Abstract
Two peptides derived from the C1B domain of protein kinase Cγ (PKCγ) were shown to associate with classical PKC isozymes and modulate their activities. These C1B peptides are designated C1B1 (amino acid residues 101–112) and C1B5 (residues 141–151). Since PKC enzyme activity is shown to be involved in colon cancer development, the effect of C1B peptides on the growth of various human colon cancer cell lines was examined in vitro and in vivo. Sub-micromolar to micromolar levels of both C1B peptides induced approximately 60–70% growth attenuation in multiple colon cancer cell lines in a soft agar tumor colony assay; however, C1B5 peptide was not cytotoxic to normal colon epithelial cells in two dimensional culture. The effect of C1B5 peptide on colony growth of COLO205 cells was reversed by treatment with the PKCα/β inhibitor, Ro-32–0432. C1B peptide treatment attenuated COLO205 cells via two mechanisms: 1) cell cycle arrest and 2) stimulation of apoptosis. This is evident in G2 arrest and increases in levels of cleaved caspase 3 and p53 phosphorylated at serine 20. Intratumoral injection of C1B5 peptide (20 mg/kg/day, every three days) markedly attenuated the growth of subcutaneous xenografts of COLO205 cells in SCID mice by 76% compared with the control. Taken together, these results strongly suggest that C1B peptides have negligible effects on normal tissues but are potentially effective chemotherapeutic agents for colon cancer.
Introduction
In the USA, colon cancer is the second leading cause of cancer mortality. Of the estimated 102,900 new cases in 2010, 51,370 resulted in death.Citation1 Recent research indicates that the causes of colon cancers are related to non-inherited factors or environmental factors, which can cause somatic mutations of specific genes such as RB1, p16, ras or p53.Citation2 However, few targeted molecular therapies have progressed to clinical trials for the treatment of colon cancer. Therefore, it is clear that novel treatment strategies are urgently needed.
Protein kinase C (PKC) is a family of serine/threonine kinases that plays a central role in cell proliferation, differentiation, and apoptosis.Citation3-Citation5 The PKC isozymes are divided into three subfamilies based on their structure and mode of action. The classic (or conventional) PKC subfamily includes α, βI, βII and γ isozymes, which are activated by diacylglycerol (DAG) and phosphatidylserine in a calcium-dependent manner. The atypical PKCs (λ, ζ and ι) are activated by phosphatidylserine in a calcium-independent manner. The novel PKCs (δ, ε, θ and η) are activated by DAG and phosphatidylserine in a calcium-independent manner. Although PKCs have overlapping substrate specificities in vitro, they exhibit distinct patterns of tissue expression and intracellular localization. The structure of all PKCs is composed of an N-terminal regulatory region, which consists of a pseudosubstrate region and C1 and C2 domains (atypical PKCs lack the C2 domain), and a C-terminal catalytic region, which contains the C3 and C4 domains. The C1 domains in the conventional and novel PKCs are highly homologous and consist of a tandem repeat of two cysteine-rich zinc-finger motifs (C1A, C1B). These subdomains are the binding sites for the PKC activator DAG and for its functional analogs, phorbol esters, which compete with DAG for the same binding site and can activate PKC.Citation3,Citation6
Recently, Nguyen et al.Citation7 demonstrated that synthetic C1B subdomain peptides C1B1 and C1B5 bind to 14-3-3ε docking protein competitively, dissociate the docking protein and activate PKCγ. Since the C1B1 or C1B5 subdomain peptides potentially act as decoys and interact with multiple C1B domain-associated signaling proteins, they may affect cell proliferation, differentiation, and apoptosis through PKC regulation. However, the relationship between C1B subdomain peptide-dependent PKC regulation and carcinogenesis has not been rigorously studied.
The roles of the PKC family in cell growth, apoptosis, invasion, and differentiation are known to occur by transduction of mitogenic signaling pathways.Citation8 The initial study showing that PKC is activated by phorbol esters suggested that PKCs are tumor promoters.Citation3,Citation9 However, it has been reported that different PKC isozymes have distinct and sometimes opposing effects.Citation10 For example, PKCδ expression has decreased azoxymethane-induced rat colon tumors and sporadic human colon tumors.Citation11,Citation12 In addition, PKCδ activation in the human colon cancer cell line Caco-2 has been reported to induce differentiation and attenuate both anchorage-dependent and independent cell growth.Citation13 In contrast to the observations suggesting that PKCδ attenuates tumor growth, studies suggest that PKCβII promotes tumor growth. For example, PKCβII is progressively induced in azoxymethane-induced colon carcinogenesis in mice.Citation14 Recent studies using transgenic mice overexpressing PKCβΙI show hyperproliferation of the colonic epithelium and high susceptibility to carcinogen-induced colon cancer.Citation15,Citation16 These studies also revealed that PKCβII-dependent hyperproliferation of the colonic epithelium is associated with reduction of glycogen synthase kinase 3β activity and increased β-catenin expression. These results indicate that PKCβII induces colonic epithelial hyperproliferation through activation of a PKCβII/β-catenin/T-cell factor (TCF; Wnt signaling) axis.Citation15 In addition, inhibition of PKCβII-mediated hyperproliferation in the colonic epithelium by N-3 fatty acids has been shown to be associated with attenuated tumor growth.Citation16 Therefore, PKCβII is suggested to be pro-oncogenic in colon tumorigenesis.
Due to the complexity of the relationship between PKC regulation and carcinogenesis, the present study was undertaken to determine whether synthetic C1B peptides regulate growth of human colon carcinoma cells. Here, we report that synthetic C1B peptide significantly attenuates the growth of human colon carcinoma cells in vitro and in vivo. Interestingly, C1B5 peptide was found to be associated with phosphorylation of PKCα/β II, which led to the induction of apoptosis of colon carcinoma cells. Expression of PKCγ in human colon carcinoma cell lines was negligible; hence, C1B5 peptide-induced growth regulation of colon carcinoma cells seems independent from PKCγ activation.
Results
C1B peptides inhibit anchorage independent growth of colon cancer cell lines
Colony formation by COLO205 cells in soft agar was significantly inhibited by sub-micromolar to micromolar concentrations of C1B1 or C1B5 peptides (). However, colony growth attenuation by C1B5 treatment was stronger than C1B1 treatment at 0.1 µM. Since nonspecific scrambled peptide did not affect colony formation (), C1B domain peptide-dependent attenuation of colony formation is suggested to be sequence-specific. We also tested the effect of C1B5 peptide on colony growth attenuation in different colon carcinoma cell lines. As shown in , C1B5 peptide at 1μM significantly attenuated the colony formation of two other human colon carcinoma cell lines, SW620 and Caco-2. In contrast, micromolar concentrations of C1B5 peptide did not show significant effects on cell viability of either COLO205 or normal intestinal epithelial cells in standard two-dimensional (2D) cell culture ().
Figure 1. C1B peptides significantly inhibit anchorage-independent growth of human colon cancer cells. Colonies over 50µm diameter were counted by an automated colony counter as described in the Materials and Methods. COLO205 cells (2 × 104) were cultured in soft agar in the presence or absence of various concentrations of C1B5 (A) or C1B1 peptide (B) for 7 d. (C) Colony size was also significantly smaller in the C1B peptide-treated group. Original magnification is x100. (D) C1B5 peptide (1 μΜ) effectively attenuated colony growth in two other human colon cancer cell lines, SW620 and Caco-2. Data are expressed as mean ± SE. Asterisks indicate significantly different values (p < 0.05) when compared with the control.
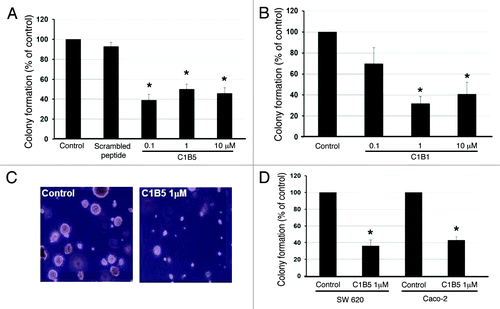
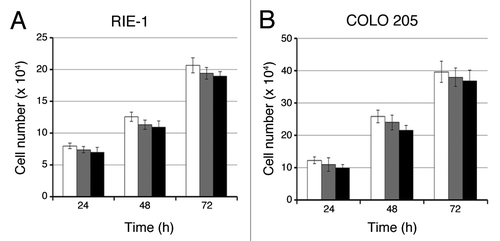
Figure 2. C1B5 peptide did not affect the growth of RIE-1 normal intestinal epithelial cells or COLO 205 cells in 2D culture. Cells were cultured in the absence (white bars) or presence of 0.1 μM (gray bars) or 1 μM (black bars) C1B5 peptide for various time periods as indicated in the figure. Dead cells were stained with trypan blue and the cell numbers were analyzed using a hemocytometer. Data are expressed as mean ± SE.
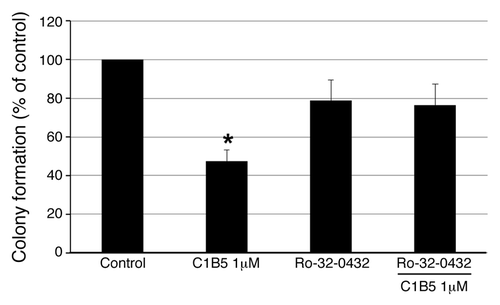
The C1B5 peptide-dependent inhibitory action was blocked by PKCα/β inhibitor
As shown in , C1B5 treatment significantly attenuated the colony growth of COLO205 cells. Treatment with the Ca2+ dependent PKCα/β isozyme inhibitor Ro-32–0432 alone did not show any effects on colony growth of COLO205 cells (). Interestingly, treatment with 1 µM Ro-32-0432 counteracted the inhibition of colony growth of COLO205 cells by C1B5 peptide (). Although the concentration of Ro-32–0432 used was high enough to inhibit other PKC isozymes, such as PKCγ and possibly PKCε,Citation17 but not various other kinases,Citation18 this result suggests that classical PKC plays an important role in colony growth attenuation by C1B5 treatment.
Figure 3. Inhibition of anchorage independent growth by C1B5 peptide is reversed by PCKα/β-specific inhibitor. As in , a soft agar assay was used and COLO205 colonies were counted with an automated colony counter. Pretreatment with the PKCα/β- inhibitor, Ro-32-0432 (1 µM), counteracted C1B5-induced colony growth attenuation. These data are representative of two independent analyses with triplicate determinations. Data are expressed as mean ± SE. An asterisk indicates significantly different value (p < 0.05) when compared with the control.
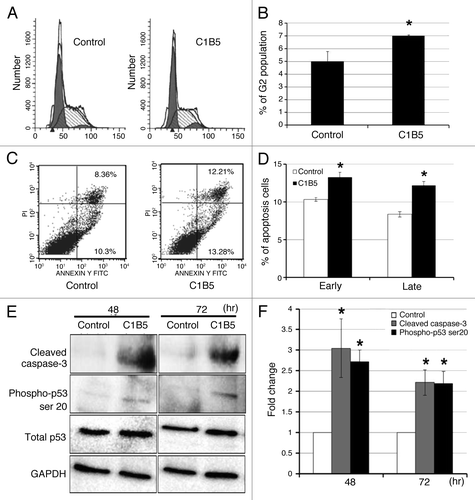
The C1B5 peptide induces G2 arrest of the cell cycle and apoptosis via phosphorylation of p53 at serine 20, which is followed by reduction of phosphorylation of PKCα/βII
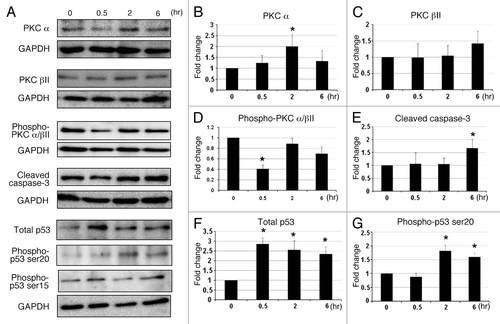
In an effort to investigate the mechanism by which C1B5 peptide treatment attenuates cell proliferation and viability of colon carcinoma cells, COLO205 cells were treated with 1 µM C1B5 peptide for 72 h and the cell cycle and apoptosis were analyzed by flow cytometry and Western blot analysis. As shown in , treatment with C1B5 significantly increased the G2 population () and the percentage of early- and late-stage apoptotic cells () as compared with the PBS treated control cells. Western blot analysis also indicated that C1B5 peptide treatment significantly increased the levels of cleaved caspase 3 and phosphorylation of p53 at serine 20 without changing total p53 levels for at least 72 h after the C1B5 treatment (). These results clearly indicate that C1B5 peptide treatment causes G2 arrest and stimulates apoptosis of COLO205 cells. Although the enzymatic activity of PKCγ is suggested to be activated by C1B5 treatment,Citation7 Western blot analysis in the present study revealed that PKCγ expression in COLO205 cells was barely detectible and was unaltered by C1B5 treatment (data not shown). In contrast, it has been shown that PKCβII plays an important role in tumorigenesis of colon cancer; the inhibition of PKCβII can cause the induction of apoptosis in glioma and colon cancer cells.Citation19
Figure 4. C1B5 peptide treatment significantly increased the G2 population (panels A and B), both early and late-stage apoptosis (panels C and D), and cleaved caspase 3 and phosphorylated p53 of COLO205 cells (panels E and F). COLO205 cells were incubated for 72 h with 1 μM C1B5 peptide. Cells were dispersed by trypsinization and incubated with either propidium iodide for cell cycle analysis or annexin V-FITC for apoptosis analysis by flow cytometry as described in Materials and Methods. These data are representative of two independent analyses with triplicate determinations. Both early apoptosis (as shown in lower right quadrant) and late apoptosis (upper right quadrant) increased significantly with the addition of C1B5 (panel D). Samples for the Western blot analysis were collected either at 48 or 72 h. Sample preparation and Western blot analysis were performed three times with duplicate determinations. The pictures in panel E represent typical blotting results. An asterisk indicates a significantly different value (p < 0.05) when compared with the control.
To better understand the mechanism by which C1B5 treatment induces apoptosis in COLO205 cells, the effect of C1B5 on the early phosphorylation of PKCα/βII and p53 at serines 15 and 20, the levels of total PKCα, total PKCβII, total p53, and cleaved caspase-3 were analyzed by Western blot. As shown in , C1B5 treatment significantly reduced phosphorylation of PKCα/βII in COLO205 without altering the expression of PKCβII at 30 min after C1B5 treatment. In contrast, the expression of PKCα was increased as compared with the control group (). The level of cleaved caspase-3 was increased by C1B5 treatment at the 6-h time point (). Phosphorylation of p53 at serine 20, but not at serine 15, was increased in a time-dependent manner with upregulation of total p53 levels (). These results indicate that C1B5 not only affects cell cycle by inducing G2 arrest but reduces phosphorylation of PKCα/βII, which may lead to apoptosis via phosphorylation of p53 at serine 20.Citation20
Figure 5. The effect of C1B5 treatment on the expression of various PKC isozymes, PKC phosphorylation, cleaved caspase-3, total p53, and phosphorylated p53 in COLO205 was analyzed by Western blot analysis. C1B5 significantly decreased levels of phosphorylated PKCα/βII after 30 min of exposure (Panels A and D). In contrast, the levels of non-phosphorylated PKCα and βwere increased by the C1B5 treatment (Panels A, B and C). The level of cleaved caspase-3 was significantly increased at 6 h (Panels A and E). The expression levels of total p53 and phosphorylated p53 at serine 20 were significantly increased after C1B5 treatment (Panels A, F, and G). Samples were prepared and subjected to Western blot analysis as described in Materials and Methods. Sample preparation and Western blot analysis were performed three times with duplicate determinations. The pictures in Panel A represent typical blotting results. Asterisks indicate significantly different values (p < 0.05) when compared with the 0 h time point.
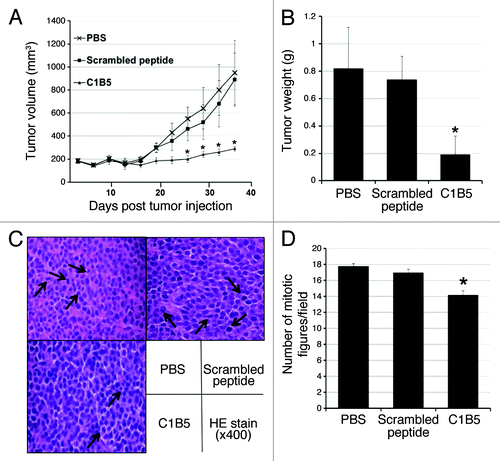
The intratumoral injection of C1B5 peptide significantly attenuated tumor growth
COLO205 cells were inoculated subcutaneously to both flanks of SCID mice (1 × 107cells per site). After allowing the tumors to grow to an average size of 150 mm3, C1B5 or a scrambled peptide solution (10 mg/ml saline at 20 mg/kg body weight) was injected into the tumors every three days, while control mice received PBS injections. While both the PBS- and the scrambled peptide-injected tumors showed rapid growth during the treatment, the tumors treated with the C1B5 peptide showed markedly smaller increases in volume (PBS, 950.7 mm3; scrambled peptide, 890.7 mm3; C1B5, 290.2 mm3 at day 36, ). The average weight of C1B5 peptide treated tumors was also significantly smaller than that of PBS- and scrambled peptide-injected tumors (). Histological examination of tumor sections shows that the C1B5 treated tumors have fewer mitotic figures than the scrambled peptide and PBS control groups (). These results clearly indicate that C1B5 peptide treatment is capable of attenuating the tumor growth of the COLO 205 cell line in vivo by attenuating cell proliferation.
Figure 6. Intratumoral injection of C1B5 peptide significantly attenuated the growth of COLO205 subcutaneous xenografts in SCID mice (n = 5, panels A and B). Histologically, the number of mitotic figures was significantly smaller in the C1B5 treated group tumors than in the scrambled peptide and PBS control group tumors (panels C and D). The H&E sections show the mitotic figures (arrows, panel C, 400× magnification). In this experiment, COLO205 cells were inoculated subcutaneously to both flanks of SCID mice at 1 × 107 cells per site. When tumors reached an average volume of 150 mm3, C1B5, scrambled peptide or PBS was injected into the tumors. The treatment was initiated at day 22 after tumor cell inoculation and repeated every 3 d for 15 d. Mice were sacrificed 3 d after the final treatment. The number of mitotic figures in 10 randomly selected fields at high power field (400x) was counted and the mean number of mitotic figures was expressed. Data are expressed as mean ± SE. Asterisks indicate significantly different values (p < 0.05) when compared with the PBS control.
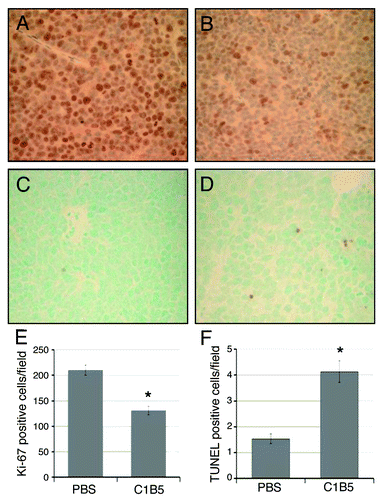
C1B5 treatment markedly attenuated cell proliferation and increased apoptosis in tumor tissues
To evaluate the effect of C1B5 treatment on cell proliferation and apoptosis in tumor cells, numbers of Ki-67 and TUNEL positive cells in tumor tissues were determined. The number of Ki-67 positive cells was significantly higher in control tumors than in C1B5-treated tumors (). The average numbers of Ki-67 positive cells in 10 randomly selected fields in PBS-treated and C1B5-treated mouse tumors were 210 ± 9.9 and 130 ± 8.5/field, respectively (p < 0.05). TUNEL positive cell numbers increased in C1B5 treated tumors (). The average numbers of TUNEL positive cells in 10 randomly selected fields in PBS-treated and C1B5-treated mouse tumors were 1.5 ± 0.2 and 4.1 ± 0.4 /field, respectively (p < 0.05). These results indicate that treatment with C1B5 attenuated cell proliferation and increased apoptosis significantly.
Figure 7. Immunohistochemical analysis of cell proliferation (panels A, B and E) and apoptosis (panels C, D, and F) in COLO205 subcutaneous xenografts in SCID mice treated intratumorally with either PBS (panels A and C) or C1B5 (panels B and D). Xenografts were collected as described in the Methods section. Cell proliferation in COLO205 xenografts was analyzed by counting the number of Ki-67 positive cells (panels A and B). Apoptosis was analyzed by counting TUNEL positive cells in the tumors (panels C and D). The average numbers of Ki-67 positive cells (E) and TUNEL positive cells (F) per high power field (40x) were determined by analyzing ten fields in each treatment group and are expressed in the bar graph. The original magnification of panels A-D was 40x. Asterisks indicate significantly different values (p < 0.05) when compared with the PBS control.
Discussion
The PKC family has been known to play important roles in cell growth, apoptosis, invasion, and differentiation.Citation8 Although PKC was considered to play a role in tumor promotion in the past,Citation3,Citation9 the role of PKC in tumorigenesis is now recognized to be very complex, with different PKC isozymes having distinct and sometimes opposing effects.Citation10,Citation21 Moreover, it has been shown that overexpression of the same PKC isozyme can lead to different biological effects depending on the character of the cell.Citation21-Citation23 For instance, HT29 or SW480 human colon carcinoma cells overexpressing PKCβI display increased doubling time, decreased saturation density, and loss of anchorage-independent growth in soft agar following exposure to phorbol ester, as well as decreased tumorigenicity in nude mice.Citation22,Citation23 In contrast, it was reported that PKCβI overexpression stimulates growth of rat embryonic fibroblasts.Citation21 However, PKCα overexpression attenuates growth of the same rat embryonic fibroblasts.Citation21 Moreover, the atypical PKC isozyme, PKCδ has been demonstrated to inhibit colon cancer cell growth and enhance differentiation and apoptosis.Citation13 Taken together, these studies suggest that while PKCs play important roles in tumorigenesis and tumor growth, their effects depend on the isozyme and cell type. Therefore, the aim of the present study was to evaluate the effect of C1B domain peptides, putative PKC activators, on the growth of human colon carcinoma cells in vivo and in vitro. The present study provides strong evidence that C1B domain peptides have therapeutic potential; they attenuate growth of colon carcinoma cells in vitro and in vivo.
In the current work, we examined the attenuation effect of C1B domain peptides, C1B1 and C1B5, on multiple human colon carcinoma cell lines. As described in the results, both peptides significantly attenuated colony growth of multiple colon carcinoma cell lines (). Comparing the growth inhibitory potential of the two peptides indicated that C1B5 peptide appears to be more potent than C1B1 peptide (). This peptide-dependent cell growth attenuation was clearly visible in 3D cell growth (colony growth in soft agar) but not in 2D cell culture (), which is a unique characteristic of this cell growth inhibition. Furthermore, C1B peptides did not affect growth of normal colon epithelial cells in 2D culture; although 3D and 2D cultures are not comparable, normal colon epithelial cells do not grow in 3D culture (). These results indicate that this C1B5 peptide may be an ideal cancer-targeted therapeutic peptide, since this peptide selectively attenuates growth of cancer cells.
In human colon cancer, PKCβII is suggested to be an important factor regulating cell growth.Citation15,Citation16 This is also supported by a study showing that a PKCβII inhibitor, enzastaurin, attenuates colon cancer cell growth by reducing expression of PKCβII.Citation24 Enzastaurin was also shown to attenuate the growth of colon cancer xenografts by inducing apoptosis.Citation19 Our findings indicate a potential mechanism by which C1B peptide attenuates cancer cell growth. In the present study, the analysis of apoptosis with flow cytometric detection of annexin V positive cells indicated that C1B5 treatment increased cell surface annexin V, an early sign of apoptosis (). The induction of apoptosis was also verified by Western blot analysis. At 48 and 72 h after C1B5 treatment, the levels of cleaved caspase-3 and phosphorylated p53 in COLO205 cells were significantly higher than those of the control (). In addition, the in vivo study showed that the number of TUNEL positive cells in the C1B5 treatment group was significantly higher than in the PBS-control group, supporting the in vitro results that C1B5 attenuated tumor growth of COLO205 through apoptosis induction.
It is well known that the tumor suppressor protein p53 plays a central role in mediating stress and DNA damage-induced growth arrest and apoptosis.Citation25 Phosphorylation of p53 at serine 20 is shown to be associated with apoptosis induction as a result of DNA damage by radiation or chemotherapy and is therefore a potential target to attenuate tumor growth.Citation20 The relationship between PKCα/βII and p53 has yet to be fully understood. However, inhibition of PKCα/βII can induce apoptosis.Citation19 Therefore, to better understand the relationship between C1B5 growth attenuation and apoptosis in COLO205 cells, the level of phosphorylation of p53 at serine 20 in COLO205 after C1B5 treatment was evaluated. Our results indicate that the phosphorylation of p53 at serine 20 was increased time dependently (). To the best of our knowledge, this is the first report that C1B5 peptide attenuates tumor cell growth by inducing apoptosis via phosphorylation of p53 at serine 20, which is followed by decreased phosphorylation and subsequent inhibition of PKCα/βII. Although this study cannot rule out a possibility that the C1B peptide also modifies PKCγ and regulates tumor cell growth, the recent discovery of the direct interaction between C1B peptide and PKCβIICitation26 strongly supports our result that C1B5 attenuated tumor growth through inhibition of phosphorylation of PKCα/βII. Negligible expression of PKCγ in COLO205 may also support the above conclusion.
Flow cytometric analysis showed that G2 arrest was induced in COLO205 cells treated with 1 µM C1B5 peptide for 72 h. This suggests that growth attenuation by C1B5 might also be associated with cell cycle arrest. Indeed, histological analysis of tumors showed significantly fewer mitotic cells in the C1B5 peptide-treated tumors than in PBS-treated tumors (). The cell cycle is known to be an important factor in tumor growth regulation. In colon cancer, Pysz et al.Citation27 revealed that transcriptional inhibition of cyclin D1 might play a role in tumor growth attenuation in association with upregulation of PKCα. PKCα, one of the classic PKC isozymes, is activated at the point of growth arrest in intestinal crypts in situ,Citation28,Citation29 and PKCα signaling promotes cell cycle withdrawal in non-transformed intestinal epithelial cells.Citation30 The tumor suppressor role of PKCα in the intestine is supported by studies using PKCα knockout mice, which showed increased proliferative activity within intestinal crypts and spontaneous intestinal adenoma formation.Citation31 The findings in the present study that C1B5 peptide treatment increased the expression of PKCα in COLO205 cells and induced G2 arrest are in strong agreement with the aforementioned reports and suggest that C1B5 peptide also affects tumor growth through upregulation of PKCα leading to G2 arrest. The G2 arrest observed in the in vitro study is supported by the in vivo immunohistochemical analysis, in which the number of Ki-67 positive cells in the C1B5 treatment group was significantly lower than in the PBS control group.
In summary, the present study demonstrates that C1B peptides significantly attenuate growth of multiple human colon carcinoma cells in three-dimensional cell culture without affecting the growth of normal intestinal epithelial cells. The C1B peptide-dependent cell growth attenuation is potentially mediated through PKCα/βII, and the mechanism is associated with phosphorylation of p53 at serine 20, and therefore with activation of intrinsic apoptosis. Treatment with C1B5 peptide significantly attenuated colon carcinoma tumor growth in a mouse xenograft study. These results clearly indicate that C1B peptides can be used as cancer-targeted therapeutics for human colon cancer.
Materials and Methods
Materials
Two C1B domain peptides, C1B1 (PKC residues 101–112; HKFRLHSYSSPT) and C1B5 (PKC residues 141–151, RCVRSVPSLCG) and a nonspecific random sequence peptide (SFGKCHLYPKV) were synthesized by the Kansas State University Biotechnology Core Facility (Kansas State University). Antibodies against PKCα, PKCβII, phospho-PKC α/βII (Thr 638 in PKCα/Thr 641 in PKC βII), and cleaved caspase-3, p53, phospho-p53 at Ser 20 were purchased from Cell Signaling Technology; and antibodies against PKCβII and GAPDH were from Santa Cruz Biotechnology. Polyclonal anti-Ki-67 antibody was from Abcam. Horseradish peroxidase-conjugated anti-rabbit IgG antibody was from GE Healthcare. Biotin-conjugated anti-rabbit IgG antibody was from Vector Laboratories. Pierce SuperSignal Western blotting substrate was from Pierce. RPMI 1640, low glucose Dulbecco's Modified Eagle Medium (DMEM) and Leibovitz's L-15 media were obtained from Mediatech, Inc. Fetal bovine serum (FBS) was from Invitrogen.
Cell lines and cell culture
Human colorectal adenocarcinoma cell lines (Caco-2, COLO 205, and SW620) and rat intestinal epithelial (RIE-1) were purchased from American Type Culture Collection. Caco-2 and COLO 205 cells were cultured with RPMI1640 medium supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin, and cultured in 5% CO2 humidified air at 37°C. SW620 cells were cultured with Leibovitz's L-15 medium supplemented with 5% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin, and cultured in 5% CO2 humidified air at 37°C. RIE-1 cells were maintained in DMEM supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin and cultured at 37°C under an atmosphere containing 5% CO2.
Effect of C1B5 on cell growth in vitro
RIE-1 or COLO205 cells were seeded at 3 × 105 cells per well in 24-well plates in 10% FBS-containing DMEM or RPMI1640, respectively. Cells were allowed to attach to culture dishes for 24 h, then C1B5 peptide (0.1 or 1 μM per well) was added to the wells. At various time points, the number of cells was counted using a hemocytometer.
Assay of anchorage-independent cell growth
The anchorage-independent growth of colon cancer cells was determined by culturing the cells in double layered soft agar. Either 2 × 104 or 5 × 104 cells in serum free Ham’s F-12 medium were mixed with 0.4% agar (total volume of 0.5 ml) and placed on top of the bottom layer of 0.8% agar (0.5 ml). Both concentrations of agar were dissolved in serum free Ham’s F-12 medium. Various concentrations of the C1B peptides in 100 µl Ham’s F-12 medium and/or 1 μM Ro-32-0432 were placed on top of the soft agar one day after starting the culture. The cells were cultured for 7 d in 5% CO2 humidified air at 37°C. Colonies with diameters over 50 μm were counted by an automated colony counter (Olympus CKX41 inverted microscope equipped with computerized motor-driven stage, ALTRA20 CMOS camera and analysis software MicroSuite FiVE).
Cell cycle analysis
To analyze the effect of C1B5 on COLO 205 cells, cell cycle analysis was performed using propidium iodide staining. In brief, 2.5 × 105 COLO205 cells were seeded in 6-well plates and left overnight. The next day, cells were treated with 1µM C1B5 and allowed to grow for 72 h. At the end of the incubation, cells were collected by trypsinization and fixed overnight in 70% pre-chilled ethanol. After collection of the cells by centrifugation, cells were incubated in PBS containing 40 µg/ml propidium iodide and 100 µg/ml RNase A for 1 h at room temperature. The fluorescence (excitation at 488 nm and emission at 585/42 nm) of 20,000 cells from each sample was measured with a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA). Data were analyzed using ModFit software and the results were displayed as histograms.
Apoptosis detection by flow cytometry
COLO205 cells were seeded and cultured for 72 h in serum-free RPMI-1640 medium with or without 1 μM C1B5 peptide. The proportion of apoptotic cells was analyzed using an Annexin V-FITC apoptosis detection kit (Biovision) with fluorescence activated cell sorting (FACS) using FACS Vantage SE flow cytometer (BD Biosciences).
Western blot analysis
Total cellular protein was prepared using RIPA buffer supplemented with protease inhibitor cocktail (Roche Diagnostics). Protein samples were separated by 10% acrylamide SDS-PAGE, electrotransferred onto nitrocellulose membrane (GE Healthcare), and blocked with 4% bovine serum albumin (BSA) in PBST (0.1% Tween20 in PBS) for 1 h at room temperature. The membranes were washed and incubated with antibodies against phospho-PKCα/βII (1:500), cleaved caspase-3 (1:1000), p53 Ser15 (1:500) and Ser20 (1:500), total p53 (1:500), PKCα (1:1000) or PKCβII (1:1000) with 0.1% BSA in PBST for 1 h at room temperature. Subsequent incubation with a horseradish peroxidase-conjugated anti-rabbit IgG antibody at a 1:2000 dilution with 0.1% BSA in PBST was done for another 1 h at room temperature. The protein expression signal was detected with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology, Rockford, IL). GAPDH was used as the loading control of sample by re-probing with an anti-GAPDH antibody at a 1:4000 dilution. The levels of the p53 phosphorylated at serine 20 was normalized by corresponding total p53 levels.
Xenografted tumor growth in SCID mice and tumor tissue collection
COLO205 human colon carcinoma cells were subcutaneously injected into 6-week-old female C.B-17 SCID mice (Charles River Laboratories, Inc.). The cancer cells (1 × 107 cells) suspended in 100 µl 0.6% agar in RPMI1640 medium were injected to the both flanks of each mouse. The tumor size was measured using a caliper every 3 d; tumor volume was estimated by the formula (length) × (width)Citation2 × 1/2. After tumor volume reached 150 mm3, C1B5 or scrambled peptide (20 mg/kg body weight) was injected intratumorally every 3 d (n = 5). Sterile PBS was injected into the control groups (n = 5). The mice were sacrificed three days after the last treatment, and the tumors were collected and weighed. All tumor tissues were fixed in 10% buffered-formalin and paraffin embedded for histologic examination. Thin sections, 4 μm thick, were stained with hematoxylin and eosin (H&E). The mean number of mitotic figures in a high power field (400×) was calculated. All experiments were performed under the approval of the Kansas State University IACUC and IBC.
Immunohistochemical determination of cell proliferation and TUNEL assay
Immunohistochemical analysis of cell proliferation in tumor tissues was performed by counting the numbers of Ki-67 positive cells in tumors. After deparaffinization of the thin sections, heat-induced epitope unmasking was performed in citrate buffer, followed by incubation with 3% H2O2/methanol for 5 min to block endogenous peroxidase activity. Sections were incubated with polyclonal anti-Ki-67 antibody at 1:100 dilution for 1 h at 37°C. Then, sections were reacted with a biotin-conjugated anti-rabbit IgG antibody (Vector Laboratories) at 1:100 dilution for 1 h at 37°C, followed by reaction with avidin–biotin peroxidase complex reagent (Vector Laboratories). Reactions were developed with 3, 3-diaminobenzodine tetrahydrochloride (Sigma) and counterstained lightly with hematoxylin.
To determine apoptosis in the tumors, the DeadEndTM colorimetric TUNEL system (Promega) was used according to the manufacturer’s protocol with slight modifications. The tissue sections were counterstained with methyl green after TUNEL staining.
The cell proliferation or apoptotic index was determined by counting either Ki-67 positive cells or TUNEL positive cells, respectively, in 10 randomly selected fields at 400x magnification. The cell proliferation or apoptotic index was expressed as the mean number of positive cells/field.
Statistical analysis
The means of the experimental groups were evaluated to confirm that they met the normality assumption. All the in vitro data were represented as mean ± standard error (SE) on graphs. Statistical analysis was performed by ANOVA and student t test as a post-hoc analysis. To evaluate the significance of overall differences in tumor size between two groups, statistical analysis was performed by ANOVA, using Fisher’s protected least significance difference (PLSD) as a post hoc test. A p-value less than 0.05 was considered significant. All the tumor size data were represented as mean ± standard error on graphs. Statistical analyses were performed by Stat View software, version 5.0.1. (JMP).
| Abbreviations: | ||
| DAG | = | diacylglycerol |
| DMEM | = | Dulbecco's Modified Eagle Medium |
| FBS | = | Fetal bovine serum |
| PKC | = | Protein kinase C |
| RIE-1 | = | rat intestinal epithelial-1 |
| TCF | = | a transcription factor in Wnt signaling |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Marla Pyle (Department of Anatomy and Physiology, Kansas State University) for critical reading and constructive comments during the preparation of the manuscript. This work was supported in part by the Kansas State University (KSU) Terry C. Johnson Center for Basic Cancer Research, KSU Provost’s Fund, KSU College of Veterinary Medicine Dean’s Fund, NIH grants P20 RR017686 and p20 RR1556, and the Kansas Bioscience Authority Collaborative Cancer Research grant.
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Fearon ER. Human cancer syndromes: clues to the origin and nature of cancer. Science 1997; 278:1043 - 50; http://dx.doi.org/10.1126/science.278.5340.1043; PMID: 9353177
- Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984; 308:693 - 8; http://dx.doi.org/10.1038/308693a0; PMID: 6232463
- Kikkawa U, Kishimoto A, Nishizuka Y. The protein kinase C family: heterogeneity and its implications. Annu Rev Biochem 1989; 58:31 - 44; http://dx.doi.org/10.1146/annurev.bi.58.070189.000335; PMID: 2549852
- Clemens MJ, Trayner I, Menaya J. The role of protein kinase C isoenzymes in the regulation of cell proliferation and differentiation. J Cell Sci 1992; 103:881 - 7; PMID: 1487501
- Blumberg PM. Protein kinase C as the receptor for the phorbol ester tumor promoters: sixth Rhoads memorial award lecture. Cancer Res 1988; 48:1 - 8; PMID: 3275491
- Nguyen TA, Takemoto LJ, Takemoto DJ. Inhibition of gap junction activity through the release of the C1B domain of protein kinase Cgamma (PKCgamma) from 14-3-3: identification of PKCgamma-binding sites. J Biol Chem 2004; 279:52714 - 25; http://dx.doi.org/10.1074/jbc.M403040200; PMID: 15459208
- Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 2007; 7:281 - 94; http://dx.doi.org/10.1038/nrc2110; PMID: 17384583
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem 1982; 257:7847 - 51; PMID: 7085651
- Mackay HJ, Twelves CJ. Protein kinase C: a target for anticancer drugs?. Endocr Relat Cancer 2003; 10:389 - 96; http://dx.doi.org/10.1677/erc.0.0100389; PMID: 14503915
- Craven PA, DeRubertis FR. Loss of protein kinase C delta isozyme immunoreactivity in human adenocarcinomas. Dig Dis Sci 1994; 39:481 - 9; http://dx.doi.org/10.1007/BF02088331; PMID: 8131683
- Wali RK, Frawley BP Jr., Hartmann S, Roy HK, Khare S, Scaglione-Sewell BA, et al. Mechanism of action of chemoprotective ursodeoxycholate in the azoxymethane model of rat colonic carcinogenesis: potential roles of protein kinase C-alpha, -beta II, and -zeta. Cancer Res 1995; 55:5257 - 64; PMID: 7585585
- Cerda SR, Bissonnette M, Scaglione-Sewell B, Lyons MR, Khare S, Mustafi R, et al. PKC-delta inhibits anchorage-dependent and -independent growth, enhances differentiation, and increases apoptosis in CaCo-2 cells. Gastroenterology 2001; 120:1700 - 12; http://dx.doi.org/10.1053/gast.2001.24843; PMID: 11375951
- Gökmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res 2001; 61:1375 - 81; PMID: 11245437
- Murray NR, Davidson LA, Chapkin RS, Clay Gustafson W, Schattenberg DGAPF, Fields AP. Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J Cell Biol 1999; 145:699 - 711; http://dx.doi.org/10.1083/jcb.145.4.699; PMID: 10330400
- Murray NR, Weems C, Chen L, Leon J, Yu W, Davidson LA, et al. Protein kinase C betaII and TGFbetaRII in omega-3 fatty acid-mediated inhibition of colon carcinogenesis. J Cell Biol 2002; 157:915 - 20; http://dx.doi.org/10.1083/jcb.200201127; PMID: 12058013
- Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J 1993; 294:335 - 7; PMID: 8373348
- Birchall AM, Bishop J, Bradshaw D, Cline A, Coffey J, Elliott LH, et al. Ro 32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J Pharmacol Exp Ther 1994; 268:922 - 9; PMID: 8114006
- Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res 2005; 65:7462 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-05-0071; PMID: 16103100
- Mroz RM, Holownia A, Chyczewska E, Chyczewski L, Braszko JJ. p53 N-terminal Ser-15 approximately P and Ser-20 approximately P levels in squamous cell lung cancer after radio/chemotherapy. Am J Respir Cell Mol Biol 2004; 30:564 - 8; http://dx.doi.org/10.1165/rcmb.2003-0077OC; PMID: 14527925
- Borner C, Ueffing M, Jaken S, Parker PJ, Weinstein IB. Two closely related isoforms of protein kinase C produce reciprocal effects on the growth of rat fibroblasts. Possible molecular mechanisms. J Biol Chem 1995; 270:78 - 86; http://dx.doi.org/10.1074/jbc.270.1.78; PMID: 7814423
- Choi PM, Tchou-Wong KM, Weinstein IB. Overexpression of protein kinase C in HT29 colon cancer cells causes growth inhibition and tumor suppression. Mol Cell Biol 1990; 10:4650 - 7; PMID: 2388620
- Goldstein DR, Cacace AM, Weinstein IB. Overexpression of protein kinase C beta 1 in the SW480 colon cancer cell line causes growth suppression. Carcinogenesis 1995; 16:1121 - 6; http://dx.doi.org/10.1093/carcin/16.5.1121; PMID: 7767974
- Fields AP, Calcagno SR, Krishna M, Rak S, Leitges M, Murray NR. Protein kinase Cbeta is an effective target for chemoprevention of colon cancer. Cancer Res 2009; 69:1643 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-08-3187; PMID: 19221092
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 10; http://dx.doi.org/10.1038/35042675; PMID: 11099028
- Leonard TA, Różycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011; 144:55 - 66; http://dx.doi.org/10.1016/j.cell.2010.12.013; PMID: 21215369
- Pysz MA, Leontieva OV, Bateman NW, Uronis JM, Curry KJ, Threadgill DW, et al. PKCalpha tumor suppression in the intestine is associated with transcriptional and translational inhibition of cyclin D1. Exp Cell Res 2009; 315:1415 - 28; http://dx.doi.org/10.1016/j.yexcr.2009.02.002; PMID: 19232344
- Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol 1994; 126:747 - 63; http://dx.doi.org/10.1083/jcb.126.3.747; PMID: 8045938
- Verstovsek G, Byrd A, Frey MR, Petrelli NJ, Black JD. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 1998; 115:75 - 85; http://dx.doi.org/10.1016/S0016-5085(98)70367-1; PMID: 9649461
- Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol 2000; 151:763 - 78; http://dx.doi.org/10.1083/jcb.151.4.763; PMID: 11076962
- Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res 2006; 66:6955 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-06-0268; PMID: 16849539