Abstract
Dysregulation of EGFR expression and signaling is well documented to contribute to disease progression and metastasis in many types of cancer including breast cancer. EGF-stimulated EGFR activation leads to receptor internalization and endocytic degradation to control EGFR-mediated signaling. This process is frequently deregulated in cancer cells, leading to increased EGFR expression and mitogenic signaling. Here, we demonstrate that Bif-1, a tumor suppressor, plays a role in EGFR endocytic degradation and chemotactic migration in MDA-MB-231 breast cancer cells. Our data reveal that suppression of Bif-1 expression delays EGFR degradation and sustains Erk1/2 activation in response to EGF stimulation. Mechanistically, loss of Bif-1 sequesters internalized EGF in Rab5-positive endosomes and delays EGFR trafficking to lysosomes. Recruitment of Rab7 to EGF-positive vesicles and the activation of Rab7 are impaired in Bif-1 knockdown cells. Additionally, intracellular pH and the localization of acidic vesicles are altered by suppression of Bif-1. Furthermore, inhibition of Bif-1 increases chemotactic cell migration in response to EGF or serum, which correlates with prolonged cytoskeletal reorganization. Importantly, the effect of Bif-1 on EGF-induced cell migration is abolished by gefitinib, an EGFR-specific inhibitor. Taken together, these data suggest a novel function for Bif-1 as a suppressor of breast cancer cell migration by promoting EGFR degradation through the regulation of endosome maturation.
Keywords: :
Introduction
Breast cancer represents the second leading cause of cancer related deaths among US women.Citation1 While scientific advances in breast cancer prevention, detection, diagnosis and treatment are currently being made, existing therapies are still lacking in their ability to effectively treat and cure this devastating disease; especially breast cancer classified as triple negative disease. Although triple negative breast cancers, which lack estrogen receptor (ER), progesterone receptor (PR) and HER2, represent only 15–20% of all breast cancers, they are associated with a disproportionate number of deaths and a high propensity of metastasis.Citation2 Metastasis represents the culmination of events leading from the primary tumor’s ability to overcome physical boundaries, disseminate into the blood or lymphatic system and colonize in distant organs, ultimately leading to organ dysfunction and death.Citation3 Cytoskeletal reorganization and cell migration represent crucial events in the metastatic cascade, as cancer cells must develop motile and invasive properties in order to progress from the primary tumor site to a secondary location. Given that ER and HER2 targeted therapies are ineffective at treating triple negative breast cancersCitation4 and existing therapies are lacking in their ability to cure metastatic disease, there is a critical need for new treatment strategies and molecular targets to be explored.
Increased expression and/or gene amplification of the epidermal growth factor receptor (EGFR) has been observed in many human cancers including triple negative breast cancer.Citation5 EGFR overexpression has been linked to increased cell proliferation, disease progression and poor prognosis.Citation6 Following EGF stimulation and receptor activation at the plasma membrane, EGFR is rapidly internalized and is delivered to early endosomes where receptor sorting to a recycling or degradative fate is initiated. During early endosome sorting, tubular fission releases recycling components from the early endosome for transport back to the plasma membrane as the process of endosome maturation begins.Citation7 The maturation process requires a host of events including Rab5 to Rab7 conversion, V-ATPase transport to early endosomes for endosome acidification, intraluminal vesicle formation mediated in part by ESCRT proteins, trafficking of endosomes along microtubules to the perinuclear region, expansion in endosome size and the acquisition of fusion machinery to promote endosome-lysosome fusion.Citation8 The process of Rab5 to Rab7 conversion involves active Rab5-GTP mediated recruitment of Rab7 onto early endosomes followed by Rab7 activation and association with effector proteins such as Rab7 Interacting Lysosomal Protein (RILP). Active Rab7 promotes EGFR degradation through association with both endosomes and lysosomes.Citation9 The interaction between Rab7 and RILP facilitates late endosome to lysosome transport and regulates the movement and positioning of endosomes and lysosomes from the cell periphery toward the perinuclear regionCitation10 through interaction with the dynein-dynactin motor complex.Citation11 In addition to their roles in mediating endocytic degradation, late endosomes also function in mediating the transport of lysosomal components including acid hydrolases from the trans-Golgi network to lysosomes to maintain the degradative nature of the lysosomal compartment.Citation8 Lysosomes represent the terminal vesicular compartment for both endocytic and autophagic trafficking.Citation12 They contain numerous acid hydrolases that function specifically at acidic pH (~5) to hydrolyze and degrade DNA, RNA, protein, polysaccharides and lipids, making the loading of V-ATPases onto endosomes a critical step in maintaining an acidic and functional lysosomal compartment.Citation13 Lysosomal degradation effectively terminates EGFR mediated signaling to downstream pathways including MEK/MAPK, JAK/STAT, Src and PI3K/Akt.Citation14 As overexpression of EGFR promotes the mitogenic signaling needed for tumor formation and metastasis, EGFR endocytic degradation represents a potential point of intervention to control downstream growth/survival-promoting signaling cascades.
Bif-1, also known as SH3GLB1 and Endophilin B1, is a tumor suppressor, which was originally identified as a pro-apoptotic Bax binding protein.Citation15,Citation16 In addition to its role in Bax activation and apoptosis, Bif-1 has been shown to function in the regulation of autophagy and intracellular membrane dynamics.Citation17 Importantly, decreased Bif-1 expression is found in various types of human cancer including gastric,Citation18 colorectal,Citation19 prostate,Citation20 pancreatic,Citation21 invasive urinary bladder and gallbladder cancers,Citation22 and loss of Bif-1 promotes tumor development in mice.Citation23 Moreover, a recent study using a mouse mammary tumor model revealed a decrease in Bif-1 expression as cells became more metastatic, suggesting a potential function for Bif-1 in breast cancer metastasis.Citation24 In this manuscript, we report a novel tumor suppressive function of Bif-1 in triple negative metastatic breast cancer. Knockdown of Bif-1 in MDA-MB-231 cells delays EGF/EGFR intracellular trafficking and lysosomal degradation, leading to sustained Erk1/2 activation. Further, suppression of Bif-1 reduces Rab7 recruitment to early endosomes, decreases Rab7 activation, and promotes chemotactic tumor cell migration toward FBS and EGF. Taken together, this study provides a novel role for Bif-1 in the regulation of EGFR endocytosis and metastatic potential in breast cancer cells.
Results
Suppression of Bif-1 delays EGFR endocytic trafficking and degradation
MDA-MB-231 cells are a TNBC cell line that represent an excellent model for studying EGFR endocytic trafficking as they overexpress EGFR but lack EGFR gene amplification.Citation25 As TNBC preferentially metastasizes to visceral organs including the lung,Citation26 a variant of the MDA-MB-231 cell line designated “LM2,” which was specifically selected to have a high propensity of lung metastasis,Citation27 was chosen for use in our studies. However, since the LM2 cells stably express GFP,Citation27 all immunostaining experiments were performed using parental MDA-MB-231 cells. To investigate the function of Bif-1 in EGFR endocytic trafficking and degradation, LM2 cells were stably transfected with a doxycycline-inducible human Bif-1 shRNA (shBif-1) lentiviral construct pTRIPz-shBif-1, which produced maximal knockdown (approximately 90%) of Bif-1 expression following 6 d of doxycycline treatment (). The pTRIPz-shBif-1construct also produced 90% knockdown in parental MDA-MB-231 cells (), and as such, 6 d of doxycycline treatment was utilized for experiments. As shown in , knockdown of Bif-1 delayed EGF-stimulated EGFR degradation and sustained receptor activation as measured by EGFR phosphorylation on Y1068. Activation of Erk1/2, an important downstream effector of EGF action, was also sustained by the suppression of Bif-1 (). Similarly, loss of Bif-1 decreases EGFR degradation rates in HeLa,Citation28 PLC-PRF-5Citation29 and HCT116 (data not shown) cancer cells, confirming that these findings are not cell type specific. Further, while Bif-1 suppression did not alter EGFR internalization, co-localization of EGFR with the lysosomal membrane protein LAMP-1 was dramatically reduced in Bif-1 knockdown cells (). Notably, EGF stimulation resulted in a peripheral distribution of EGFR in control cells at 15 min, which was followed by perinuclear localization and LAMP-1 co-localization at 30 and 60 min after EGF treatment. Conversely, knockdown of Bif-1 delayed the perinuclear trafficking of EGFR and decreased EGFR-LAMP-1 co-localization following EGF stimulation. In addition, while EGFR staining was largely diminished in control cells at 120 min, knockdown of Bif-1 delayed EGF-induced EGFR degradation and resulted in the accumulation of enlarged EGFR-positive vesicles. Taken together, these data indicate that Bif-1 plays a role in EGFR trafficking to lysosomes for degradation.
Figure 1. Establishment of doxycycline-inducible Bif-1 knockdown cell lines. (A) LM2-pTRIPz-shBif-1 and (B) MDA-MB-231-pTRIPz-shBif-1 cells were cultured in the presence of 1 μg/ml doxycycline (to induce shBif-1 expression) for the indicated number of days, harvested and subjected to western blot analysis. The immunoblotting results of Bif-1 were quantified by densitometry, normalized to β-actin and presented relative to the control.
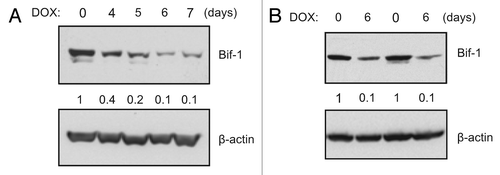
Figure 2. Suppression of Bif-1 delays EGFR endocytic degradation and sustains signaling in MDA-MB-231 cells. (A–D) LM2-pTRIPz-shBif-1 cells were cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d, serum starved for 16 h, treated with 100 ng/ml EGF for the indicated time points and subjected to western blot analysis. (B–C) The immunoblotting results of pY1068-EGFR and total EGFR were quantified by densitometry, normalized to β-actin and presented relative to the 0.5 h and 0 h time points, respectively, in graphs (mean ± s.d.; n = 4). (D) The activation of pErk1/2 by immunoblotting was quantified by densitometry, normalized to total Erk1/2 and presented relative to the 0 h time point, in graphs (mean ± s.d.; n = 4). Asterisks in (B–D) indicate significant differences (p < 0.05) between Bif-1 knockdown and control groups determined by Student’s t-test. (E) MDA-MB-231-pTRIPz-shBif-1 cells were cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d, serum starved for 16 h, treated with 100 ng/ml EGF for the indicated time points, and subjected to immunostaining using EGFR-XP (green) and LAMP-1 (red) antibodies. Scale bar: 10 μm.
Knockdown of Bif-1 decreases Rab7 activation in response to EGF
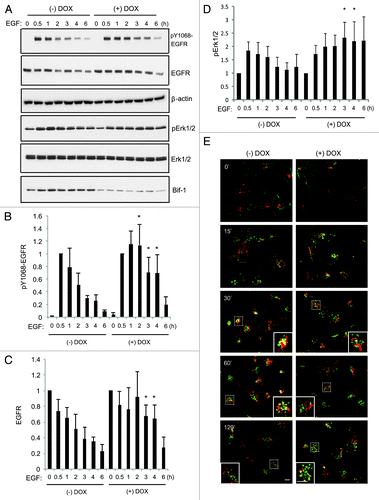
To better understand the role of Bif-1 in EGFR endocytosis, we investigated the effects of Bif-1 on Rab5 and Rab7, two small GTPases of the Ras family that play important roles in the endocytic trafficking of growth factor receptors such as EGFR. As shown in , knockdown of Bif-1 resulted in increased EGF co-localization with Rab5 and decreased EGF co-localization with Rab7 as compared with control cells. These data indicate that loss of Bif-1 suppresses Rab7 recruitment to EGF-positive vesicles and traps EGF in Rab5-positive compartments. Improper recruitment of Rab7 to the early endosome may contribute to the delay in EGF-EGFR endocytic degradation by preventing endosome maturation in cells lacking Bif-1. Further, Bif-1 has been suggested to play an important role in controlling the size of early endosomes as suppression of Bif-1 promotes the formation of enlarged early endosomes following NGF or EGF treatment.Citation29,Citation30 Consistently, our data reveal that suppression of Bif-1 promoted the accumulation of enlarged Rab5-positive endosomes (). Taken together, these data suggest that Bif-1is involved in the regulation of endosome maturation by promoting EGFR transport from early endosomal to late endosomal/lysosomal compartments. In support of this theory, EGF stimulation of control LM2 cells resulted in Rab7 activation at 15 min, which was suppressed by knockdown of Bif-1, as measured by the specific binding of activated (GTP-bound) Rab7 to its effector RILP (). Further, as Rab7 can bind to effector proteins other than RILP in response to EGF, activation of Rab7 was investigated using a nucleotide binding assay. As shown in the ratio of GTP-bound (active) to GDP-bound (inactive) Rab7 was decreased in Bif-1 knockdown cells following EGF stimulation () again suggesting that Rab7 activation is suppressed by loss of Bif-1. Taken together, these findings suggest that Bif-1 plays a positive role in EGFR endocytosis by regulating endosome maturation.
Figure 3. Knockdown of Bif-1 maintains EGF in Rab5 positive vesicles and decreases Rab7 recruitment to EGF positive vesicles. (A-B) MDA-MB-231-pTRIPz-shBif-1 cells were cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d, serum starved for 16 h, treated with 1 μg/ml Alexa-Fluor488-EGF (green) for the indicated time points, and subjected to immunostaining using Rab5 and Rab7 antibodies (red). Arrows indicate enlarged EGF-positive early endosome compartments. Colocalization was quantified as the percent of total EGF that colocalized with Rab5 or Rab7 using Slidebook software for at least 50 cells per condition. Statistical significance was determined by Student’s t-test. Scale bar: 10 μm.
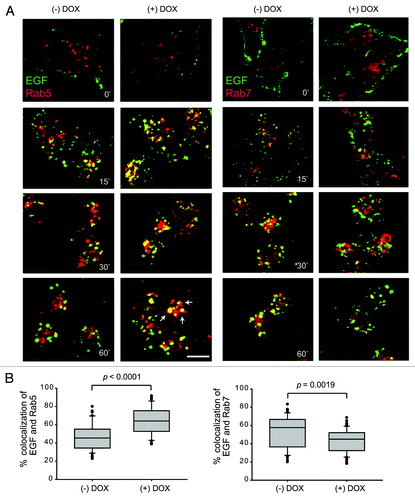
Figure 4. Knockdown of Bif-1 decreases EGF-induced Rab7 activation. (A) LM2-pTRIPz-shBif-1 cells were cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 3 d, infected with GST-RILP-RBD lentivirus for 72 h, serum starved for 16 h and treated with 100 ng/ml EGF for the indicated time points. 300 μg of TCL was incubated with Glutathione beads and Rab7 binding was measured by western blot. The amount of GST-RILP-RBD-bound Rab7 was quantified by densitometry, normalized to GST-RILP-RBD and presented relative to the value of control cells at 0 h. (B) LM2-pTRIPz-shBif-1 cells cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d were metabolically labeled with 32P orthophosphate for 5.5 h and treated with 100 ng/ml EGF for 30 min. Rab7 was immunoprecipitated and the bound GTP and GDP were determined by chromatography on PEI cellulose TLC plates. The graph represents the relative ratio of GTP to GDP normalized to the origin with the control normalized to one.
Suppression of Bif-1 alter the pH and intracellular localization of acidic vesicles
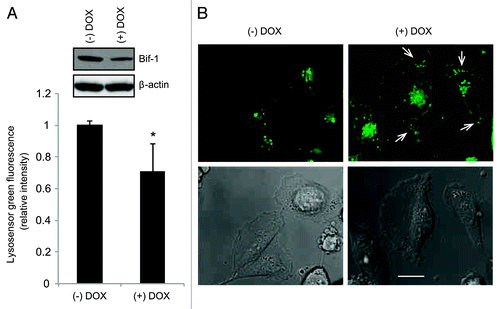
The pH of endosomes becomes increasingly more acidic during their progression through the endocytic pathway in order to support the proper functioning of acid hydrolases. To study the effects of Bif-1 suppression on lysosome localization and acidity, we used a vital dye, Lysosensor Green-DND189, which specifically accumulates in acidic vesicles and increases in fluorescent intensity as the vesicles become more acidic. As shown in , depletion of Bif-1 decreased the fluorescence intensity of Lysosensor Green-DND189, indicating that intracellular vesicles in Bif-1 knockdown cells are less acidic than those of their wild-type counterparts. Further, suppression of Bif-1 accelerated the redistribution of Lysosensor Green-DND189-positive acidic vesicles away from the perinuclear region and toward the cell periphery (). Blocking lysosomal traffic through Rab7 inhibition negatively alters lysosome intactness, acidity and proper localization to the perinuclear region of the cell.Citation31 Changes in lysosomal localization toward the cell periphery have been shown to increase metastatic potential through the secretion of lysosomal contents to degrade the extracellular matrix and promote cell motility, invasion and angiogenesis.Citation32 Based on our findings and the known tumor suppressor properties of Bif-1, we next investigated the role of Bif-1 in breast cancer cell migration.
Figure 5. Suppression of Bif-1 alters the pH and intracellular localization of acidic vesicles. (A) MDA-MB-231-pTRIPz-shBif-1 cells cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d were treated with 1 μM Lysosensor Green DND-189 for 30 min and the green fluorescent intensity was measured using the Guava flow cytometer (mean ± s.d.; n = 6). Asterisk indicates significant differences (p < 0.005) between Bif-1 knockdown and control groups determined by Student’s t-test. (B) MDA-MB-231-pTRIPz-shBif-1 cells cultured without or with 1 μg/ml doxycycline for 6 d were incubated with 20 μM z-VAD-fmk for 30 min before treatment with 1 μM Lysosensor Green DND-189 for an additional 30 min and analyzed using a fluorescence microscope. Arrows indicate the peripheral localization of acidic vesicles. Scale bar: 10 μm.
Suppression of Bif-1 promotes cytoskeletal reorganization and enhances chemotactic cell migration
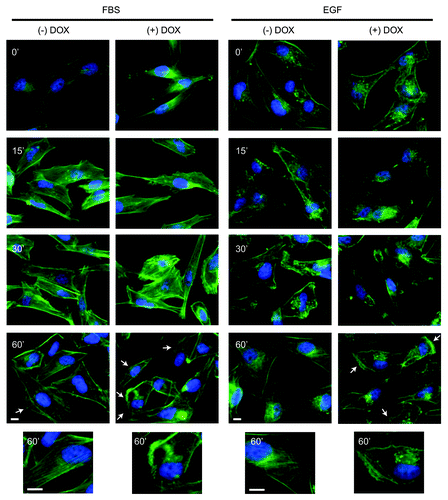
To examine the role of Bif-1 in cytoskeletal reorganization in response to growth factors, control and Bif-1 knockdown MDA-MB-231-pTRIPz-shBif-1 cells were stimulated with EGF or FBS and stained for F-actin with fluorescently labeled phalloidin. As shown in , suppression of Bif-1 increased the formation of membrane ruffling, microspikes, and filopodia projections following treatment with FBS and EGF, indicative of an increased migratory phenotype. After 60 min of stimulation with EGF or FBS, control cells predominantly reverted back to their morphology before stimulation, with the presence of stress fibers, smooth cell border staining and minimal lamellipodia. However, the presence of lamellipodia and filopodia were still observed in Bif-1 knockdown cells after 60 min of EGF or FBS stimulation, indicating that these cells maintain the morphological characteristics necessary for migration for an extended period of time compared with cells expressing Bif-1.
Figure 6. Suppression of Bif-1 promotes cytoskeletal reorganization. MDA-MB-231-pTRIPz-shBif-1 cells were cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d, serum starved for 16 h, stimulated with 10% FBS or 10 ng/ml EGF for the indicated time points and stained with Alexa-fluor-488 Phalloidin (green). Images were obtained and arrows indicate sites of sustained cytoskeletal reorganization. Scale bar: 10 μm.
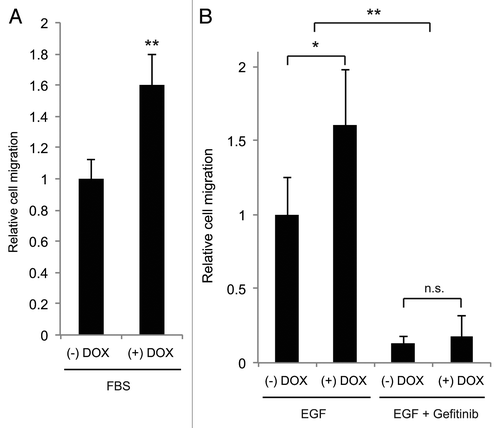
Exposure of cells to a chemotactic gradient of growth factors such as EGF or FBS causes cells to translocate along the gradient and enhance cell invasion, intravasation and metastasis.Citation33 To study the role of Bif-1 in breast cancer cell migration in response to a chemotactic gradient, a transwell chemotactic cell migration chamber was used to evaluate LM2-pTRIPz-shBif-1 cell migration in response to EGF and FBS. While the addition of doxycycline alone did not alter the migration of parental LM2 cells (data not shown), reveals that suppression of Bif-1 increased chemotactic cell migration toward FBS and EGF. Treatment of cells with the EGFR tyrosine kinase inhibitor, gefitinib, significantly blocked EGF-induced cell migration in both control and Bif-1 knockdown cells () without altering cell survival at concentrations up to 5 µM (data not shown). Taken together, these data support a tumor suppressive role for Bif-1 in breast cancer progression through suppression of chemotactic cell migration.
Figure 7. Suppression of Bif-1 promotes chemotactic cell migration toward serum and EGF. LM2-pTRIPz-shBif-1 cells cultured without or with 1 μg/ml doxycycline (to knockdown Bif-1) for 6 d were plated onto the apical side of 8 μm pore membranes and allowed to migrate toward 10% FBS or 10 ng/ml EGF in the basolateral chamber for 22 or 42 h, respectively. Where indicated, the cells were pretreated with DMSO or 1 μM Gefitinib for 30 min before exposure to EGF. For quantification, 3–5 representative sections were manually counted per membrane. Statistical significance was determined by (A) Student's t-test or (B) one-way ANOVA; *p < 0.01; **p < 0.001.
Discussion
In this study, we describe a novel function for Bif-1 in mediating endosome maturation during EGF induced EGFR endocytosis, which contributes to the suppression of breast cancer cell chemotaxis. Upon EGF stimulation, EGFR undergoes rapid internalization into clathrin-coated pits through a process involving membrane invagination and vesicle scission. Endophilin family members serve important roles in mediating membrane dynamics and structurally contain an N-BAR (Bin/Amphiphysin/Rvs) domain, which binds to lipids and induces membrane curvature, and a C-terminal SH3 domain, which complexes with proteins containing a proline rich region.Citation17 Endophilin A positively regulates membrane curvature and vesicle scission at the plasma membrane during endocytosis.Citation34,Citation35 Based on the structural similarities between Endophilin A and Bif-1, we reasoned that Bif-1 might be involved in EGFR internalization. However, suppression of Bif-1 did not affect the uptake of a fluid phase marker, horseradish peroxidase (data not shown), and EGF colocalization with Rab5 positive early endosomes, suggesting that Bif-1 is unlikely to play a role in EGFR internalization at the plasma membrane.
Early endosomes undergo a maturation process which involves the conversion of Rab5 to Rab7, changes in the movement and positioning of endosomes, decreased endosomal pH, alterations in fusion machinery and cargo internalization into intraluminal vesicles (ILV). The generation of ILVs and multi-vesicular bodies (MVB) serves as a critical step in endosome maturation and requires endosomal membrane invagination and internal vesicle budding. Cargo internalization into ILVs requires the actions of ESCRT proteins and serves to effectively terminate receptor signaling. Interestingly, Bif-1 interacts with the proapoptotic protein Bax which triggers Bif-1 oligomerization and alters the size and morphology of giant unilamellar vesicles by inducing massive vesiculation of liposomes, resulting in MVB-like structures.Citation36 Based on this observation, we investigated whether Bif-1 plays a role in inducing ILV formation using a trypsin protection assay. However, we found that loss of Bif-1 did not suppress the in vitro formation of MVBs (data not shown), indicating that Bif-1 is not required for the generation of ILVs.
Bif-1 is known to interact with Beclin1 through UVRAG to stimulate the activation of Vps34/PI3KC3, which is involved in both autophagy induction and the regulation of vesicle transport, including endocytic trafficking.Citation23 UVRAG positively regulates the class C-Vps complex to promote EGFR degradation and endosomal fusion.Citation37 Through its GEF activity on Rab7, C-Vps proteins promote endosome maturation by regulating Rab5 to Rab7 conversion.Citation38 Consistent with its ability to interact with UVRAG, suppression of Bif-1 prolongs EGF presence in Rab5-positive endosomes, reduces Rab7 recruitment to EGF-positive vesicles, decreases Rab7 activation and delays EGFR trafficking to lysosomes, suggesting a possible connection of Bif-1 to C-Vps functions via UVRAG. Further, Bif-1 has recently been reported to promote EGFR trafficking and cytokinesis through a process that is independent of ATG14L and its role in autophagy, as a consequence of interactions with VPS15, VPS34, Beclin 1 and UVRAG.Citation28 Taken together, it is conceivable that Bif-1 functions as a positive regulator of endosome maturation through interaction with UVRAG to activate the C-Vps complex, thereby promoting Rab5 to Rab7 conversion and endosomal fusion.
Additionally, Bif-1 may function in a distinct complex along with TIP30 and ASCL4 to regulate the trafficking of Rab5 and V-ATPases from the Golgi apparatus to EEA1-positive endosomes in response to EGF in hepatoma cells.Citation29 V-ATPase trafficking to early endosomes is proposed to enhance early endosome acidification, EGF-EGFR dissociation and EGFR endocytosis.Citation29 While our findings in MDA-MB-231 cells suggest a dispensable role of Bif-1 in mediating Rab5 recruitment to EGF-positive vesicles, we did observe an increase in intracellular pH and altered acidic vesicle localization in Bif-1 knockdown cells. In contrast, Bif-1 negatively regulates endocytic trafficking of the NGF/TrkA receptor in neuronal PC12 cells through an interaction with EEA1, suggesting an alternate function for Bif-1 in neurons.Citation30 Undoubtedly, further studies are needed to elucidate the precise function(s) of Bif-1 in regulating EGFR endocytosis.
Alterations in EGFR expression and signaling occur in many types of cancer and contribute to disease progression and poor prognosis. EGFR overexpression in breast cancer cells induces migration, suggesting an important role for EGFR in cell motility.Citation39
Cell migration is involved in both physiological and pathophysiological processes including normal embryonic development, the inflammatory response, wound healing and tumor metastasis.Citation40 Migration serves as a necessary component of the metastatic cascade as cancer cells must develop the ability to detach from the primary tumor, migrate into the blood or lymphatic systems, survive detachment induced apoptosis and migrate out of the circulatory system into a secondary site where distal metastasis can form and survive.Citation3 Migration is initiated in response to extracellular stimuli including growth factors such as EGF and requires lamellipodium extension at the leading edge, focal adhesion complex formation, protease secretion, cell body contraction and rear tail detachment.Citation41 Our findings reveal that in response to EGF stimulation, depletion of Bif-1 increases cell migration in metastatic breast cancer cells, which is abolished by treatment with the EGFR inhibitor gefitinib. These findings imply that EGFR signaling serves as a key factor to regulate MDA-MB-231 cell migration in response to EGF, which is mediated, at least in part, by Bif-1. EGF mediated activation of EGFR initiates downstream signaling through multiple pathways including Ras/Raf/MEK/ERK. ERK activation leads to the phosphorylation of substrates such as MLCK, which in turn increases the phosphorylation of MLC, promotes the formation of membrane protrusions at the leading edge and enhances cell migration.Citation42,Citation43 In response to EGF, suppression of Bif-1 delays EGFR degradation, sustains the activation of Erk1/2 and prolongs the formation of migratory structures including lamellipodia and filopodia. These findings suggest the potential involvement of EGFR signaling through ERK to enhance cell migration when Bif-1 is suppressed.
Interestingly, we also show that depletion of Bif-1 alters the localization of acidic vesicles toward the cell periphery. Changes in lysosomal localization toward the cell periphery has been shown to increase metastatic potential.Citation32 The release of lysosomal proteases from peripherally localized lysosomes can lead to extracellular matrix degradation and ultimately promote cell motility, invasion and angiogenesis.Citation32 Additionally, our data demonstrate that suppression of Bif-1 increases intracellular pH. Such alterations in pH may negatively impact the function of acid hydrolases within the lysosomal compartment and result in decreased lysosomal function and a reduction in the degradation of internalized cargo. Further studies are necessary to determine whether loss of Bif-1 alters ECM degradation and lysosome function.
Taken together, these studies reveal a novel inhibitory role for Bif-1 in breast cancer cell migration by promoting EGFR degradation at the stage of endosome maturation. Based on our findings and the known roles of Bif-1 in intracellular membrane dynamics, we propose a model whereby Bif-1 functions in endosome maturation through interaction with UVRAG at the early endosome to recruit and activate the C-Vps complex to induce Rab5-Rab7 conversion and endosomal fusion. Further, sustained cytoskeletal reorganization and increased cell migration that results from Bif-1 suppression may potentially be due to prolonged EGFR signaling to ERK. These findings indicate that further studies are warranted to gain a greater understanding of the molecular mechanisms through which Bif-1 regulates EGFR endocytic degradation and metastatic potential. These studies will be of great importance as they may lead to more effective treatment strategies for triple negative breast cancer.
Materials and Methods
Plasmids and cell lines
The doxycycline-inducible human Bif-1 shRNA (shBif-1) lentiviral vector pTRIPz-shBif-1 (RHS4696–99682777) was obtained from Open Biosystems and was used to generate stable doxycycline-inducible shBif-1-expressing MDA-MB-231 cell lines as described.Citation23 MDA-MB-231 and HEK293T/17 cell lines were obtained from ATCC and the MDA-MB-231 variant cell line, LM2, selected for a high propensity of lung metastasis was a generous gift from Dr. Joan Massague at Memorial Sloan Kettering.Citation27
Antibodies and reagents
The following antibodies were used: pY1068-EGFR (2234), EGFR (2232), EGFR-XP (4267), phospho-Erk1/2 (9101), Erk1/2 (9102), and Rab7 polyclonal (9367) antibodies were purchased from Cell Signaling Technology. Antibodies against Rab7 monoclonal (R8779) and β-actin (A2228) were purchased from Sigma. Bif-1 (IMG265A), GST-HRP (A190–121P), and LAMP-1 (BDB555798) antibodies were purchased from Imgenex, Bethyl, and BD Biosciences, respectively. EGF (PHG0311), Alexa-Fluor488-EGF (E13345), Alexa-Fluor488-Phalloidin (A12379), and Lysosensor Green DND-189 (L7535) were purchased from Invitrogen. Gefitinib was obtained from Active Biochem (A-1024).
Immunoblot analysis
LM2-pTRIPz-shBif-1 cells cultured without or with 1 µg/ml doxycycline for 6 d (to knockdown Bif-1) were serum starved for 16 h and treated with 100 ng/ml EGF in Dulbecco's Modified Eagle Medium (DMEM) containing 0.2% BSA and 20 mM HEPES for the indicated time points. Cell lysates were prepared in lysis buffer (50 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 25 mM NaF, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 2 µg/μl aprotinin, 2 µg/ml leupeptin, 100 µg/ml phenylmethylsufonyl fluoride, 1 mM dithiothreitol, 20 mM p-nitrophenyl phosphate and 1% Triton X-100) and subjected to western blot analysis with antibodies specific for EGFR, pY1068-EGFR, Bif-1, β-actin, phospho-Erk1/2, and Erk1/2.
Immunofluorescent microscopy
MDA-MB-231-pTRIPz-shBif-1 cells cultured without or with 1 µg/ml doxycycline for 6 d were seeded on chamber slides overnight and serum starved for 16 h. Cells were treated with 100 ng/ml EGF or 1 µg/ml AlexaFluor488-EGF in DMEM containing 0.2% BSA and 20 mM HEPES for the indicated time points, fixed in 4% paraformaldehyde, permeabilized with 100 µg/ml digitonin, and incubated with the indicated primary antibodies, followed by incubation with fluorescent conjugated secondary antibodies. Fluorescent images were obtained using an OLYMPUS IX81 deconvolution microscope and analyzed using SlideBook 5.0 software (Intelligent Imaging Innovations).
Rab7 activation assays
GST-RILP/Rab7 Co-IP
GST-RILP-RBDCitation37 was subcloned into pCDH1-MCS1-EF1-Puro vector and lentivirus was produced in HEK293T/17 cells. LM2-pTRIPz-shBif-1 cells cultured without or with 1 µg/ml doxycycline for 3 d were infected with GST-RILP-RBD lentivirus. 72h post-infection, cells were serum starved for 16 h and stimulated with 100 ng/ml EGF for 0, 15 and 30 min. Cell lysates were prepared in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA and 1% Triton X-100) containing protease and phosphatase inhibitor cocktails (Sigma) and 300 µg of total protein lysate was incubated with Glutathione Sepharose 4B beads (GE Healthcare) for 1 h at 4°C. Beads were washed with lysis buffer, boiled in Laemmli sample buffer, and subjected to western blot analysis using a monoclonal Rab7 antibody (Sigma).
Nucleotide-binding
LM2-pTRIPz-shBif-1 cells cultured without or with 1 µg/ml doxycycline for 6 d were incubated in phosphate-free DMEM containing sodium pyruvate, 0.2% BSA and 20 mM HEPES for 1 h at 37°C. Then, 0.2 mCi/ml 32P orthophosphate was added to the media for 5.5 h at 37°C, followed by addition of 100 ng/ml EGF for 30 min. Cell lysates were prepared in RIPA buffer, precleared with protein A beads, and incubated overnight with a Rab7 antibody (Cell Signaling). The immunocomplexes were precipitated with protein A beads and the associated nucleotides were eluted in 20 µl of 2 mM EDTA, 2 mM DTT and 0.2% SDS at 58°C for 20 min. An aliquot (5 μl) of the elution was spotted onto PEI cellulose TLC plates along with GTP and GDP standards, resolved in 0.75 M KH2PO4 (pH 3.5) buffer for 1.5 h, and subjected to autoradiography.
Labeling of acidic compartments
MDA-MB-231-pTRIPz-shBif-1 cells treated without or with 1 µg/ml doxycycline for 6 d were treated with 1 µM Lysosensor Green DND-189 for 30 min to selectively label acidic compartments. Cells were washed with PBS, trypsinized, and subjected to flow cytometry to evaluate intracellular acidity as measured by green fluorescent intensity. For microscopic examination, the cells were pretreated for 30 min with 20 µM Z-VAD-fmk (to prevent cell detachment) before adding 1 µM Lysosensor Green DND-189. Fluorescent images were obtained using an OLYMPUS IX81 deconvolution microscope.
Actin reorganization
MDA-MB-231-pTRIPz-shBif-1 cells treated without or with 1 µg/ml doxycycline for 6 d were serum starved for 16 or 24 h and stimulated with DMEM containing 10% FBS or 10 ng/ml EGF respectively, for the indicated time points. Cells were fixed in 4% paraformaldehyde, stained with Alexa-Fluor488-Phalloidin for 30 min, and mounted with DAPI. Fluorescent images were obtained using an OLYMPUS IX81 deconvolution microscope.
Cell migration assay
LM2-pTRIPz-shBif-1 cells cultured without or with 1 µg/ml doxycycline for 6 d were plated onto control insert membranes (BD Biosciences) in serum free DMEM containing 0.2% BSA and 20 mM HEPES. Media containing 10% FBS or 10 ng/ml EGF was added to the basolateral chamber and cells were allowed to migrate for 22 or 42 h. Cells on the apical side of the membrane were removed and cells that had migrated to the basolateral side of the membrane were fixed with 100% methanol and stained with 10% Giemsa. Where indicated, cells were treated with 1 µM gefitinib or DMSO for 30 min prior to the addition of EGF. The cells were mounted onto slides and the number of migrated cells was manually counted from 3–5 fields per membrane.
| Abbreviations: | ||
| ECM | = | extracellular matrix |
| EEA1 | = | early endosome antigen 1 |
| EGF | = | epidermal growth factor |
| EGFR | = | epidermal growth factor receptor |
| ESCRT | = | endosomal sorting complex required for transport |
| FBS | = | fetal bovine serum |
| ILV | = | intraluminal vesicle |
| LAMP-1 | = | lysosomal associated membrane protein 1 |
| LM2 | = | lung metastasis 2 |
| MLC | = | myosin light chain |
| MLCK | = | myosin light chain kinase |
| MVB | = | multivesicular body |
| NGF | = | nerve growth factor |
| RILP | = | Rab7 interacting lysosomal protein |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge Dr. Xingcong Ren for assistance with Guava flow cytometry and Dr. Chengyu Liang for the GST-RILP-RBD plasmid. This work is supported by the grants from the National Institute of Health (CA82197 and CA129682) to H.G.W.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10 - 29; http://dx.doi.org/10.3322/caac.20138; PMID: 22237781
- Nanda R. “Targeting” triple-negative breast cancer: the lessons learned from BRCA1-associated breast cancers. Semin Oncol 2011; 38:254 - 62; http://dx.doi.org/10.1053/j.seminoncol.2011.01.007; PMID: 21421115
- Chiang AC, Massagué J. Molecular basis of metastasis. N Engl J Med 2008; 359:2814 - 23; http://dx.doi.org/10.1056/NEJMra0805239; PMID: 19109576
- Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application?. J Clin Oncol 2005; 23:7350 - 60; http://dx.doi.org/10.1200/JCO.2005.03.3845; PMID: 16145060
- Kruger JS, Reddy KB. Distinct mechanisms mediate the initial and sustained phases of cell migration in epidermal growth factor receptor-overexpressing cells. Mol Cancer Res 2003; 1:801 - 9; PMID: 14517342
- Morris C. The role of EGFR-directed therapy in the treatment of breast cancer. Breast Cancer Res Treat 2002; 75 Suppl 1:S51-5; discussion S7-9.
- Mesaki K, Tanabe K, Obayashi M, Oe N, Takei K. Fission of tubular endosomes triggers endosomal acidification and movement. PLoS One 2011; 6:e19764; http://dx.doi.org/10.1371/journal.pone.0019764; PMID: 21572956
- Huotari J, Helenius A. Endosome maturation. EMBO J 2011; 30:3481 - 500; http://dx.doi.org/10.1038/emboj.2011.286; PMID: 21878991
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731 - 43; http://dx.doi.org/10.1016/j.molcel.2011.04.024; PMID: 21700220
- Wang T, Ming Z, Xiaochun W, Hong W. Rab7: role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal 2011; 23:516 - 21; http://dx.doi.org/10.1016/j.cellsig.2010.09.012; PMID: 20851765
- Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol 2001; 11:1680 - 5; http://dx.doi.org/10.1016/S0960-9822(01)00531-0; PMID: 11696325
- Fehrenbacher N, Jäättelä M. Lysosomes as targets for cancer therapy. Cancer Res 2005; 65:2993 - 5; PMID: 15833821
- Sun-Wada GH, Wada Y, Futai M. Lysosome and lysosome-related organelles responsible for specialized functions in higher organisms, with special emphasis on vacuolar-type proton ATPase. Cell Struct Funct 2003; 28:455 - 63; http://dx.doi.org/10.1247/csf.28.455; PMID: 14745137
- Roepstorff K, Grøvdal L, Grandal M, Lerdrup M, van Deurs B. Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer. Histochem Cell Biol 2008; 129:563 - 78; http://dx.doi.org/10.1007/s00418-008-0401-3; PMID: 18288481
- Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem 2001; 276:20559 - 65; http://dx.doi.org/10.1074/jbc.M101527200; PMID: 11259440
- Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics 2001; 71:222 - 34; http://dx.doi.org/10.1006/geno.2000.6378; PMID: 11161816
- Takahashi Y, Meyerkord CL, Wang HG. Bif-1/endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ 2009; 16:947 - 55; http://dx.doi.org/10.1038/cdd.2009.19; PMID: 19265852
- Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, et al. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 2006; 38:312 - 5; http://dx.doi.org/10.1080/00313020600820880; PMID: 16916719
- Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, Wang HG. Down-regulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer 2008; 113:2665 - 70; http://dx.doi.org/10.1002/cncr.23892; PMID: 18833585
- Coppola D, Oliveri C, Sayegh Z, Boulware D, Takahashi Y, Pow-Sang J, et al. Bax-interacting factor-1 expression in prostate cancer. Clin Genitourin Cancer 2008; 6:117 - 21; http://dx.doi.org/10.3816/CGC.2008.n.018; PMID: 18824435
- Coppola D, Helm J, Ghayouri M, Malafa MP, Wang HG. Down-regulation of Bax-interacting factor 1 in human pancreatic ductal adenocarcinoma. Pancreas 2011; 40:433 - 7; http://dx.doi.org/10.1097/MPA.0b013e318205eb03; PMID: 21283040
- Kim SY, Oh YL, Kim KM, Jeong EG, Kim MS, Yoo NJ, et al. Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathology 2008; 40:553 - 7; http://dx.doi.org/10.1080/00313020802320440; PMID: 18752120
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142 - 51; http://dx.doi.org/10.1038/ncb1634; PMID: 17891140
- Ho J, Kong JW, Choong LY, Loh MC, Toy W, Chong PK, et al. Novel breast cancer metastasis-associated proteins. J Proteome Res 2009; 8:583 - 94; http://dx.doi.org/10.1021/pr8007368; PMID: 19086899
- Davidson NE, Gelmann EP, Lippman ME, Dickson RB. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol Endocrinol 1987; 1:216 - 23; http://dx.doi.org/10.1210/mend-1-3-216; PMID: 3502607
- Rakha EA, Chan S. Metastatic triple-negative breast cancer. Clin Oncol (R Coll Radiol) 2011; 23:587 - 600; http://dx.doi.org/10.1016/j.clon.2011.03.013; PMID: 21524569
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005; 436:518 - 24; http://dx.doi.org/10.1038/nature03799; PMID: 16049480
- Thoresen SB, Pedersen NM, Liestøl K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res 2010; 316:3368 - 78; http://dx.doi.org/10.1016/j.yexcr.2010.07.008; PMID: 20643123
- Zhang C, Li A, Zhang X, Xiao H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J Biol Chem 2011; 286:9373 - 81; http://dx.doi.org/10.1074/jbc.M110.207720; PMID: 21252234
- Wan J, Cheung AY, Fu WY, Wu C, Zhang M, Mobley WC, et al. Endophilin B1 as a novel regulator of nerve growth factor/ TrkA trafficking and neurite outgrowth. J Neurosci 2008; 28:9002 - 12; http://dx.doi.org/10.1523/JNEUROSCI.0767-08.2008; PMID: 18768694
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell 2000; 11:467 - 80; PMID: 10679007
- Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta 2000; 291:113 - 35; http://dx.doi.org/10.1016/S0009-8981(99)00224-7; PMID: 10675719
- Wang W, Goswami S, Sahai E, Wyckoff JB, Segall JE, Condeelis JS. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol 2005; 15:138 - 45; http://dx.doi.org/10.1016/j.tcb.2005.01.003; PMID: 15752977
- Ringstad N, Nemoto Y, De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci U S A 1997; 94:8569 - 74; http://dx.doi.org/10.1073/pnas.94.16.8569; PMID: 9238017
- Gad H, Ringstad N, Löw P, Kjaerulff O, Gustafsson J, Wenk M, et al. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron 2000; 27:301 - 12; http://dx.doi.org/10.1016/S0896-6273(00)00038-6; PMID: 10985350
- Rostovtseva TK, Boukari H, Antignani A, Shiu B, Banerjee S, Neutzner A, et al. Bax activates endophilin B1 oligomerization and lipid membrane vesiculation. J Biol Chem 2009; 284:34390 - 9; http://dx.doi.org/10.1074/jbc.M109.021873; PMID: 19805544
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10:776 - 87; http://dx.doi.org/10.1038/ncb1740; PMID: 18552835
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005; 122:735 - 49; http://dx.doi.org/10.1016/j.cell.2005.06.043; PMID: 16143105
- Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett 1998; 425:145 - 50; http://dx.doi.org/10.1016/S0014-5793(98)00224-5; PMID: 9541025
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell 1996; 84:359 - 69; http://dx.doi.org/10.1016/S0092-8674(00)81280-5; PMID: 8608589
- Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 2010; 8:23; http://dx.doi.org/10.1186/1478-811X-8-23; PMID: 20822528
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 1997; 137:481 - 92; http://dx.doi.org/10.1083/jcb.137.2.481; PMID: 9128257
- Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev 2003; 22:395 - 403; http://dx.doi.org/10.1023/A:1023781114568; PMID: 12884914