Abstract
Interferon-gamma (IFNγ) is a cytokine with roles in immune responses as well as in tumor control. Interferon is often used in cancer treatment together with other therapies. Here we report a novel approach to enhancement of cancer cell killing by combined treatment of IFNγ with ionizing radiation.
We found that IFNγ treatment alone in HeLa cells induced phosphorylation of Chk1 in a time- and dose-dependent manner, and resulted in cell arrest. Moreover IFNγ treatment was correlated with attenuation of Chk1 as the treatment shortened protein half-life of Chk1. As Chk1 is an essential cell cycle regulator for viability after DNA damage, attenuation of Chk1 by IFNγ pre-treatment in HeLa cells resulted in increased cell death following ionizing radiation about 2-folds than ionizing radiation treatment alone whereas IFNγ treatment alone had little effect on cell death.
X-linked inhibitor of apoptosis-associated factor 1 (XAF1), an IFN-induced gene, seems to partly regulate IFNγ-induced Chk1 destabilization and radiation sensitivity because transient depletion of XAF1 by siRNA prevented IFNγ-induced Chk1 attenuation and partly protected cells from IFNγ-enhanced radiation cell killing.
Therefore the results provide a novel rationale to combine IFNγ pretreatment and DNA-damaging anti-cancer drugs such as ionizing radiation to enhance cancer cell killing.
Introduction
Interferons (IFNs) are a family of cytokines that can induce diverse biological functions, such as antiviral, antitumor and immunomodulatory activities.Citation1 IFNs can be divided by type I, II, and III. Type I IFNs consist of seven classes, mainly IFN-α and IFN-β, whereas type II IFN has only one, IFNγ.Citation1 IFNγ has important immunoregulatory functions including activation of macrophages as well as antiproliferative effects on transformed cells.Citation2
IFNs mediates antitumorigenic effects indirectly by modulating immunomodulatory responses or directly by regulating tumor cell proliferation and differentiation.Citation3 IFN-induced apoptosis involves the FADD/caspase-8 signaling pathway, activation of the caspase cascade, release of mitochondrial cytochrome c, disruption of mitochondrial membrane potential, changes in plasma membrane symmetry and DNA fragmentation.Citation4-Citation6
Many of the inhibitory effects of IFNγ appear to be mediated by IFN regulatory factor-1 (IRF-1). IFNγ induces attenuation of telomerase activity and of human telomerase reverse transcriptase (hTERT) expression via IRF-1.Citation7 IRF-1 also regulates expression of several genes involved in apoptosis, such as the pro-apoptotic protein Bax, the tumor suppressor p53, caspase-1, and PKR.Citation8
The anti-proliferative action of IFNγ depends in part on STAT1, which induces the expression of the cell cycle inhibitor, CDKN1A (p21CIP1), preventing entry into the S phase of the cell cycle.Citation9 IFNγ-induced growth suppression can be rescued by blocking Ras GTPase,Citation10 and reducing p53 or the ATM kinase protein levels.Citation11
Checkpoint kinase 1 (Chk1) is the major mediator in the activation of cell-cycle checkpoints in response to a variety of genotoxic stresses. Chk1 has a highly conserved N-terminal kinase domain and a less conserved C-terminal regulatory domain.Citation12,Citation13 Following DNA damage and subsequent replication inhibition, Chk1 is phosphorylated by ATR and ATM at specific C-terminal residues, including the highly conserved S317 and S345 sites.Citation14 Phosphorylation of S345 is commonly used as a biomarker of Chk1 activation and is associated with p53-dependent cell cycle arrest following radiation.Citation15 Moreover the S345 phosphorylation seems to change Chk1 conformation and trigger ubiquitination and degradation of Chk1.Citation12,Citation13
Since Chk1 is essential for cell viability after DNA damage, combined treatment of cancer cells with Chk1 inhibitor and various anti-cancer agents was tried to enhance cancer cell killing.Citation16,Citation17 Considering an essential role of checkpoint regulators in cell cycle regulation and viability following DNA damage, targeting of the regulators by inhibition or depletion would sensitize cancer cells to chemotherapy. Or the approach of combining DNA damage with checkpoint inhibitors can be used to selectively eliminate p53-defective cancer cells.Citation18
We explored the possibility whether IFNγ could be combined with radiation to enhance cancer cell killing. IFNγ treatment of p53-defective HeLa cells induced activation and destabilization of Chk1. Pretreatment of the cells with IFNγ and the following ionizing irradiation (IR) dramatically increased cell killing. Thus pretreatment of p53-defective HeLa cells with IFNγ attenuated Chk1 which resulted in enhanced radiation sensitivity following IR. These results may provide a rationale for combining IFNγ and DNA-damaging anti-cancer agents to enhance cancer cell killing.
Results
IFNγ induces cell cycle arrest but not cell death
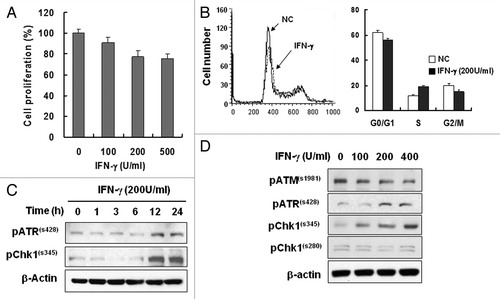
To confirm the role of IFNγ in tumor suppression, we treated HeLa cells with recombinant human IFNγ proteins and cell proliferation was measured by MTT assays or by cell counting. Cells were initially treated with IFNγ for 4 d. Proliferation decreased slightly with IFNγ treatment in a dose-dependent manner (). Distribution of cell cycle phase exhibited moderate increase of S phase of the cell cycle following IFNγ treatment, which suggests delayed S-phase transition (). Significant cell death was not observed by the doses of IFNγ treatment as shown by the cell cycle analysis ().
Figure 1. IFNγ treatment induced S-phase delay and ATR/Chk1 phosphorylation. (A) HeLa cells were treated with the indicated concentrations of IFNγ for 4 d, and cell proliferation was measured with the MTT assay. (B) Cells were treated with 200 U/ml IFNγ for 2 d, and cell cycle profiles were obtained by flow cytometry. Values are means ± standard deviation (SD) of three independent experiments. (C) IFNγ treatment activates Chk1 via ATR. HeLa cells were treated with 200 U/ml IFNγ up to 24 h, and protein expression was determined by western blot analysis. β-actin was used as a loading control. (D) Cells were treated with 0, 100, 200, or 400 U/ml of IFNγ for 12 h, and ATM, ATR and Chk1 phosphorylation was determined by western blot analysis.
IFNγ treatment activates Chk1 via ATR
Various mechanisms have been associated with IFNγ-induced tumor suppression. In addition to the direct effect of IFNγ on tumor cell killing, IFNγ inhibits cellular proliferation. Such anti-proliferative activity of IFNγ is mediated by STAT1, IRF1, p21 or p27.Citation7,Citation9,Citation19 We wished, however, to examine relatively unknown mechanism of IFNγ-induced anti-proliferative action and noted involvement of ATR and Chk1 in S checkpoint.Citation20 IFNγ treatment induced phosphorylation of ATR and Chk1 which suggests activation of ATR and Chk1 signaling following the treatment (). ATR, a kinase upstream of Chk1, was phosphorylated at serine 428 whereas Chk1 was phosphorylated at serine 345 after a 12 h IFNγ treatment. Since the phosphorylation sites are associated with activation of the kinases,Citation21,Citation22 ATR-Chk1 signaling might be associated with IFNγ-induced responses. Phosphorylation of both ATR and Chk1 increased in the presence of IFNγ in a dose-dependent manner whereas ATM phosphorylation was not increased by the conditions ( and ). Although both ATM and ATR have capacity for Chk1 activation, the result of ATR phosphorylation but not that of ATM indicates that ATR might be involved in Chk1 activation following IFNγ treatment. Therefore, IFNγ treatment induced activation of ATR-Chk1 signaling and this in turn might be involved in the IFN- γ responses as in .
IFNγ treatment destabilizes Chk1
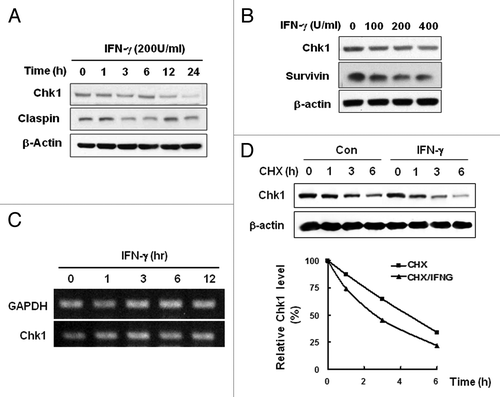
As an essential kinase for checkpoint, activity of Chk1 is regulated by phosphorylation and by protein stability.Citation21,Citation23 Moreover as the regulation of Chk1 protein stability is closely associated with its phosphorylation by which proteosomal degradation of Chk1 is triggered,Citation13 we examined Chk1 stability following IFNγ treatment. IFNγ treatment decreased Chk1 protein levels in a time-dependent manner (). Levels of Claspin, a Chk1-regulatory protein in checkpoint, were fluctuating by IFN- γ treatment during the incubation period (). Dose-dependent decrease of Chk1 protein was also observed following IFN- γ treatment (). However the treatment did not alter Chk1 transcript levels (). Thus the results suggest that transcriptional regulation is not responsible for the Chk1 decrease by IFNγ treatment. We examined Chk1 stability by measuring its half life in the presence or absence of IFN- γ (). IFN- γ treatment decreased half-life of Chk1 indicating that Chk1 protein was destabilized following IFN- γ treatment.
Figure 2. IFNγ treatment induces instability of Chk1 protein. (A) HeLa cells were treated with 200 U/ml IFNγ up to 24 h, and Chk1 expression was measured by western blot analysis. (B) Cells were treated with 0, 100, 200, or 400 U/ml of IFNγ for 12 h, and Chk1 protein levels were determined by western blot analysis. (C) Cells were treated with 200 U/ml IFNγ for 0, 1, 3, 6, or 12 h, and Chk1 mRNA levels were determined by RT-PCR. (D) Cells were treated with 100 μg/ml cyclohexamide (CHX) and cultured for the indicated times after 12 h treatment with 200 U/ml IFNγ. Chk1 protein levels were determined by western blot analysis. β-actin was used as a loading control. The figure shows representative data from three independent experiments.
IFNγ treatment increases radiation sensitivity
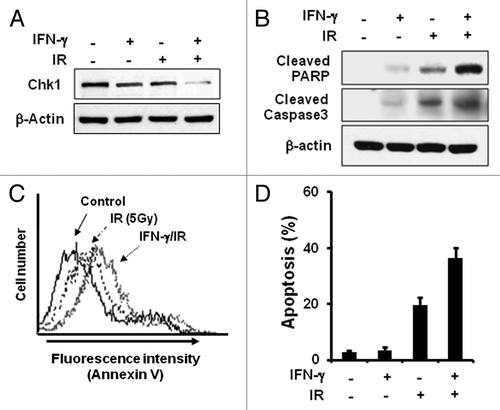
The current data may provide a novel scheme to enhance radiation sensitivity of cancer cells in that IFNγ treatment decreased Chk1, an essential regulator in cellular radiation responses. To test the idea, we treated HeLa cells with IFNγ for 12 h or with 5Gy ionizing radiation (IR) alone or together. Whereas IR or IFNγ treatment alone decreased Chk1 compared with the untreated control sample, combined treatment with IFNγ and IR significantly decreased more Chk1 protein (). As expected, the combined treatment enhanced apoptosis compared with single treatment alone as shown by increased cleavage of PARP and caspase-3 (). Moreover annexin V staining of cells showed more cell death by the combined treatment (). The combined treatment increased cancer cell killing about 2-folds than IR treatment alone (). IFNγ treatment alone had little effect on cell death. Therefore, the results show that combination of IFNγ and IR can enhance radiation sensitivity of cancer cells by downregulating Chk1 protein levels.
Figure 3. IFNγ treatment increases radiosensitivity. (A) HeLa cells were treated with 200 U/ml IFNγ for 12 h before irradiation (5Gy), and the levels of indicated proteins were determined by western blotting. (B) Cleavage of PARP and Caspase3 was measured after treatment with IFNγ and IR by western blotting. (C) Cells were treated with 200 U/ml IFNγ and IR for 2 d and stained with Annexin V-FITC and propidium iodide (PI). The fluorescence intensity of Annexin V-FITC was quantified by flow cytometry. (D) The percentage of the population undergoing apoptosis was calculated by flow cytometry with Annexin V-FITC and PI. Values are means ± SD of three independent experiments.
IFNγ-induced downregulation of Chk1 is XAF1-dependent
X-linked inhibitor of apoptosis-associated factor 1 (XAF1), an IFNγ-induced gene, is a cell cycle regulator that interacts with Chk1 in gastrointestinal cancer cells.Citation24 In order to identify which cellular factors regulate IFNγ-induced Chk1 decrease, we examined involvement of XAF1 in Chk1 destabilization. IFNγ treatment induced IRF-1 expression beginning at 1 h and XAF1 at 8 h after the treatment (). Since IRF-1 regulates XAF1 expression following IFNγ treatment,Citation25 we examined whether IRF-1 depletion has effect on IFNγ-induced XAF1 expression as well as on Chk1 protein levels. Depletion of IRF-1 by siRNA treatment eliminated IFNγ-induced XAF1 expression confirming that IRF-1 mediates IFNγ-induced XAF1 expression (). Interestingly, IFNγ-induced Chk1 attenuation was lost by IRF-1 depletion (). Whereas IFNγ treatment decreased Chk1 protein level about 30%, depletion of IRF-1 restored Chk1 level comparable to that of IFNγ-untreated control. This result suggests that IRF-1 is involved in the regulation Chk1 protein levels following IFNγ treatment.
Figure 4. XAF1 is involved in IFNγ-induced Chk1 downregulation. (A) HeLa cells were treated with 200 U/ml IFNγ for the indicated times, and IRF-1 and XAF1 protein levels were measured by western blot analysis. (B) HeLa cells were transfected with IRF-1 siRNA and treated with 200 U/ml IFNγ for 12 h. The levels of IRF1, XAF1 and Chk1 were analyzed by western blotting. (C) Cells were transfected with XAF1, or a control vector and incubated for 36 h. The level of Chk1 was analyzed by western blotting. (D) Cells were transfected with XAF1 siRNA and treated with 200 U/ml IFNγ for 12 h. The level of Chk1 was analyzed by western blotting.
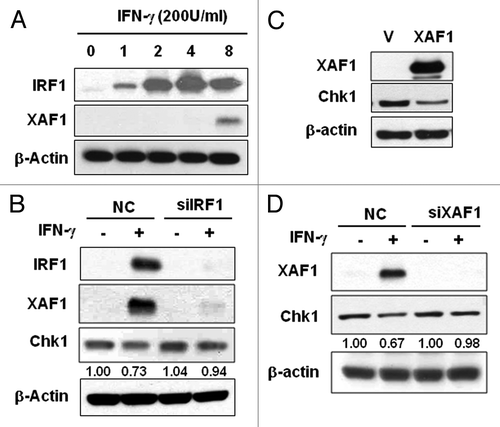
We further examined the role of XAF1 in the IFNγ-induced responses. XAF1 overexpression alone decreased expression level of Chk1 protein, which indicates involvement of XAF1 in downregulation of Chk1 (). In order to further confirm XAF1 involvement in the regulation of Chk1 protein level, we depleted XAF1 expression by siRNA and examined Chk1 expression level in the absence or presence of IFNγ treatment. Depletion of XAF1 prevented IFNγ-induced Chk1 decrease (). The abundance of Chk1 protein in XAF1-depleted cells was comparable to that of IFNγ-untreated control. These results suggest that XAF1 participates in IFNγ-induced Chk1 downregulation.
IFNγ enhances radiation sensitivity of cancer cells
In order to examine effect of XAF1 on IFNγ-induced radiosensitization, we depleted XAF1 expression by siRNA and measured cell viability following ionizing radiation. XAF1 depletion prevented Chk1 downregulation by IFNγ treatment either in the presence or absence of ionizing radiation (). Irradiation of XAF1-depleted cells following IFNγ-pretreatment resulted in more cell survival than un-depleted IFNγ-pretreated control (). The viability following irradiation of IFNγ-pretreated XAF1-depleted cells was less than that of IFNγ-untreated and XAF1-un-depleted cells. The result suggests that hitherto unknown factors as well as XAF1 are involved in the IFNγ-induced radiosensitization.
Figure 5. XAF1 is involved in IFNγ-induced radiosensitization. (A) HeLa cells were transfected with XAF1 siRNA and treated with 200 U/ml IFNγ for 12 h before irradiation (5Gy). The levels of indicated proteins were determined by western blotting. (B) Cell viability of the cells after irradiation was measured with MTT assay. Results were expressed as a percent cell survival compared with the control. Values are means ± SD of three experiments. *p < 0.05 vs. control. (C) Clonogenic cell survival assay. HeLa cells were transfected with control vector (V), XAF1 expression vector (XAF1), Chk1 expression vector (Chk1) or Chk1 siRNA (siChk1) and incubated for 36 h. Control for Chk1 siRNA is indicated as C. After each of cells reseeded and incubated for 24 h, cells were irradiated with 0, 1, 2 and 4 Gy of IR. Number of colonies were counted and expressed in comparison to 0 Gy control sample. Values are means ± SD of three independent experiments.
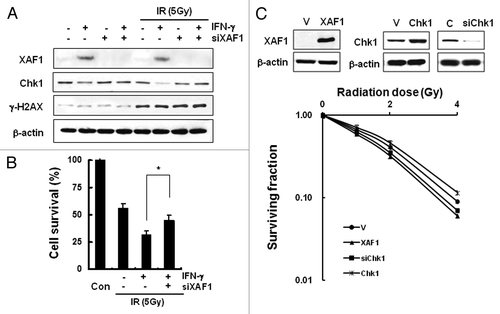
To examine the effect of Chk1 or XAF1 on colony forming ability following ionizing radiation, HeLa cells with transient overexpression of Chk1 or XAF1 as well as downregulation of Chk1 respectively were subjected to clonogenic assay after irradiation (). Chk1 overexpression increased clonogenic survival whereas XAF1 overexpression or Chk1 depletion by siRNA decreased clonogenic cell survival compared with vector-transfected control. Taken together, the data suggest that enhanced radiation sensitivity by IFNγ pre-treatment is at least partly attributable to downregulation of Chk1 and to IFNγ-induced XAF1.
To examine whether IFNγ potentiate cell death in other cancer cell types in combination with radiation, lung cancer cell lines such as A549, H460 and H1299 were treated with IFNγ. The lung cancer cell lines induced XAF1 following IFN treatment (not shown) and showed increased cell death by combination of IFNγ and IR as summarized in . It is well known that lung cancer cells become resistant to cell death by IR. Our findings imply that IFNγ treatment before ionizing radiation may enhance radiation sensitivity of lung cancer cells and provide effective measures to overcome resistance to IR.
Table 1. Cell death measurement of lung cancer cell lines following combined treatment with IFNγ and IR
Discussion
Our data demonstrated that IFNγ induced ATR/Chk1 signaling and that this could be employed to enhance radiation sensitivity by combining IFNγ and ionizing radiation.
IFNγ treatment of HeLa cells induced activation of ATR and Chk1 (). ATR and Chk1 are essential regulators of cell cycle checkpoints.Citation26 Upon DNA damaging stress, ATR and its downstream kinase Chk1 are activated. Once activated, these kinases phosphorylate various substrates initiating a signaling cascade that results in cell cycle arrest and facilitates DNA repair for cell survival. One of the cell cycle checkpoints, S-phase checkpoint, which blocks cell cycle progression into S phase, is induced by proteolytic degradation of CDC25A, which is facilitated by phosphorylation of CDC25A by activated Chk1.Citation26-Citation28 The IFNs usually arrest the cell cycle at the G1/S checkpoint.Citation29,Citation30 IFNβ or -γ induces cellular senescence through ATM and p53-dependent DNA damage signaling.Citation11,Citation31 In our data, ATM activation was not observed () and HeLa is a p53 non-functional cancer cell line. Therefore ATM and p53-dependent pathway might not be responsible for the delay in S phase.
Activation and destabilization of Chk1 by IFNγ treatment ( and ) provide a novel scheme to enhance radiation cell killing of cancer cells. Chk1 activation by phosphorylation is coupled to its degradation.Citation13 Phosphorylation of Chk1 serine 345 leads to Chk1 ubiquitination and proteasome-mediated degradation of Chk1. In our system, Chk1 phosphorylation at serine 345 residue was increased by IFNγ treatment and Chk1 stability significantly decreased ( and ). Moreover combination of IFNγ pretreatment and IR decreased further the level of Chk1 (). In light of the findings that Chk1 plays an essential role for cell cycle checkpoint and cell survival after DNA damage, diminished Chk1 level by IFNγ pretreatment might render the cells vulnerable to DNA damages caused by IR (). Such scheme which combines Chk1 inhibition (or depletion) and DNA damage has been explored to enhance cancer cell killing.Citation32 For instance Chk1 inhibitor UCN-01 was combined with topotecan or irinotecan in the clinical trials. Chk1 inhibition or destabilization might be more effective to treat advanced cancers with defective p53 because such cells lack normal G1 checkpoint and heavily rely on Chk1-regulated G2 checkpoints for survival.Citation16,Citation17 Tumor cells with Chk1 inhibition in combination with chemo- or radiotherapy enter mitosis inappropriately and ultimately undergo mitotic cell death.
The identity of Chk1-destabilizing activity is unclear from our data although they suggest involvement of XAF1. Chk1 undergoes proteasome-dependent degradation which requires its ubiquitination by the Skp1-Cul1-Fbx6 or Cul4A-DDB1 E3 ligases.Citation13,Citation23,Citation33 Those studies propose that phosphorylation of Chk1 by ATR induces conformational changes from a closed inactive form to an open active structure that renders Chk1 susceptible to degradation by ubiquitin ligases. Thus activation-induced degradation of Chk1 may serve to limit the duration of stress-induced checkpoint signaling. Our findings suggest involvement of XAF1 in IFNγ-induced destabilization of Chk1. Activated Chk1 by phosphorylation might be targeted for destruction by XAF1-associated E3 ligase(s) to limit the duration of Chk1 signaling following IFNγ treatment. It has been reported that XAF1 interacts with XIAP, an E3 ligase, and that XIAP-XAF1 complex induces downregulation of Survivin.Citation34 The report also shows that IFN-β−mediated Survivin decrease requires XAF1 induction. Because we find it similar to our findings, we speculate that IFNγ-induced XAF1 in complex with XIAP might mediate downregulation of Chk1. Interestingly, it has been reported that XIAP interacts with Chk1 and that the interaction requires N-terminal sequences of Chk1 which is not a domain for phosphorylation.Citation35 Considering XIAP’s ability to bind Chk1 as well as XAF1, we may expect XIAP-Chk1 complex in the absence of stress and then gradually increased formation of XAF1-XIAP-Chk1 complex following IFNγ treatment with Chk1 S345 phosphorylated. Phosphorylation of Chk1 might trigger conformational changes in Chk1 which might render the protein susceptible to degradation by XAF1-XIAP E3 ligase activity. Or we may speculate additional model to explain Chk1 downregulation following IFNγ treatment. Since Chk1 is degraded by Cullin-based E3 ligasesCitation13,Citation23,Citation33 under both normal and stress conditions, functional association of XAF1, XIAP or XAF1-XIAP complex with Chk1-targeting Cullin-based E3 ligases might be expected. Or XAF1-XIAP complex might downregulate Chk1 under IFNγ treatment condition, whereas Cullin-based E3 ligases might target Chk1 in other conditions. Selective depletion of respective E3 ligase components and treatment of the depleted cells with various stress conditions may elucidate the issues.
Taken together, IFNγ activated ATR-Chk1 signaling in cancer cells and destabilized Chk1. These properties of IFNγ on Chk1 could be utilized to enhance IR-induced cancer cell killing when the cells are treated with IFNγ before IR. The results may provide a new rationale to combine IFNγ and DNA-damaging anti-cancer drugs to enhance cancer cell killing.
Materials and Methods
Materials
Sequence-specific primers for Chk1 (forward, aaggccccgagtcacttc; reverse, catgtgggctgggaaaag) and GAPDH (forward, catggagaaggctggggctc; reverse, cgccagtagaggcagggatg) were obtained from Bioneer Inc. Recombinant human IFNγ was purchased from R&D System Inc. Antibodies against Chk1, XAF1, IRF-1 and Survivin were purchased from Santa Cruz Biotechnology, Inc. Antibodies against cleaved PARP, cleaved Caspase3, phospho-ATR (S428), phospho-Chk1 at serine 280 and serine 345, and phospho-ATM at serine 1981 were obtained from Cell Signaling Technology, Inc. siRNAs against IRF-1, XAF1, and ATR were purchased from Dharmacon, Inc. pCMV-XAF1 construct was obtained from Open Biosystems Inc.
Cell culture, treatment, and protein extraction
HeLa cells were cultured in DMEM medium and lung cancer cell lines A549, H460 and H1299 were done in RPMI 1640 medium supplemented with 10% fetal bovine serum. All cells were incubated in a humidified incubator of 5% CO2 at 37°C at a seeding density of 5 x 105 cells per 100-mm culture dish. For IFNγ treatment, cells were plated in 60 mm dishes and incubated for 12 h. Cells were treated with DMEM medium containing 200 U/ml IFNγ for 12 h before γ-irradiation. Cells were washed with ice-cold PBS, lysed in 50 µl ice-cold RIPA buffer (25 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.5% SDS, 1 mM Na3VO4, 5 mM NaF and 1 mM phenylmethylsulfonyl fluoride) and centrifuged at 13,600g for 15 min. Protein concentrations in the supernatants were quantified with the bicinchoninic acid (BCA) method (Pierce Biotechnology, Inc.) using bovine serum albumin as a standard, and the volumes of the supernatants were adjusted for protein concentration.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from IFN-treated cells using Trizol (Invitrogen) according to the manufacturer’s protocol and quantified by measuring absorbance at 260 nm. RNA was reverse-transcribed using 2.5 µM oligo-dT primers, 1 mM dNTPs and Superscript II reverse transcriptase (Promega Corp.), and the resulting cDNAs were amplified with Ex TaqTM HS DNA polymerase (TaKaRa Bio). GAPDH primers were used to normalize the amount of RNA in each sample. PCR products were resolved by electrophoresis on 1.5% (w/v) agarose gels stained with ethidium bromide.
Western blot analysis
Proteins (30 μg) were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes, which were blotted with specific antibodies. The proteins were visualized using an enhanced chemiluminescence detection system. The membranes were then re-probed with the anti-β-actin antibody to control for loading.
Cell proliferation and viability
Cell proliferation was measured with the MTT assay. Cells were seeded in 96-well plates at a density of 1 × 103 cells/well. After treatments, the cells were incubated with 1 mg/ml MTT (3-(4, 5-dimethylthiazol-2yl)-2, 5-diphenyltetrazolium bromide) solution for 2 h. The medium was aspirated and the resulting formazan product was solubilized with 100 μl of dimethyl sulfoxide. Viability was assessed by measuring absorbance at 570 nm with a BioRad microplate reader. Cell clonogenic survival assay was performed by following standard protocols.Citation36
Flow cytometric analysis for apoptosis and cell cycle
Apoptosis induction was analyzed by Annexin V-FITC staining (BD Biosciences), according to the manufacturer's instructions. Cells were seeded at a seeding density 3 × 105 per 60-mm dish and incubated overnight. Cells were treated with IFNγ and irradiation for 2 d and then stained with Annexin V-FITC and propidium iodide (PI) in the dark. The FITC/PI fluorescence intensity was measured using a Becton-Dickinson FACS Calibur flow cytometer. Cell cycle profiles were obtained by staining cells with PI. Cells were seeded at a seeding density 3 x 105 per 60-mm dish and incubated overnight. Cells were treated with IFNγ for 0, 1, or 2 d, then harvested, washed twice with PBS, and fixed with 70% ethanol at -20°C for 1 h. A minimum of 10,000 cells in each sample was sorted using fluorescence activated cell sorting with PI detection on a Becton-Dickinson FACS Calibur flow cytometer, and cell cycle profiles were analyzed using the Cell Quest software.
| Abbreviations | ||
| IFNγ | = | interferon gamma |
| IR | = | ionizing radiation |
| XAF1 | = | X-linked inhibitor of apoptosis-associated factor 1 |
| IRF-1 | = | interferon regulatory factor-1 |
| ATM | = | ataxia-telangectasia mutated |
| ATR | = | ataxia-telangectasia and Rad3-related protein |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by nuclear research and development program of the national research foundation of Korea funded by the Korea government.
Related Research Data
References
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004; 202:8 - 32; http://dx.doi.org/10.1111/j.0105-2896.2004.00204.x; PMID: 15546383
- Leon ML, Zuckerman SH. Gamma interferon: a central mediator in atherosclerosis. Inflamm Res 2005; 54:395 - 411; http://dx.doi.org/10.1007/s00011-005-1377-2; PMID: 16283107
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003; 8:237 - 49; http://dx.doi.org/10.1023/A:1023668705040; PMID: 12766484
- Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res 2001; 7:1821 - 31; PMID: 11410525
- Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem 2001; 276:24965 - 70; http://dx.doi.org/10.1074/jbc.M101161200; PMID: 11337497
- Ossina NK, Cannas A, Powers VC, Fitzpatrick PA, Knight JD, Gilbert JR, et al. Interferon-gamma modulates a p53-independent apoptotic pathway and apoptosis-related gene expression. J Biol Chem 1997; 272:16351 - 7; http://dx.doi.org/10.1074/jbc.272.26.16351; PMID: 9195941
- Lee SH, Kim JW, Lee HW, Cho YS, Oh SH, Kim YJ, et al. Interferon regulatory factor-1 (IRF-1) is a mediator for interferon-gamma induced attenuation of telomerase activity and human telomerase reverse transcriptase (hTERT) expression. Oncogene 2003; 22:381 - 91; http://dx.doi.org/10.1038/sj.onc.1206133; PMID: 12545159
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004; 75:163 - 89; http://dx.doi.org/10.1189/jlb.0603252; PMID: 14525967
- Chin YE, Kitagawa M, Su WC, You ZH, Iwamoto Y, Fu XY. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996; 272:719 - 22; http://dx.doi.org/10.1126/science.272.5262.719; PMID: 8614832
- Prasanna SJ, Saha B, Nandi D. Involvement of oxidative and nitrosative stress in modulation of gene expression and functional responses by IFNgamma. Int Immunol 2007; 19:867 - 79; http://dx.doi.org/10.1093/intimm/dxm058; PMID: 17606979
- Kim KS, Kang KW, Seu YB, Baek SH, Kim JR. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev 2009; 130:179 - 88; http://dx.doi.org/10.1016/j.mad.2008.11.004; PMID: 19071156
- Tapia-Alveal C, Calonge TM, O’Connell MJ. Regulation of chk1. Cell Div 2009; 4:8; http://dx.doi.org/10.1186/1747-1028-4-8; PMID: 19400965
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell 2005; 19:607 - 18; http://dx.doi.org/10.1016/j.molcel.2005.07.019; PMID: 16137618
- Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A 2008; 105:20752 - 7; http://dx.doi.org/10.1073/pnas.0806917106; PMID: 19091954
- Tian H, Faje AT, Lee SL, Jorgensen TJ. Radiation-induced phosphorylation of Chk1 at S345 is associated with p53-dependent cell cycle arrest pathways. Neoplasia 2002; 4:171 - 80; http://dx.doi.org/10.1038/sj.neo.7900219; PMID: 11896572
- Suganuma M, Kawabe T, Hori H, Funabiki T, Okamoto T. Sensitization of cancer cells to DNA damage-induced cell death by specific cell cycle G2 checkpoint abrogation. Cancer Res 1999; 59:5887 - 91; PMID: 10606229
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004; 3:513 - 9; PMID: 15078995
- Mukhopadhyay UK, Senderowicz AM, Ferbeyre G. RNA silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res 2005; 65:2872 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-04-2502; PMID: 15805289
- Hobeika AC, Etienne W, Torres BA, Johnson HM, Subramaniam PS. IFN-gamma induction of p21(WAF1) is required for cell cycle inhibition and suppression of apoptosis. J Interferon Cytokine Res 1999; 19:1351 - 61; http://dx.doi.org/10.1089/107999099312812; PMID: 10638704
- Ashwell S, Zabludoff S. DNA damage detection and repair pathways--recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res 2008; 14:4032 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-07-5138; PMID: 18593978
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Chandris P, Giannouli CC, Panayotou G, Kletsas D. Compromise in mRNA processing machinery in senescent human fibroblasts: implications for a novel potential role of Phospho-ATR (ser428). Biogerontology 2010; 11:421 - 36; http://dx.doi.org/10.1007/s10522-010-9261-z; PMID: 20084458
- Leung-Pineda V, Huh J, Piwnica-Worms H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res 2009; 69:2630 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-08-3382; PMID: 19276361
- Wang J, Gu Q, Li M, Zhang W, Yang M, Zou B, et al. Identification of XAF1 as a novel cell cycle regulator through modulating G(2)/M checkpoint and interaction with checkpoint kinase 1 in gastrointestinal cancer. Carcinogenesis 2009; 30:1507 - 16; http://dx.doi.org/10.1093/carcin/bgp155; PMID: 19628579
- Wang J, Zhang W, Zhang Y, Chen Y, Zou B, Jiang B, et al. c-Jun N-terminal kinase (JNK1) upregulates XIAP-associated factor 1 (XAF1) through interferon regulatory factor 1 (IRF-1) in gastrointestinal cancer. Carcinogenesis 2009; 30:222 - 9; http://dx.doi.org/10.1093/carcin/bgn271; PMID: 19056926
- Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007; 6:953 - 66; http://dx.doi.org/10.1016/j.dnarep.2007.02.015; PMID: 17531546
- Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem 2003; 278:21767 - 73; http://dx.doi.org/10.1074/jbc.M300229200; PMID: 12676925
- Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3:247 - 58; http://dx.doi.org/10.1016/S1535-6108(03)00048-5; PMID: 12676583
- Balkwill F, Taylor-Papadimitriou J. Interferon affects both G1 and S+G2 in cells stimulated from quiescence to growth. Nature 1978; 274:798 - 800; http://dx.doi.org/10.1038/274798a0; PMID: 683317
- Lundblad D, Lundgren E. Block of glioma cell line in S by interferon. Int J Cancer 1981; 27:749 - 54; http://dx.doi.org/10.1002/ijc.2910270604; PMID: 6169666
- Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell 2006; 17:1583 - 92; http://dx.doi.org/10.1091/mbc.E05-09-0858; PMID: 16436515
- Choudhury A, Cuddihy A, Bristow RG. Radiation and new molecular agents part I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Semin Radiat Oncol 2006; 16:51 - 8; http://dx.doi.org/10.1016/j.semradonc.2005.08.007; PMID: 16378907
- Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, et al. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell 2009; 35:442 - 53; http://dx.doi.org/10.1016/j.molcel.2009.06.030; PMID: 19716789
- Arora V, Cheung HH, Plenchette S, Micali OC, Liston P, Korneluk RG. Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J Biol Chem 2007; 282:26202 - 9; http://dx.doi.org/10.1074/jbc.M700776200; PMID: 17613533
- Galvan V, Kurakin AV, Bredesen DE. Interaction of checkpoint kinase 1 and the X-linked inhibitor of apoptosis during mitosis. FEBS Lett 2004; 558:57 - 62; http://dx.doi.org/10.1016/S0014-5793(03)01488-1; PMID: 14759516
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1:2315 - 9; http://dx.doi.org/10.1038/nprot.2006.339; PMID: 17406473