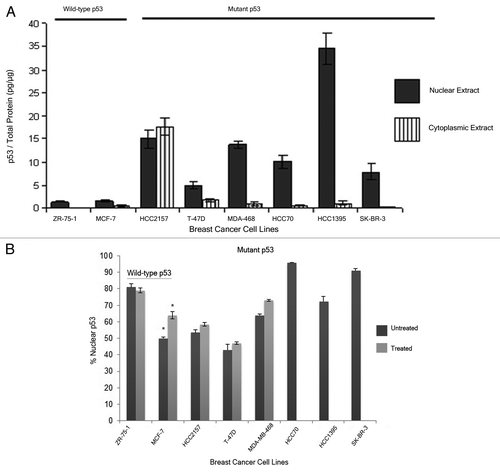
The level of p53 in the nuclear and cytoplasmic fractions of these breast cancer cell lines was analyzed using ELISA and western blot analysis (data not shown). The two methods were expected to detect both wild-type and mutant forms of the protein. It was seen that the breast cancer cell lines with wild-type p53, ZR-75–1 and MCF-7, displayed low total p53 in the cells, while the level of this protein in the breast cancer cell lines encoding mutant p53 was consistently higher (). Upon treatment with H2O2, the level of nuclear p53 in the MCF-7, ZR-75–1 and HCC2157 cells went up 4, 2 and 6 times, respectively compared with the untreated extracts, while this treatment did not change the p53 levels in T-47D and MDA-MB-468 cells significantly (; Table S2). The sub-cellular distribution of the p53 protein was estimated based on the amount of p53 found in the cytoplasmic and the nuclear fractions from the individual breast cancer cell lines from ELISA. In the breast cancer cell lines expressing the wild-type form of p53, ZR-75-1 cells retained most of the protein in the nucleus whereas MCF-7 displayed an equivalent amount of p53 in the nuclear and cytoplasmic fractions (). When these cells were exposed to oxidative stress, this resulted in a small increase in nuclear p53 in the MCF-7 cells, but did not alter the distribution of wild-type p53 in ZR-75-1. A similar analysis was done in the breast cancer cell lines expressing different mutant p53 proteins. We observed that the mutant p53 protein was mostly retained in the nucleus, except in the HCC2157 and T-47D cells. The former cells displayed an equal distribution of the tumor suppressor protein in the cytoplasm and the nucleus, while the T-47D cells had a higher percentage of p53 in the cytoplasm (). Interestingly, cells with the same mutation in the TP53 gene did not show similar accumulation or subcellular distribution of the p53 protein (compare the levels in HCC2157 and HCC70 cells or the levels in SK-BR-3 and HCC1395 cells) (; Table S2). H2O2 treatment did not alter the distribution of mutant p53 in any of the treated cell lines.
Figure 5. Analysis of p53 levels in different breast cancer cell lines. Nuclear and cytoplasmic extracts were prepared using a published methodCitation32 for each of the 8 cell lines some treated with H2O2 (Treated). Some of the cell lines carrying mutant p53 (HCC70, HCC1395 and SK-BR-3) were not treated with H2O2. The level of p53 was determined using ELISA and the total protein using bincinchoninic acid (see Materials and Methods). The distribution of p53 in the nuclear and cytoplasmic extracts of the breast cancer cell lines normalized for the total protein measured in that extract (Panel A) or the percentage of p53 in the nuclear extracts of the breast cancer cell lines normalized for the total p53 measured in the cytoplasmic and nuclear extracts (Panel B) were plotted. The error bars shown for the values are the SEM detected in duplicate samples from the same extract and from at least 2 extracts of independent passages of the cell lines. The * indicates a significant difference between treated and untreated levels at p < 0.05 as determined by two tailed t-test. Supplemental provides the actual values for the p53 in each fraction.
Discussion
Both wild-type and mutant p53 undergo a series of different post-translational modifications that have been linked to the stabilization, activation and sequence specific DNA binding of this protein in cellsCitation7,Citation24,Citation25 A challenge with understanding these changes is the fact that modifications of p53 are cell type and treatment specific. Moreover, how each modification affects p53 stability and function is widely different. The present research aims at elucidating the importance of phosphorylation of serine 15 to sequence specific DNA binding on wild-type and mutant p53 proteins from different breast cancer cell lines. This modification is linked to the activation and gain of function of this tumor suppressor.Citation26 The phosphorylation status of the serine 15 on wild-type p53 from MCF-7 and ZR-75–1 cells was significantly different. We did not detect any measurable amount of phosphorylation at this site in unstressed or stressed MCF-7 cells, but, there was an increase in phosphorylation of the serine 15 residue in the nuclear p53 obtained from the ZR-75–1 cells. A previous study has shown that wild-type p53 in ZR-75–1 does not have a phosphorylated serine 15 whereas the protein gains this modification upon exposure to certain types of carcinogenic compounds such as benzo(α) pyrene.Citation27 The protein can accumulate independent of this modification in the published study and in the one we present here. We observed a significant increase in the serine 15 phosphorylation upon induction of oxidative stress in HCC2157, T-47D and MDA-MB-468 cells harboring mutant p53. There appeared to be a basal level of serine 15 phosphorylated p53 in these three cell lines which might explain the higher levels of the p53 protein. An earlier report by our groupCitation14 has shown a basal level of serine 15 phosphorylation in the accumulated mutant p53 from ARO and WRO cell lines which does not change following oxidative stress. The presence of this modification on the mutant p53 protein from ARO and WRO cells is consistent with our present study of breast cancer cells.
Serine 15 phosphorylation has been implicated in disrupting the interaction between p53 and Mdm-2 or JNK, resulting in the stabilization of p53.Citation28,Citation29 Matsuoka and IgisuCitation30 showed that wild-type p53 in MCF-7 cells acquires serine 15 phosphorylation following treatment with cadmium, whereas treatment with wortmannin or caffeine suppresses any phosphorylation at that residue. Our present study indicates that stabilization of wild-type p53 in MCF-7 cells in response to H2O2 treatment can occur independent of serine 15 phosphorylation, but whether other post-translational modifications occur following oxidative damage in these cells remains to be shown. Although an increase in the serine 15 phosphorylation of the wild-type p53 in the ZR-75–1 cells was observed upon exposure to oxidative stress, it has not been confirmed from our studies if phosphorylation at this particular residue is necessary or sufficient for p53 stabilization. This modification may be responsible for the comparatively high cellular concentration of mutant p53 in the several breast cancer cell lines we examined.
Changes in post-translational modifications have been implicated in altering the DNA binding activity of p53. Phosphorylation of the serine 15 residue on wild-type p53 is essential for recruitment of acetylases such as CBP and p300 required for sequence specific DNA binding by the p53 protein in response to gamma irradiation.Citation31 A recent article reported that phosphorylation of serine 15 on p53 renders cells more resistant to radiation treatment, indicative of the requirement of this type of modification to endow cells with a growth advantage.Citation18 In this study, p53 null cell lines were transformed with genes producing a p53 protein with aspartic acid in place of the serines at positions 15, 20 or 46 to mimic constitutive phosphorylation at these sites. Okaichi and colleagues observed that expression of the S15D mutant lead to greater survival of the cells following radiation.Citation18 However, Ashcroft and colleaguesCitation32 showed that even in the absence of serine 15 phosphorylation (following actinomycin D treatment), transcriptional activation of p21 was observed. In another study, blockage of DNA replication by hydroxyurea resulted in serine 15 phosphorylation on p53 but did not cause increased expression of p53 target genes such as p21 in HCT116 cells containing wild-type p53.Citation33 Our work is consistent with the two latter studies as an increase in p53 binding to a number of gene regulatory sequences after MCF-7 cells were exposed to stress did not accompany detectable serine 15 phosphorylation. There is some similarity between the wild-type p53 protein from ZR-75–1 cells and the mutant p53 protein in the breast cancer cells used for this research as the presence of serine 15 modification is associated with low levels of DNA binding. The effect of phosphorylation of mutant p53 on residues known to be modified in wild-type p53 is controversial. Earlier studies on the phosphorylation of mutant p53 at serine 15 have revealed the necessity of this modification for gain-of-function of the mutant protein. Sugikawa and colleaguesCitation17 showed that inhibition of serine 15 phosphorylation restored the wild-type consensus sequence specific DNA binding function of the p53 mutant R175H. We have found that HCC2157, T-47D and MDA-MB-468 cells with mutant p53 show serine 15 phosphorylated p53, and the level of phosphorylated protein increases following treatment with H2O2. But, the protein from these extracts did not bind to any of the DNA sequences we tested (the p53 consensus sequence was not tested). We conclude that either the basal level of serine 15 phosphorylation of mutant p53 prevents it from binding to DNA or this modification has no effect on DNA binding. Other modifications of the p53 (or lack of them) in these cell lines may explain the lack of DNA binding. It has been speculated that the mutant p53 proteins may bind and regulate other genes distinct from those regulated by the wild-type protein. This so-called gain-of-function may explain the ability of mutant p53 proteins to promote tumorigenesis and progression of cancer.Citation34 We have not yet examined the binding of the mutant p53 proteins to other gene sequences than the 12 described here. This will have to be explored in the future.
Weinberg and colleaguesCitation35,Citation36 using heterologously expressed full length and truncated p53 proteins observed that wild-type p53 bound with high affinity to the regulatory sites from cell cycle arrest, DNA repair and anti-angiogenesis genes, but with a 100-fold difference in affinity to the regulatory region of some of the genes controlling apoptosis. Our observations indicate that the wild-type p53 from ZR-75-1 and MCF-7 cells showed differential affinity for gene regulatory sequences. The ZR-75-1 cells produce a p53 protein that binds with moderate affinity to most of the sequences tested, generally having higher affinity for the DNA than the p53 from MCF-7 cells. In comparing our results to that published by Weinberg and colleagues,Citation35 the p53 produced in bacteria binds with highest affinity to the gene regulatory sequences from the p21, pcna, cyclin G and noxa genes, and shows a much lower affinity to one of the gene regulatory sequences from the puma gene (called puma BS1 in Weinberg et.alCitation35). This is generally consistent with the relative affinities noted for the p53 we analyzed from the ZR-75–1 and H2O2 treated MCF-7 cells (; Table S1). Our study differed in that the p53 from the breast cancer cells had a higher affinity for the bax regulatory sequence relative to the sequence from the kai1 gene, while the bacterially produced protein had the affinities reversed. Also, the affinity of the p53 from the ZR-75-1 and untreated MCF-7 cells was low for the regulatory sequence from the mdm2 gene, while the p53 from bacteria or from H2O2 treated MCF-7 cells had generally moderate affinity for this sequence. Chromatin immunoprecipitation (ChIP) has been used extensively to view the DNA sites where transcription factors are bound to chromatin,Citation37-Citation39 although one should be cautious about using it to quantitate the level of chromatin binding since the method of detection of genes (PCR or DNA sequencing) may not be particularly quantitative for the level of protein bound. In any case, there are findings with ChIP that are consistent with our work. Two papers have shown that there is an apparent increase in the chromatin binding by p53 to the p21 regulatory sequence following gamma radiation, consistent with our work.Citation37,Citation39 ChIP analysis of p53 bound to the regulatory sequences of the mdr1 and survivin genes shows the transcription factor is associated with these sites, but the amplification of the survivin regulatory region does not change following gamma radiation.Citation38,Citation39 This is not consistent with our work, as we found an increase in DNA binding of the p53 from the MCF-7 cells to these sequences following oxidative stress. But as the methods of analysis, the stress applied and the system used (xenografts of MCF-7 cells in mice in some cases) are not the same, there are many possible explanations for this difference.
The p53 protein is known to regulate a variety of different genes in several cellular pathways (2) (). Some of the regulated genes repress these pathways while others promote them. The regulation by p53 sometimes activates transcription of a gene, while for other genes, transcription is repressed; thus, it is not trivial to build a model for how the changes in DNA binding may be manifested as changes in cellular functions. We analyzed the p53 DNA binding to the regulatory regions of 5 genes involved in controlling apoptosis in cells (bax, fas, noxa, puma and survivin). The current work and studies previously from our laboratoryCitation13,Citation14 have shown that wild-type p53 from MCF-7 cells binds with moderate affinity to the survivin gene regulatory sequence, but after exposure to oxidative stress, the protein binding increases to this and other sequences (bax, fas, noxa), but binding actually goes down with the regulatory sequence from the puma gene (; Table S1). Normally the bax, fas and noxa gene products promote cell death while the protein encoded by the survivin gene prevents apoptosis.Citation40,Citation41 Our observation that the DNA binding by wild-type p53 from MCF-7 cells to the regulatory sequences for all 4 of these genes following oxidative stress suggests all are further regulated by this tumor suppressor. On the contrary, our data indicate that p53 from ZR-75–1 cells does not show changes in binding to the pro-apoptotic gene regulatory sequences following stress but the affinity toward the regulatory region of the survivin gene goes down. As the pro-apoptotic genes are upregulated by p53 and promote apoptosis, while the survivin gene is downregulated by p53 and inhibits apoptosisCitation42 this change in DNA binding should be reflected in an increase in cell death in the MCF-7 cells and an increase in cell survival of the ZR-75–1 cells following oxidative stress. The puma gene product which promotes apoptosis is upregulated by p53,Citation41 so a reduction in binding by the tumor suppressor from MCF-7 cells following oxidative stress is not consistent with p53 promoting cell death via the other genes. The p53 protein also regulates the cell cycle and we monitored the changes in binding of the wild-type protein to 3 genes controlling this pathway, p21, cdc25C, and cyclin G. The DNA binding of p53 to the p21 and cdc25C regulatory sequences should both cause a repression of the cell cycle, as the p21 protein blocks cell cycle progression and is upregulated by p53, while the Cdc25C protein promotes the cell cycle but is downregulated by p53.Citation43 The increased affinity of wild-type p53 from MCF-7 cells for the regulatory region of these cell cycle regulatory genes following stress indicates that there is functional wild-type p53 in this cell line. Our observation that binding of wild-type p53 from nuclear extracts of ZR-75–1 cells to the regulatory regions of the p21 and cdc25C genes goes down significantly could imply a loss of cell cycle arrest and enhanced cell division through mitosis in this cells. The cyclin G gene is upregulated by p53, and the encoded protein promotes the cell cycle.Citation44,Citation45 An increased affinity of wild-type p53 from MCF-7 cells to the cyclin G regulatory region results in a distinctly different effect than that predicted for the p21 and cdc25C genes. Thus, with the cell cycle, as with the control of apoptosis, there appears to be a complicated regulation by p53 DNA binding that may be involved. Interestingly, there is no change in the DNA binding by the wild-type p53 (from MCF-7 or ZR-75–1 cells) to the regulatory sequence from the pcna gene following oxidative stress. But, this gene is known to be both up- and downregulated by p53 depending on the level of this transcription factor.Citation46,Citation47 As the wild-type p53 protein is at a relatively low concentration in the untreated MCF-7 and ZR-75–1 cells, it is expected to upregulate the pcna gene. Following oxidative stress, the p53 protein accumulates () which may cause the transcription factor to downregulate this gene in MCF-7 cells, although binding affinity of wild-type p53 from both cell types remained largely unchanged to pcna after exposure to stress. Thus, as there is already regulation of this gene based on the level of p53, it may not be necessary to alter the DNA binding by this transcription factor to the regulatory region as well.
From this and other work, it appears that the mutation status of the TP53 gene alone cannot predict the activity of this tumor suppressor since cell lines with the same genetic information do not show the same properties of this protein. The wild-type protein from the MCF-7 and ZR-75-1 cells binds well to many different DNA sequences albeit with different specificity but in one case the affinity goes up following oxidative stress and in the other, it goes down. We also observed differences in the R273H mutant p53 from two different cell lines, one of which produces p53 protein that binds to DNA sequencesCitation14and the other which does not (from this present study). Often, the genetic changes alone have been used to predict the function of this tumor suppressor or correlate with response to treatment and clinical outcome.Citation10 As well, the level and localization of the p53 protein, particularly in IHC, have been used to predict mutation status and functionality of this tumor suppressor.Citation48,Citation49 These attributes of p53 are also variable depending on the cell line as demonstrated in MCF-7 and ZR-75–1, as well as HCC2157 and HCC70 cells in our study (;B; Table S2). As we have found cells expressing the same genetic form of p53 to display different properties of the protein, measures of transcription activity or DNA binding in cells must be done in order to correlate the functionality of p53 with clinical outcome in tumors. Our study provides a platform to choose from the wide array of p53 regulated genes that should be studied in cancer cells to explore the contribution of p53 to regulation of several cellular pathways. An initial in vitro study using the streptavidin assay can prove to be a cost effective way to screen for the genes that could be studied in vivo. The streptavidin magnetic bead assay done here may be a more quantitative method to assess differences in levels of DNA binding which is not necessarily achieved with EMSA. When these studies are completed, we expect a clearer picture of how that information can be used to improve treatment of patients with breast cancer as well as other diseases of this type.
| Abbreviations: | ||
| ATCC | = | American Type Culture Collection |
| BRCA1 | = | breast cancer type 1 susceptibility protein |
| ChIP | = | chromatinimmunoprecipitation |
| ELISA | = | Enzyme linked immunosorbent assay |
| BSA | = | Bovine Serum Albumin |
| HRP | = | Horse radish peroxidase |
| IHC | = | Immunohistochemistry |
| PBST | = | phosphate buffered saline plus Tween |
| PVDF | = | Polyvinylidene fluoride |
| SDS-PAGE | = | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| TBE | = | Tris borate EDTA |
Additional material
Download Zip (218 KB)Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank Dr. Dennis McGee (Binghamton University) for the use of his tissue culture facilities for part of the work, Dr. Gokul Das (Roswell Park Cancer Institute) for providing some of the cells, Nancy Monteith for assistance with some of the tissue culture, and Matthew T. Balmer and Rebecca S. Young for helpful discussions. The work was supported by grants from the Upstate New York Translational Research Network and the Bella Fund, both to S.G., a grant from the NIH (R15 GM-093941) to support D.R., and a grant to the State University of New York at Binghamton from the Howard Hughes Medical Institute through the Precollege and Undergraduate Science Education Program to support K.R.M.
References
- Lane DP. p53, guardian of the genome. Nature 1992; 358:15 - 6; http://dx.doi.org/10.1038/358015a0; PMID: 1614522
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 12; http://dx.doi.org/10.1038/nrm2395; PMID: 18431400
- Terzian T, Suh Y-A, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 2008; 22:1337 - 44; http://dx.doi.org/10.1101/gad.1662908; PMID: 18483220
- Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004; 4:793 - 805; http://dx.doi.org/10.1038/nrc1455; PMID: 15510160
- Komarova EA, Zelnick CR, Chin D, Zeremski M, Gleiberman AS, Bacus SS, et al. Intracellular localization of p53 tumor suppressor protein in γ-irradiated cells is cell cycle regulated and determined by the nucleus. Cancer Res 1997; 57:5217 - 20; PMID: 9393737
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001; 268:2764 - 72; http://dx.doi.org/10.1046/j.1432-1327.2001.02225.x; PMID: 11358490
- Arbely E, Natan E, Brandt T, Allen MD, Veprintsev DB. Robinson, C.V.et al. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Nat Acad Sci USA 2011;108: 8251-8256.
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A 2004; 101:2259 - 64; http://dx.doi.org/10.1073/pnas.0308762101; PMID: 14982997
- Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ 2010; 17:901 - 11; http://dx.doi.org/10.1038/cdd.2010.35; PMID: 20379196
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr., Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250:1233 - 8; http://dx.doi.org/10.1126/science.1978757; PMID: 1978757
- Done SJ, Eskandarian S, Bull S, Redston M, Andrulis IL. p53 missense mutations in microdissected high-grade ductal carcinoma in situ of the breast. J Natl Cancer Inst 2001; 93:700 - 4; http://dx.doi.org/10.1093/jnci/93.9.700; PMID: 11333292
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 2011; 7:285 - 95; http://dx.doi.org/10.1038/nchembio.546; PMID: 21445056
- Chandrachud U, Gal S. Three assays show differences in binding of wild-type and mutant p53 to unique gene sequences. Technol Cancer Res Treat 2009; 8:445 - 53; PMID: 19925028
- Ray D, Balmer MT, Gal S. The functionality of p53 in thyroid cancer. [ed.] Fahey T. J. Updates in the Understanding and Management of Thyroid Cancer, Intech Publishers, Rijeka, Croatia, 2012, pp. 33-52.
- Park DJ, Nakamura H, Chumakov AM, Said JW, Miller CW, Chen DL, et al. Transactivational and DNA binding abilities of endogenous p53 in p53 mutant cell lines. Oncogene 1994; 9:1899 - 906; PMID: 8208536
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002; 2:594 - 604; http://dx.doi.org/10.1038/nrc864; PMID: 12154352
- Sugikawa E, Yazaki N, Tsunoda S, Nakanishi N, Ohashi M. Inhibition of mutant p53 phosphorylation at serine 15 or serine 315 partially restores the function of wild-type p53. Biochem Biophys Res Commun 1999; 261:256 - 63; http://dx.doi.org/10.1006/bbrc.1999.1019; PMID: 10425175
- Okaichi K, Nose K, Kotake T, Izumi N, Kudo T. Phosphorylation of p53 modifies sensitivity to ionizing radiation. Anticancer Res 2011; 31:2255 - 8; PMID: 21737649
- Wolff A, Technau A, Ihling C, Technau-Ihling K, Erber R, Bosch FX, et al. Evidence that wild-type p53 in neuroblastoma cells is in a conformation refractory to integration into the transcriptional complex. Oncogene 2001; 20:1307 - 17; http://dx.doi.org/10.1038/sj.onc.1204251; PMID: 11313875
- Ostermeyer AG, Runko E, Winkfield B, Ahn B, Moll UM. Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc Natl Acad Sci U S A 1996; 93:15190 - 4; http://dx.doi.org/10.1073/pnas.93.26.15190; PMID: 8986786
- Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, et al. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998; 16:1713 - 21; http://dx.doi.org/10.1038/sj.onc.1201932; PMID: 9582019
- Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene 2001; 20:3348 - 53; http://dx.doi.org/10.1038/sj.onc.1204438; PMID: 11423985
- Jagelská E, Brázda V, Pospisilová S, Vojtěšek B, Palecek E. New ELISA technique for analysis of p53 protein/DNA binding properties. J Immunol Methods 2002; 267:227 - 35; http://dx.doi.org/10.1016/S0022-1759(02)00182-5; PMID: 12165443
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006; 13:941 - 50; http://dx.doi.org/10.1038/sj.cdd.4401925; PMID: 16601750
- Ito T, Kaneko K, Makino R, Ito H, Konishi K, Kurahashi T, et al. Prognostic value of p53 mutations in patients with locally advanced esophageal carcinoma treated with definitive chemoradiotherapy. J Gastroenterol 2001; 36:303 - 11; http://dx.doi.org/10.1007/s005350170095; PMID: 11388392
- Zerbini LF, Wang Y, Correa RG, Cho JY, Libermann TA. Blockage of NF-kappaB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005; 4:1247 - 53; http://dx.doi.org/10.4161/cc.4.9.1966; PMID: 16082226
- Huovinen M, Loikkanen J, Myllynen P, Vähäkangas KH. Characterization of human breast cancer cell lines for the studies on p53 in chemical carcinogenesis. Toxicol In Vitro 2011; 25:1007 - 17; http://dx.doi.org/10.1016/j.tiv.2011.03.018; PMID: 21457773
- Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res 2003; 1:1001 - 8; PMID: 14707283
- Fuchs SY, Adler V, Buschmann T, Yin Z, Wu X, Jones SN, et al. JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes Dev 1998; 12:2658 - 63; http://dx.doi.org/10.1101/gad.12.17.2658; PMID: 9732264
- Matsuoka M, Igisu H. Cadmium induces phosphorylation of p53 at serine 15 in MCF-7 cells. Biochem Biophys Res Commun 2001; 282:1120 - 5; http://dx.doi.org/10.1006/bbrc.2001.4700; PMID: 11302731
- Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 1998; 273:33048 - 53; http://dx.doi.org/10.1074/jbc.273.49.33048; PMID: 9830059
- Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 2000; 20:3224 - 33; http://dx.doi.org/10.1128/MCB.20.9.3224-3233.2000; PMID: 10757806
- Gottifredi V, Shieh SY, Taya Y, Prives C. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci U S A 2001; 98:1036 - 41; http://dx.doi.org/10.1073/pnas.021282898; PMID: 11158590
- Göhler T, Jäger S, Warnecke G, Yasuda H, Kim E, Deppert W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res 2005; 33:1087 - 100; http://dx.doi.org/10.1093/nar/gki252; PMID: 15722483
- Weinberg RL, Freund SM, Veprintsev DB, Bycroft M, Fersht AR. Regulation of DNA binding of p53 by its C-terminal domain. J Mol Biol 2004; 342:801 - 11; http://dx.doi.org/10.1016/j.jmb.2004.07.042; PMID: 15342238
- Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J Mol Biol 2004; 341:1145 - 59; http://dx.doi.org/10.1016/j.jmb.2004.06.071; PMID: 15321712
- Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, et al. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J Biol Chem 2006; 281:9837 - 40; http://dx.doi.org/10.1074/jbc.C600001200; PMID: 16469747
- Liu W, Ip MM, Podgorsak MB, Das GM. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat 2009; 115:43 - 50; http://dx.doi.org/10.1007/s10549-008-0044-z; PMID: 18481172
- Sayeed A, Konduri SD, Liu W, Bansal S, Li F, Das GM. Estrogen receptor α inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res 2007; 67:7746 - 55; http://dx.doi.org/10.1158/0008-5472.CAN-06-3724; PMID: 17699779
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577 - 90; http://dx.doi.org/10.1016/S1097-2765(03)00050-9; PMID: 12667443
- Huntington ND, Labi V, Cumano A, Vieira P, Strasser A, Villunger A, et al. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim sustains B lymphopoiesis in the absence of IL-7. Int Immunol 2009; 21:715 - 25; http://dx.doi.org/10.1093/intimm/dxp043; PMID: 19454543
- Formby B, Wiley TS. Bcl-2, survivin and variant CD44 v7-v10 are downregulated and p53 is upregulated in breast cancer cells by progesterone: inhibition of cell growth and induction of apoptosis. Mol Cell Biochem 1999; 202:53 - 61; http://dx.doi.org/10.1023/A:1007081021483; PMID: 10705995
- Le Gac G, Estève P-O, Ferec C, Pradhan S. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J Biol Chem 2006; 281:24161 - 70; http://dx.doi.org/10.1074/jbc.M603724200; PMID: 16807237
- Zauberman A, Lupo A, Oren M. Identification of p53 target genes through immune selection of genomic DNA: the cyclin G gene contains two distinct p53 binding sites. Oncogene 1995; 10:2361 - 6; PMID: 7784084
- Smith ML, Kontny HU, Bortnick R, Fornace AJ Jr.. The p53-regulated cyclin G gene promotes cell growth: p53 downstream effectors cyclin G and Gadd45 exert different effects on cisplatin chemosensitivity. Exp Cell Res 1997; 230:61 - 8; http://dx.doi.org/10.1006/excr.1996.3402; PMID: 9013707
- Mercer WE, Shields MT, Lin D, Appella E, Ullrich S. J Growth suppression induced by wild-type p53 protein is accompanied by selective down-regulation of proliferating-cell nuclear antigen expression. Proceesings of the National Academy of Sciences (USA) 1996; 88:1958 - 62; http://dx.doi.org/10.1073/pnas.88.5.1958
- Morris GF, Bischoff JR, Mathews MB. Transcriptional activation of the human proliferating-cell nuclear antigen promoter by p53. Proc Natl Acad Sci U S A 1996; 93:895 - 9; http://dx.doi.org/10.1073/pnas.93.2.895; PMID: 8570655
- Bodner SM, Minna JD, Jensen SM, D’Amico D, Carbone D, Mitsudomi T, et al. Expression of mutant p53 proteins in lung cancer correlates with the class of p53 gene mutation. Oncogene 1992; 7:743 - 9; PMID: 1565469
- Allred DC, Clark GM, Elledge R, Fuqua SAW, Brown RW, Chamness GC, et al. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J Natl Cancer Inst 1993; 85:200 - 6; http://dx.doi.org/10.1093/jnci/85.3.200; PMID: 8423624