Abstract
The use of combinations of chemotherapy and natural products has recently emerged as a new method of cancer therapy, relying on the capacity of certain natural compounds to trigger cell death with low doses of chemotherapeutic agents and few side effects. The current study aims to evaluate the modulatory effects of curcumin (CUR), Nigella sativa (NS) and taurine on etoposide (ETP) cytotoxicity in a panel of cancer cell lines and to identify their underlying mechanisms.
CUR alone showed potent antitumor activity, but surprisingly, its interaction with ETP was antagonistic in four out of five cancer cell lines. Neither taurine nor Nigella sativa affect the sensitivity of cancer cells to ETP. Examination of the DNA damage response machinery (DDR) showed that both ETP and CUR elicited DNA double-strand breaks (DSB) and evoked γ-H2AX foci formation at doses as low as 1 µg/ml. Cell cycle analysis revealed S phase arrest after ETP or CUR application, whereas co-treatment with ETP and CUR led to increased arrest of the cell cycle in S phase (MCF-7 cells) or the accumulation of cells in G2/M phases (HCT116, and HeLa cells). Furthermore, cotreatment with ETP and CUR resulted in modulation of the level of DNA damage induction and repair compared with either agent alone. Electron microscopic examination demonstrated that different modalities of cell death occurred with each treatment. CUR alone induced autophagy, apoptosis and necrosis, whereas ETP alone or in combination with CUR led to apoptosis and necrosis.
Conclusions: Cotreatment with ETP and CUR resulted in an antagonistic interaction. This antagonism is related, in part, to the enhanced arrest of tumor cells in both S and G2/M phases, which prevents the cells from entering M-phase with damaged DNA and, consequently, prevents cell death from occurring. This arrest allows time for the cells to repair DNA damage so that cell cycle -arrested cells can eventually resume cell cycle progression and continue their physiological program.
Introduction
In cancer therapy, the phenomenon of chemotherapy resistance has been the focus of intensive research with the aim of understanding its mechanisms and finding novel therapies to overcome it.Citation1 One of the strategies that are used to increase the sensitization of tumor cells to chemotherapeutic agents is combination therapy using natural products and chemotherapy.Citation2
In the last few decades, natural products have gained increasing importance as a source of potential anticancer agents. The use of natural agents or dietary factors possessing anticancer properties together with other chemotherapeutic drugs at lower doses is a novel approach to reduce the side effects of anticancer agents.Citation3
In this study, we investigated the combinatorial effect of one chemotherapeutic drug and three natural compounds. Curcumin (CUR), the major yellow pigment extracted from turmeric (Curcuma longa) and commonly used as a flavouring or food additive, exhibits potent anticancer properties.Citation4 Curcumin’s anticancer properties are attributed mainly to its ability to activate cell death in a wide variety of tumor cells with low toxicity to normal cells.Citation5 Nigella sativa (NS), or black cumin seed, is an herb used in traditional medicine for many centuries in Middle Eastern and Asian countries to treat a broad array of diseases. It has been shown to have antineoplastic activities in different types of cancer.Citation6 The third natural compound is taurine, a sulfur-containing amino acid that is manufactured in the body from the amino acids, methionine and cysteine. Taurine has been suggested as a potential treatment for epilepsy, cardiovascular disease, diabetes, alcoholism, cystic fibrosis and Alzheimer’s disease.Citation7 ().
Figure 1. (A)(a) Chemical structures of compounds used in this study. (b) Whole plant extracts of Nigella sativa were used in this study. (B) Cytotoxic effect of ETP alone (■) or in the presence of (a) NS (▲) or (b) taurine (▲) on MCF-7, HepG2, U251, HeLa and HCT116 cell lines. Tumor cell lines were treated with different concentrations of ETP ± 50 µg/ml NS or ± 100 µg/ml taurine. Cell counts were performed after 48 h of exposure to drugs. Each point is the mean ± SEM of three separate experiments.
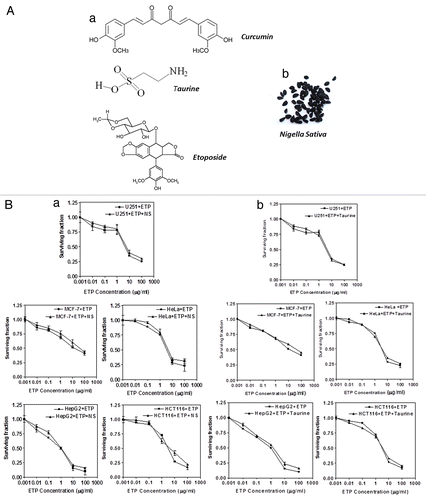
Etoposide (ETP) is a potent inducer of DNA double-strand breaks (DSBs) via the inhibition of the enzyme, topoisomerase II. It is used as an effective form of chemotherapy for malignancies such as Ewing's sarcoma, lung cancer, testicular cancer, lymphoma, non-lymphocytic leukemia, and glioblastoma multiforme.Citation8 Acquired ETP resistance remains an important barrier to effective treatment. Combining the most active chemotherapeutic drugs with natural agents that target specific pathways is a powerful approach to cancer treatment and may counteract the many ways that human cancer cells can become drug resistant.Citation9
The current study was designed to examine the interaction between ETP and several natural agents, aiming to sensitize tumor cells to ETP and consequently decrease its dose and side effects. We found that curcumin had a potent antiproliferative effect and induced three types of programmed cell death (autophagy, apoptosis and necrosis) in a panel of human tumor cell lines. We investigated the type of DNA lesions formed by CUR in more detail and found that doses as low as 10µg/ ml produced DNA double-strand breaks (DSBs) that led to the arrest of the cell cycle in S- or G2/M- phase. We further studied the effects of cotreatment with ETP and CUR, which showed an antagonistic interaction in four out of five cancer cell lines, blocking the cells in S-phase or arresting the cell cycle in both S and G2/M phases. In addition, combined treatment with ETP and CUR, modulated DNA damage induction and repair compared with either drug alone.
Results
Cytotoxic effects of ETP alone and in combination with NS and Taurine in human cancer cells
Cell viability was expressed as the percentage of surviving, treated cells relative to untreated control cells. The sulphorhodamine B (SRB) assay revealed that incubation of MCF-7, HepG2, U251, HCT116 and HeLa cell lines with different concentrations of ETP (0.01–100 µg/ml) led to a dose-dependent reduction in cell survival. Neither taurine nor NS alone reduced the survival of any of the tested cell lines (data not shown). The simultaneous addition of taurine or NS with ETP to the five tumor cell lines did not alter their sensitivity to ETP ().
Antiproliferative and cytotoxic effects of etoposide and curcumin, alone and in combination
MCF-7, HepG2, HCT116, HeLa and U251 cells were treated with CUR (0.1–100 µg/ml) for 48 h. As shown in (), CUR significantly reduced the proliferation of all cell lines in a concentration-dependent manner, although to different extents. Overall, 10μg/ml CUR was chosen for the combination experiments. It is interesting to note that cotreatment with different concentrations of ETP and 10 μg/ml CUR was found to have an antagonistic interaction. The treatment with 10 μg/ml CUR alone was found to reduce the survival of the treated cells to about 5–20% of the control cells (except for the U251 cells in which CUR reduced the survival only to 60% of untreated control cells). The combination of CUR (10 μg/ml) with the highest concentration of ETP (10 μg/ml) significantly decreased cytotoxicity, with 20–50% of treated cells surviving, which clearly reflects the antagonistic interaction between both agents.
Figure 2. Cell survival after treatment of tumor cell lines with ETP and/or CUR. MCF-7, HepG2, HCT116, HeLa and U215 cells were cultured in micro-well plates. Twenty-four hours later, the cells were treated with different concentrations of ETP and/or CUR. After 48 h of exposure to the drugs, the cells were fixed, stained with Sulphorhodamine-B (SRB) and the optical density was measured spectrophotometrically at 564 nm. Survival fraction = OD564 of drug treated sample/OD564 of untreated control sample. (a) Statistical significance compared with ETP alone using an unpaired t-test, p < 0.01. It should be noted that, although the effect of the combination may be significantly high er than ETP alone but it is still lower than the sum of the effects of either agent alone which indicates antagonism
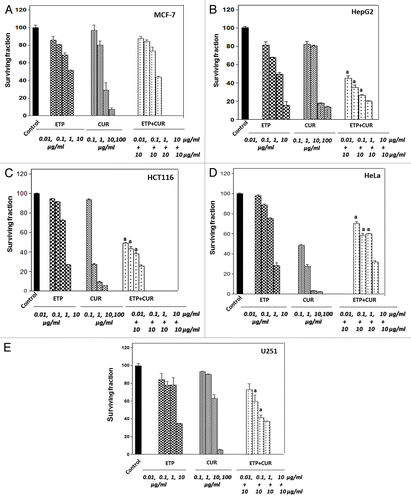
To verify the type of interaction between ETP and CUR, isobologram analysis was conducted, which revealed that the interaction was antagonistic in the MCF-7, HepG2, HCT116 and HeLa cell lines with an interaction index (I) of 2.5, 3.3, 67 and 19, respectively, whereas in U251 cells both agents (ETP + CUR) showed a synergistic interaction (I = 0.59) ().
Table 1. Effect of CUR on ETP cytotoxicity in MCF-7, HepG2, HeLa, HCT116 and U251 cells
DNA damage induction and repair after treatment with ETP and curcumin, alone and in combination
To investigate the molecular mechanisms underlying the antagonistic interaction between ETP and CUR, we first studied the type and the amount of DNA damage induced by ETP and/or CUR and their repair. ETP has been shown to induce double-strand breaks (DSBs) by stabilizing the cleavable complex between DNA and topoisomerase II,Citation16,Citation17 whereas the type of DNA damage induced by CUR is not clear. Here we used MCF-7, HepG2, HCT116 and HeLa cell lines to look for CUR-induced DNA damage. γ-H2AX foci are formed upon DNA damage and are often taken as a marker of DNA DSBs.Citation18 We found that doses as low as 10µg/ml CUR could induce γ-H2AX foci (). It is not clear exactly which type of DNA damage induces γ-H2AX foci, so we used constant field gel electrophoresis (CFGE) to look directly at DSB formation.Citation14 By conventional ethidium bromide staining, DSBs were detectable after treatment with 1–100 µg/ml CUR ().
Figure 3. Formation of γ-H2AX foci after treatment with ETP and/or CUR in HeLa cells. (A) An example of γ-H2AX foci (white) and DNA (green) staining with and without treatment. (B) Quantification of γ-H2AX foci, MCF-7, HepG2, HCT116 and HeLa Cells were treated with ETP (1 µg/ml) and/or CUR (10 µg/ml) for 24 h and subjected to immunofluorescence detection of γ-H2AX foci assay. Results are the average and standard deviation of three repeats; on each occasion at least 50 cells were counted for each condition. (C) HeLa cells were treated with 10µg/ml CUR for 48 h, and morphological changes were observed using an inverted microscope (100×). (a) Significantly different from ETP alone at p < 0.05. It should be noted that, although the effect of the combination may be significantly high er than ETP alone but it is still lower than the sum of the effects of either agent alone which indicates antagonism
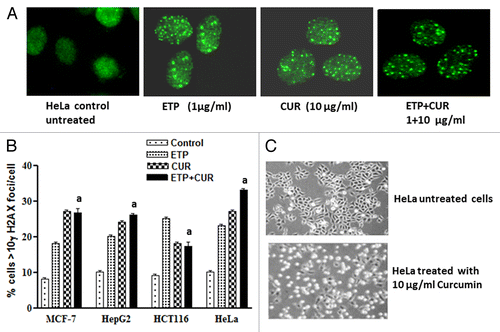
Figure 4. Induction and repair of DNA double-strand breaks (DSB) induced by ETP and/or CUR in human tumor cell lines. (A) Ethidium bromide staining of DNA double-strand breaks visualized with constant field gel electrophoresis (CFGE) after 48 h treatment with increasing doses of CUR in HeLa cells. (B) Induction of DSBs measured by CFGE. MCF-7, HepG2, HCT116 and HeLa cells were treated with ETP (1 µg/ml) and/or CUR (10–50 µg/ml) for 48 h, immediately followed by the measurement of DSB by CFGE. Constant field gel electrophoresis was run at 0.6 V cm−1 for 30 h. The fluorescence intensity of each band in each lane was recorded with a gel documentation system and video camera. (C) The band intensities in ethidium bromide-stained gels were quantified, and the fraction of DNA released (FDR) at each drug concentration was calculated. The FDR released into the gel, corresponding to fragmented DNA, was calculated according to the following equation: FDR = FDR rel / (FDR plug +FDR rel). FDR rel: the fraction of DNA released outside the well. FDR plug: the fraction of DNA remaining in the well. Data represent the results of three independent experiments. (a) Significantly different from ETP alone at p < 0.05.
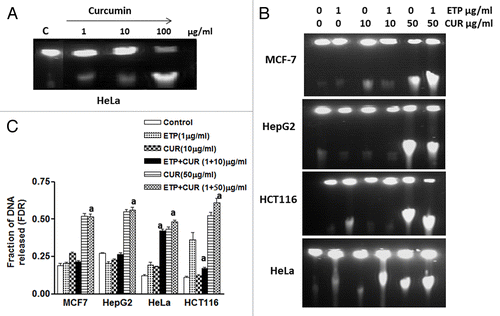
shows the dose-dependent separation of ETP- and/or CUR-induced DNA fragments in MCF7, HepG2, HCT116 and HeLa cells using CFGE. All released DNA fragments were in the form of a single well-defined band that separated after treatment with ETP and/or CUR. The fraction of DNA released (FDR), as determined by CFGE for all tumor cell lines was found to increase with treatment dose (). Although clear differences in the number of DSB between the different cell lines exist; however, no significant increase in the induction of DSB was detected after treatment of the tumor cell lines with the combined treatment compared with either drug alone. One possibility is that the repair of DSBs in the tested cell lines was enhanced after cotreatment with ETP and CUR. Because we incubated the cells at 37°C, both induction and repair of DSB occurred concurrently.
Evaluation of cell cycle regulation after treatment with ETP and curcumin, alone and in combination
To gain further insight into the mechanism(s) of antagonism between CUR and ETP, MCF-7, HepG2, HCT116 and HeLa cells were treated with 1 µg/ml ETP and/or 10 µg/ml CUR for 48 h and analyzed for cell cycle progression. As shown in and , as compared with vehicle-treated cells, 1 µg/ml ETP treatment caused significant arrest of cells in S phase. In HCT116 cells, the accumulation of cells in S phase increased from approximately 9–34%, at the expense of G0/G1 phase cells. The same finding was observed in MCF-7, HepG2 and HeLa cells where 1 µg/ml ETP induced 18.19, 22.23 and 29.64% of cells to accumulate in S phase as compared with the controls, which had 2.6, 15.49 and 21.91% of cells in S phase, respectively.
Figure 5. Cell cycle distribution analysis of the human tumor cell lines treated with ETP and/or CUR. (A) MCF-7, (B) HepG2, (C) HCT116 and (D) HeLa cells were treated with ETP (1 µg/ml) and/or CUR (10 µg/ml) for 48 h. The cells (control and treated) were collected by trypsinisation, washed, incubated with RNase and treated with propidium iodide; the DNA content was then measured by flow cytometry. ETP or CUR treatment blocked the cells at the intra-S-phase checkpoint, whereas combined treatment (ETP + CUR) arrested the tumor cells in the S and/or G2/M phases. The percentage of cells in the subG1, G1, S and G2/M phases are detailed in graphs on the right panel and in . Data refer to three experiments.
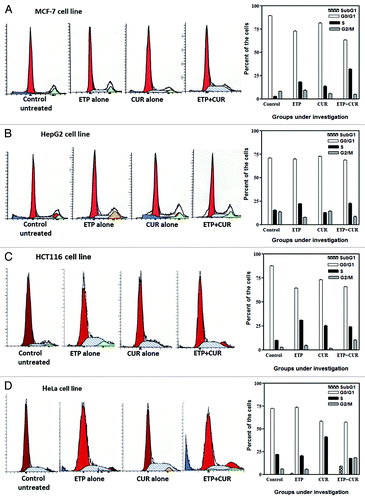
Table 2. Cell cycle modifications induced by ETP, CUR and their combination in MCF-7, HepG2, HCT116 and HeLa cells after 48 h of treatment
In a similar manner, analysis of cell cycle distribution after 10 µg/ml CUR treatment presented evidence of a preferential arrest of MCF-7, HCT116 and HeLa cancer cells in S phase. On the other hand, HepG2 cells did not show any change in the S phase fraction, as compared with control cells (, ).
Interestingly, DNA histograms from cells incubated with a combination of ETP (1 µg/ml) and CUR (10 µg/ml) for 48 h revealed a significant change in cell cycle distribution compared with ETP or CUR alone. In the MCF-7 cell line, the fraction of cells in S phase increased from 2.60 ± 0.01% to 31.98 ± 0.5%. In addition to enhanced arrest in S phase, combined treatment also induced accumulation of HCT116 and HeLa cells in the G2/M phase of the cell cycle.
Effects of ETP, CUR and their cotreatment on the morphology of human cancer cells and the induction of cell death
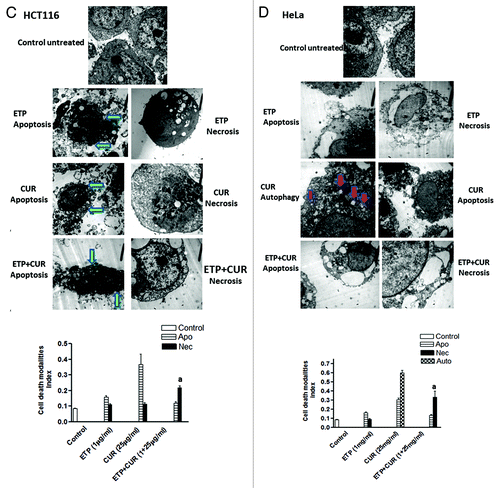
To assess the morphological changes induced by ETP and/or CUR that may reflect the mode of cell death, MCF-7, HepG2, HCT116 and HeLa cells were cultured for 24 h in the presence of the DMSO vehicle alone, 1 µg/ml ETP and/or 25 µg/ml CUR. Electron microscopic analysis showed that ETP treatment induced some cellular morphological changes consistent with apoptosis (programmed cell death type I), including chromatin condensation, cell shrinkage, cytoplasmic blebbing and fragmentation, and necrosis (programmed cell death type 3), evidenced by swelling and large clear vesicles with ruptured plasma membranes (). On the other hand, CUR treatment induced morphological changes consistent with apoptosis, necrosis and autophagy (programmed cell death type II), manifested by the appearance of numerous membrane-bound vesicles (autophagic vacuoles) that occupied most of the cytoplasmic space and frequently contained electron-dense materials from cytoplasmic fragments and organelles. These autophagic vacuoles were markedly increased in HepG2 and HeLa cells treated with 25µg/ml CUR but not in the control cells (). Furthermore, cotreatment with ETP and CUR led to manifestations of apoptosis and necrosis (Fig. 6). Altogether, programmed cell death (PCD), categorised as apoptotic, autophagic or necrotic, was detected after treatment of tumor cell lines with ETP or CUR alone and in combination.
Figure 6A and B. Representative electron micrographs of (A) MCF-7, (B) HepG2, (C) HCT116 and (D) HeLa cells treated with DMSO (control) or ETP (1 µg/ml) and/or CUR (25 µg/ml) for 24 h. Different modalities of programmed cell death (PCD) were observed after treatment with ETP and CUR, alone and in combination. CUR induced autophagy, apoptosis and necrosis in tumor cell lines. HepG2 and HeLa cells showed autophagic vesicles after treatment with 25 µg/ml CUR. Arrows indicate autophagic vacuoles. MCF-7 and HCT116 cells showed signs of apoptosis, including chromatin condensation, cell shrinkage, cytoplasmic blebbing and fragmentation. ETP alone induced apoptosis and necrosis. Cotreatment with ETP and CUR caused apoptosis and necrosis. Bars represent mean ± SD in each treatment group. The mean density for autophagic, apoptotic and necrotic was determined by counting the number of cells showing these modes of cell death in both ETP and /or CUR-treated cells and control cells in ten low power fields (2000X) of each section in a blinded fashion. (a) Significantly different from ETP alone at p < 0.05. It should be noted that, although the effect of the combination may be significantly high er than ETP alone but it is still lower than the sum of the effects of either agent alone which indicates antagonism.

Figure 6C and D. See previous page for figure legend.
Discussion
A minority of cancer patients are cured, partly due to the development of increased resistance to conventional anticancer agents in different types of cancer.Citation19,Citation20 Accordingly, intensive research has been initiated to explore the feasibility of using natural products as effective chemotherapeutic agents for treating cancer.Citation21,Citation22 Another potential application of natural products is their use as an adjunct to other commonly used chemotherapeutic agents. Their co-administration would be preferable to chemotherapy alone, as this would involve the use of reduced doses and, consequently, reduced toxicity.Citation23,Citation24 Therefore, the purpose of the current study is to investigate the modulatory effects of taurine, NS, and CUR on ETP cytotoxicity in a panel of human cancer cell lines.
We first studied the sensitivity of MCF-7, HepG2, U251, HeLa and HCT116 cells to the cytotoxic effect of ETP and its modulation by taurine, NS and CUR. Dose survival curves showed that cotreatment of cancer cells with ETP plus taurine or NS did not enhance ETP cytotoxicity. This is the first demonstration that taurine, which is used as a potent treatment for epilepsy, Alzheimer disease, and cardiovascular diseases, is not an effective cancer therapy, alone or in combination with other anticancer drugs. Our results with NS contradict many previous studiesCitation25-Citation28 that found that the antitumor activity of different types of chemotherapeutic agents is augmented by thymoquinone (the pure, bioactive constituent of the volatile oil of black seed Nigella sativa) in a panel of cancer cells. This finding could be explained by the fact that we did not use pure thymoquinone, but instead the whole plant extract, which may contain a low concentration of the bioactive component along with a wide range of potent antioxidants (free radical scavengers). This extract could lead to an antioxidant function rather than antiproliferative effects, due to the protection of DNA from genotoxic insults.Citation29,Citation30 In addition, we may speculate that thymoquinone may have dual functions. Thymoquinone may be an antioxidant at low concentrations, but have antiproliferative effects at high concentrations.
CUR, the third natural product that was used in this study, was found to reduce the survival of MCF-7, HepG2, HCT116 and HeLa cells alone, which is in accordance with previous studies.Citation31,Citation32 We next studied the modulatory effect of CUR on the cytotoxicity of ETP on tumor cell lines. Unexpectedly, the interaction of ETP and CUR was antagonistic. This finding, although confirms some previous investigations,Citation33 contradicts with many others which reported curcumin-induced enhancement of the cytotoxic effects of different anticancer drugs on variable types of cancer cells.Citation34-Citation38
Induction of DNA DSB as shown in the present study by constant-field gel electrophoresis and formation of γ-H2AX foci is thought to be the initiator of curcumin-induced reduction in cell viability. A similar finding was reported previously by some investigators who reported that DNA damage induced by topoisomerase II poisoning is a possible mechanism by which curcumin initiates apoptosis.Citation39,Citation40
The fact that CUR induces its effects through its action on topoisomerase II enzymeCitation39-Citation41 which is the target of etoposide may partly explain the antagonistic interaction between CUR and ETP. Having the same target (ie topoisomeraseII) both agents may compete together and partially decreasing the effect of one another. This explanation can be supported by the data presented in and where the combined treatment with ETP + CUR resulted in a number of DSB that is less than the sum of the number of DSB induced by both agents when each one was given alone which indicates antagonism.
To counter DNA damage (DSBs) induced by ETP and/or CUR, cells have evolved mechanisms, collectively termed the DNA-damage response (DDR), to detect DNA lesions, signal their presence and promote their repair.Citation42-Citation44 To gain further insight into the antagonistic effects that resulted from cotreatment with ETP and CUR in the examined cell lines, we tested several different mechanisms that have an essential role in resistance to genotoxic stresses: Cell cycle arrest, DNA repair and cell death. Analysis of DNA ploidy by flow cytometry revealed that treatment with CUR alone, like ETP, accumulated the cells in S phase and arrested the cells in either S or G2/M phases, when combined with ETP. This arrest increases the time available for the cells to repair damaged DNA before replication or mitosis ensues, and the increased accumulation of cells in S and G2/M phases as well as increased DNA repair may explain the antagonism phenomenon. This is in line with previous investigation showing that curcumin significantly induces DNA damage and mediates S and G2/M phase arrestCitation45 and enhances DNA repair by modulating the expression of DNA repair proteins like PARP-1, XRCC1, XRCC4, DNA ligaseIII, DNA-PKcs and DNA ligase IV.Citation46 In addition, our results are consistent with previous studies that attribute the resistance of MCF7/DOX cells to doxorubicin, compared with MCF7/WT cells, to the G1/S and G2/M phase arrest seen in MCF7/DOX, but not in MCF7/WT cells.Citation47 Moreover, a recent review by Jackson and BartekCitation48 confirmed that the major biological function of DNA damage checkpoints is to allow time to repair the damage so that check point-arrested cells can eventually resume cell cycle progression and continue their physiological program. It should, however, be mentioned that, unlike ionizing radiation, it is not possible to separate DNA damage induction and repair processes upon treatment with anticancer drugs because both events occur concomitantly during the period of incubation with the anticancer drugs. Therefore, it is not clear whether decreased induction or increased repair of DSB or both is responsible for the antagonism between ETP and CUR.
Cancer cells can die via different mechanisms, and this cell death is accompanied by distinct morphological changes, depending on the precise cause of death.Citation49 Electron microscopic examinations were performed and morphometric analysis revealed different modalities of cell death, which can be classified according to phenomenological and ultrastructural changes into type 1, 2 and 3 cell deaths, corresponding to apoptosis, autophagic cell death and necrosis, respectively.Citation50,Citation51 In the present study, treatment of cancer cells with CUR alone led to autophagy, apoptosis and necrosis. These results agree with those of Aoki et al.Citation52 and Saiki et al.Citation53 who found that curcumin-induced autophagy was associated with inhibition of the Akt pathway and activation of ERK1/2, whereas inhibition of the ERK1/2 pathway inhibited curcumin-induced autophagy and induced apoptosis in malignant gliomas and HeLa cells. In addition, treatment with ETP alone or as a cotreatment with CUR induced apoptosis and necrosis. Our morphometric analysis results raise an important question regarding whether different forms of programmed cell death occur independently in parallel pathways or represent successive events in a cell death cascade. Our results are consistent with recent studies reporting that autophagy, apoptosis and necrosis are closely intertwined processes. Cells manifest autophagy before they undergo apoptosis or necrosis, and apoptosis is generally followed by secondary necrosis.Citation50,Citation54
In conclusion, the interaction of CUR with ETP is antagonistic which raises a question about the clinical application of this combination. Cell cycle arrest, DNA damage induction and repair and similarity of the target (ie the topoisomerase II) are possible mechanisms of this antagonistic interaction. In addition, different modalities of programmed cell death involving apoptotic, autophagic, and necrotic cell death could be induced by ETP and/or CUR.
Materials and methods
Cell lines and culture conditions
The following human cancer cell lines were used in this study: breast carcinoma (MCF-7), cervix carcinoma (HeLa), colon carcinoma (HCT116), liver carcinoma (HepG2), and glioblastoma (U251) cells. The cell lines, frozen in liquid nitrogen, were obtained from Kranken House Eppendorf, Hamburg University, Germany and the American Type Culture Collection. The tumor cell lines were maintained in the National Cancer Institute, Cairo, Egypt by serial subculturing. The cells were grown as monolayers in RPMI-1640 or DMEM medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 2 mg/ml streptomycin. Cells from each cell line were divided into four treatment groups. The first group served as the control. The second and third groups of cells were treated with either ETP or CUR alone while the last group was treated with a combination of ETP and CUR.
Drugs and chemicals
Etoposide [Calbiochem, 2 mg/ml in dimethyl sulphoxide (DMSO)] was serially diluted with medium and used at concentrations from 1 µg/ml to 1000 µg/ml. The final DMSO concentration was 0.02% or less.
Curcumin (diferuloylmethane) with purity greater than 98% was purchased from Sigma, Germany and prepared as a stock solution at 50 mg/ml in dimethyl sulfoxide. The stock solution was further diluted in cell culture medium and used at concentrations from 1–1,000 µg/ml.
Nigella sativa seeds were purchased from a local market in Cairo, Egypt. Air-dried seeds of Nigella sativa were powdered in a mixer, and an extraction was performed with methanol. The extract was concentrated by distilling off the solvent and then evaporated to dryness using a vacuum drier. Dried methanolic extract was dissolved in (DMSO) with a concentration of 0.02% or less before the experiments.
Taurine was purchased from Fluka (Germany). Taurine was freshly dissolved in sterile water for injection to yield a stock solution of 50 mg/ml, then serially diluted with RPMI-1640 or DMEM medium and used at concentrations from 1–1,000 µg/ml.
Chemosensitivity assay
Cytotoxicity was determined using the Sulphorhodamine-B (SRB) method, as previously described by El-awady et al.Citation10 Cells were seeded in 96-well microtiter plates at a concentration of 5 × 104–105 cells/well in RPMI-1640 or DMEM supplemented medium. After 24 h, cells from each cell line were incubated for 48 h with 0.01–100 µg/ml etoposide (3 wells for each concentration). After 48 h of treatment, the cells were fixed with 50% trichloroacetic acid for 1 h at 4οC. Wells were washed five times with water, stained for 30 min at room temperature with 0.4% SRB dissolved in 1% acetic acid and then washed four times with 1% acetic acid. The plates were air-dried, and the dye was solubilised with 100 ml/well of 10 mM tris base (pH 10.5) for 5 min on a shaker at 1,600 rpm. The optical density (OD) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (Meter tech. S960, USA). The IC50 values were calculated using sigmoidal concentration–response curve fitting models (Graph Pad, Prizm software).
Evaluation of drug interactions
Dose-response curves for curcumin alone in each cell line were first generated. The extent of the effect of the combined treatment was analyzed applying the isobologram equation,Citation11 I = d1/D1 + d2/D2, where d1 and d2 are the respective concentrations of ETP and CUR used in the combination required to produce a fixed level of inhibition, IC50 and D1 and D2 are the concentrations of each treatment alone that are able to produce the same magnitude of effect (50% inhibition of cell growth). If I (interaction index) is less than 1, the effect of combination is synergistic, whereas if I = 1 or I > 1 the effect is additive or antagonistic, respectively.
Cell cycle distribution analysis
The analysis was performed according to the method described by El-awady et al.Citation12 Cells were plated at a density of 5 × 106 cells/ml in supplemented medium. After 24 h, the medium was replaced with fresh medium containing ETP (1 µg/ml) and/or CUR (10 µg/ml), except for the control cells, and all flasks were incubated at 37°C. The cells were harvested after 48 h and washed twice with phosphate buffered saline (PBS), then fixed with 70% (v/v) ethanol. The sample was concentrated by removing ethanol and resuspended in 500 µl PBS + 500 µl RNase solution (2.7 mg/ml). The mixture was incubated at 37°C for 30 min. Then, 50 µl propidium iodide solution (1 mg/ml) was added and flow cytometric DNA ploidy analysis was performed by acquiring a minimum of 200,000 nuclei using FACScan (Becton Dickenson). Cell cycle analysis was performed with the percentages of cells with DNA content corresponding to the SubG1, G1, S and G2/M phases were computed by planimetry of the histogram and then compared for each cell line.
Immunofluorescence
Induction of double-strand breaks was studied by examining the formation of γ-H2AX foci.Citation13 2 × 105 cells were seeded on glass slides placed in 10 cm Petri dishes for two days and then treated with 1 μg/ml etoposide and/or 10 μg/ml curcumin for 24 h. The slides were washed twice with PBS. Cells were directly fixed in 2% formaldehyde/ PBS solution for 15min at room temperature and washed 3 times for 10 min in PBS. Fixed slides were incubated at –20°C until further processing. Cells were permeabilised by 0.2% Triton-X100/PBS for 5 min on ice and blocked for 1 h at room temperature (RT) in 3% bovine serum albumin (BSA) in PBS. Next, 50 μl anti γ-H2AX antibody (# 05–636, Upstate, USA) (dilution 1:100 in PBS/1%BSA) was added to each slide, which was then incubated in a humidified atmosphere for 1 h at RT. After washing with PBS once, cells were incubated with the Alexa Fluor-594-conjugated goat anti-mouse secondary antibody (1:600) (Molecular probes) for 1 h at RT and washed 4 times for 10 min each in PBS/0.5%Tween 20. Cells were stained with 4, 6-diamidino-2-phenylindol (DAPI)/antifade mixture, and the number of γ-H2AX foci was counted using a fluorescence microscope (Axioplan 2, Zeiss, Germany). At least 50 cells were analyzed for each group.
Constant-field gel electrophoresis of DNA
Double-strand break (DSB) induction and repair were measured by constant-field gel electrophoresis (CFGE) using exponentially growing cells, as described by El-awady et al.Citation14 The cells were treated with 1 µg/ml ETP alone, 10 or 50 µg/ml CUR alone, 1 µg/ml ETP + 10 µg/ml Cur or 1 µg/ml ETP + 50 µg/ml Cur at RT. After treatment, the cells were incubated for 48 h at 37οC. The cells were detached with trypsin, and the resulting cell suspension was mixed with an equal amount of 1.6% low melting point agarose (Bio-Rad), pipetted into a 180 ml mold and left on ice to solidify to form agarose cell plugs. Plugs were incubated in lysis buffer (0.4 M ethylene diamine tetraacetic acid, EDTA, 2% N-Lauryl sarcosine, 1 mg ml−1 proteinase K; all Sigma) for 30 min on ice and then overnight at 37οC. Plugs were washed three times with Tris-EDTA buffer, and a piece of the plug containing about 105 cells was inserted into a preformed 0.8% agarose gel. The gel was then covered with a thin overlayer of 0.8% agarose to avoid light fraction artifacts upon optical imaging. Electrophoresis was run for 36 h at 0.6V cm−1 in 0.5 × Tris-Borate-EDTA buffer (45 mM Tris base, 45 mM boric acid, 2 mM EDTA) in a conventional apparatus (Subcell, Bio-Rad). After completion of electrophoresis, the gel was stained overnight in 0.5 µg ml−1 ethidium bromide, destained overnight in distilled water and the fraction of DNA released (FDR) after each treatment was determined using a gel documentation system (Biometra).
Electron microscopy
The interior structure of the cells was examined for each cell line after treatment with ETP alone, CUR alone or ETP + CUR. The cells were prepared according to the method of Kovacs et al.Citation15 and were seeded at a density of 5 × 106 in 75 cm2 cell culture flasks and treated 24 h later. The cells were divided into four groups: control untreated cells, cells treated with 1 µg/ml ETP alone, cells treated with 25 µg/ml CUR alone and cells treated with 1 µg/ml ETP + 25 µg/ml CUR. All flasks were incubated at 37°C. After 24 h, the flasks were trypsinised and exposed to the primary fixative, 3% glutaraldehyde/PBS (pH 7.4), for 4 h at 4οC. The cells were washed twice with PBS and treated with the second fixative, 1% aqueous OsO4/PBS (pH 7.4), for 1.3 h at 4οC. The cells were washed twice with PBS at 4οC and then dehydrated by different concentrations of ethanol (50, 70, 90 and 100%) at RT. After dehydration, the cells were exposed to the transitional solvent (propylene oxide) at RT for 30 min. The cells were infiltrated in two steps. First, the cells were treated with propylene oxide/araldite 2:1 for 1 h, then with propylene oxide/araldite 1:1 overnight. Second, the cells were treated with 100% araldite (resin). Embedding was achieved by placing the araldite tissue at the apex or on an inverted BEEM capsule filled with liquid resin while the polymerisation step was done by heating capsules at 60οC overnight, then the capsules were stored or cut and examined via electron microscope (JEOL, JEM-100S). Autophagy, apoptosis and necrosis index was determined by counting the number of cells showing these modes of cell death in both ETP and /or CUR-treated cells and control cells in ten low power fields (2000X) of each section in blinded fashion.
Statistics
Each experiment was repeated three times, and the data are presented as mean ± SEM. Unpaired t-tests were used to compare two different treatment groups. Multiple comparisons were conducted using one-way analysis of variance (ANOVA) followed by LSD (least significant difference) for post hoc analysis. p < 0.05 was considered statistically significant. Statistical analysis, data fitting and graphics were performed using Prism software (GraphPad Software).
| Abbreviations: | ||
| CUR | = | Curcumin |
| NS | = | Nigella sativa |
| ETP | = | eteposide |
| DDR | = | DNA damage response machinery |
| DSBs | = | double strand breaks |
| γ-H2AX | = | phosphorylated histone type 2AX |
| Apo | = | apoptosis |
| Auto | = | autophagy |
| Nec | = | necrosis |
Acknowledgments
We thank all the members of the Cancer Biology Department, ENCI for their stimulating ideas and practical support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Notes
† These authors contributed equally to this work.
References
- Raguz S, Yagüe E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer 2008; 99:387 - 91; http://dx.doi.org/10.1038/sj.bjc.6604510; PMID: 18665178
- Oneschuk D, Younus J. Natural health products and cancer chemotherapy and radiation therapy. Oncol Rev 2008; 1:233 - 42; http://dx.doi.org/10.1007/s12156-008-0028-6
- Shervington A, Pawar V, Menon S, Thakkar D, Patel R. The sensitization of glioma cells to cisplatin and tamoxifen by the use of catechin. Mol Biol Rep 2009; 36:1181 - 6; http://dx.doi.org/10.1007/s11033-008-9295-3; PMID: 18581255
- Weissenberger J, Priester M, Bernreuther C, Rakel S, Glatzel M, Seifert V, et al. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin Cancer Res 2010; 16:5781 - 95; http://dx.doi.org/10.1158/1078-0432.CCR-10-0446; PMID: 21138870
- Syng-Ai C, Kumari AL, Khar A. Effect of curcumin on normal and tumor cells: role of glutathione and bcl-2. Mol Cancer Ther 2004; 3:1101 - 8; PMID: 15367704
- Hassan MI, Mabrouk GM, Shehata HH, Aboelhussein MM. Antineoplastic Effects of Bee Honey and Nigella sativa on Hepatocellular Carcinoma Cells. Integr Cancer Ther 2010; In press http://dx.doi.org/10.1177/1534735410387422; PMID: 21147814
- Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615 - 22; PMID: 8915402
- Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int J Cancer 2003; 105:472 - 9; http://dx.doi.org/10.1002/ijc.11106; PMID: 12712436
- Yang AK, He SM, Liu L, Liu JP, Wei MQ, Zhou SF. Herbal interactions with anticancer drugs: mechanistic and clinical considerations. Curr Med Chem 2010; 17:1635 - 78; http://dx.doi.org/10.2174/092986710791111279; PMID: 20345351
- El-Awady RA, Ali MM, Saleh EM, Ghaleb FM. Apoptosis is the most efficient death-pathway in tumor cells after topoisomerase II inhibition. Saudi Med J 2008; 29:558 - 64; PMID: 18382799
- Berenbaum MC. What is synergy?. Pharmacol Rev 1989; 41:93 - 141; PMID: 2692037
- El-Awady RA, Saleh EM, Dahm-Daphi J. Targeting DNA double-strand break repair: is it the right way for sensitizing cells to 5-fluorouracil?. Anticancer Drugs 2010; 21:277 - 87; http://dx.doi.org/10.1097/CAD.0b013e328334b0ae; PMID: 20075715
- Ying S, Myers K, Bottomley S, Helleday T, Bryant HE. BRCA2-dependent homologous recombination is required for repair of Arsenite-induced replication lesions in mammalian cells. Nucleic Acids Res 2009; 37:5105 - 13; http://dx.doi.org/10.1093/nar/gkp538; PMID: 19553191
- El-Awady RA, Dikomey E, Dahm-Daphi J. Radiosensitivity of human tumour cells is correlated with the induction but not with the repair of DNA double-strand breaks. Br J Cancer 2003; 89:593 - 601; http://dx.doi.org/10.1038/sj.bjc.6601133; PMID: 12888835
- Kovacs K, Lloyd R, Horvath E, Asa SL, Stefaneanu L, Killinger DW, et al. Silent somatotroph adenomas of the human pituitary. A morphologic study of three cases including immunocytochemistry, electron microscopy, in vitro examination, and in situ hybridization. Am J Pathol 1989; 134:345 - 53; PMID: 2464941
- Zeng Z, Cortés-Ledesma F, El Khamisy SF, Caldecott KW. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J Biol Chem 2011; 286:403 - 9; http://dx.doi.org/10.1074/jbc.M110.181016; PMID: 21030584
- Sunter NJ, Cowell IG, Willmore E, Watters GP, Austin CA. Role of Topoisomerase IIβ in DNA Damage Response following IR and Etoposide. J Nucleic Acids 2010; 2010: 1-8.
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858 - 68; http://dx.doi.org/10.1074/jbc.273.10.5858; PMID: 9488723
- Azab SS, Salama SA, Abdel-Naim AB, Khalifa AE, El-Demerdash E, Al-Hendy A. 2-Methoxyestradiol and multidrug resistance: can 2-methoxyestradiol chemosensitize resistant breast cancer cells?. Breast Cancer Res Treat 2009; 113:9 - 19; http://dx.doi.org/10.1007/s10549-008-9898-3; PMID: 18228136
- Nielsen D, Maare C, Skovsgaard T. Cellular resistance to anthracyclines. Gen Pharmacol 1996; 27:251 - 5; http://dx.doi.org/10.1016/0306-3623(95)02013-6; PMID: 8919638
- Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell Div 2008; 3:3 - 14; http://dx.doi.org/10.1186/1747-1028-3-14; PMID: 18211690
- Yi T, Cho SG, Yi Z, Pang X, Rodriguez M, Wang Y, et al. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol Cancer Ther 2008; 7:1789 - 96; http://dx.doi.org/10.1158/1535-7163.MCT-08-0124; PMID: 18644991
- Effenberger-Neidnicht K, Schobert R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother Pharmacol 2011; 67:867 - 74; http://dx.doi.org/10.1007/s00280-010-1386-x; PMID: 20582416
- Hour TC, Chen J, Huang CY, Guan JY, Lu SH, Pu YS. Curcumin enhances cytotoxicity of chemotherapeutic agents in prostate cancer cells by inducing p21(WAF1/CIP1) and C/EBPbeta expressions and suppressing NF-kappaB activation. Prostate 2002; 51:211 - 8; http://dx.doi.org/10.1002/pros.10089; PMID: 11967955
- Arafa SA, Zhu Q, Shah ZI, Wani G, Barakat BM, Racoma I, et al. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat Res 2011; 706:28 - 35; http://dx.doi.org/10.1016/j.mrfmmm.2010.10.007; PMID: 21040738
- Gurung RL, Lim SN, Khaw AK, Soon JF, Shenoy K, Mohamed Ali S, et al. Thymoquinone induces telomere shortening, DNA damage and apoptosis in human glioblastoma cells. PLoS One 2010; 5:e12124; http://dx.doi.org/10.1371/journal.pone.0012124; PMID: 20711342
- Jafri SH, Glass J, Shi R, Zhang S, Prince M, Kleiner-Hancock H. Thymoquinone and cisplatin as a therapeutic combination in lung cancer: In vitro and in vivo. J Exp Clin Cancer Res 2010; 29:87; http://dx.doi.org/10.1186/1756-9966-29-87; PMID: 20594324
- Banerjee S, Padhye S, Azmi A, Wang Z, Philip PA, Kucuk O, et al. Review on molecular and therapeutic potential of thymoquinone in cancer. Nutr Cancer 2010; 62:938 - 46; http://dx.doi.org/10.1080/01635581.2010.509832; PMID: 20924969
- Burits M, Bucar F. Antioxidant activity of Nigella sativa essential oil. Phytother Res 2000; 14:323 - 8; http://dx.doi.org/10.1002/1099-1573(200008)14:5<323::AID-PTR621>3.0.CO;2-Q; PMID: 10925395
- Abdel-Wahhab MA, Aly SE. Antioxidant property of Nigella sativa (black cumin) and Syzygium aromaticum (clove) in rats during aflatoxicosis. J Appl Toxicol 2005; 25:218 - 23; http://dx.doi.org/10.1002/jat.1057; PMID: 15856529
- Collett GP, Campbell FC. Curcumin induces c-jun N-terminal kinase-dependent apoptosis in HCT116 human colon cancer cells. Carcinogenesis 2004; 25:2183 - 9; http://dx.doi.org/10.1093/carcin/bgh233; PMID: 15256484
- Srivastava RK, Chen Q, Siddiqui I, Sarva K, Shankar S. Linkage of curcumin-induced cell cycle arrest and apoptosis by cyclin-dependent kinase inhibitor p21(/WAF1/CIP1). Cell Cycle 2007; 6:2953 - 61; http://dx.doi.org/10.4161/cc.6.23.4951; PMID: 18156803
- Hsu JL, Chiang PC, Guh JH. Tunicamycin induces resistance to camptothecin and etoposide in human hepatocellular carcinoma cells: role of cell-cycle arrest and GRP78. Naunyn Schmiedebergs Arch Pharmacol 2009; 380:373 - 82; http://dx.doi.org/10.1007/s00210-009-0453-5; PMID: 19777212
- Dhandapani KM, Mahesh VB, Brann DW. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J Neurochem 2007; 102:522 - 38; http://dx.doi.org/10.1111/j.1471-4159.2007.04633.x; PMID: 17596214
- Ikeda R, Vermeulen LC, Lau E, Jiang Z, Pomplun M, Kolesar JM. Establishment and characterization of irinotecan-resistant human non-small cell lung cancer A549 cells. Mol Med Report 2010; 3:1031 - 4; PMID: 21472350
- Lee CK, Ki SH, Choi JS. Effects of oral curcumin on the pharmacokinetics of intravenous and oral etoposide in rats: possible role of intestinal CYP3A and P-gp inhibition by curcumin. Biopharm Drug Dispos 2011; 32:245 - 51; http://dx.doi.org/10.1002/bdd.754; PMID: 21506134
- Yu LL, Wu JG, Dai N, Yu HG, Si JM. Curcumin reverses chemoresistance of human gastric cancer cells by downregulating the NF-κB transcription factor. Oncol Rep 2011; 26:1197 - 203; PMID: 21811763
- Qian H, Yang Y, Wang X. Curcumin enhanced adriamycin-induced human liver-derived Hepatoma G2 cell death through activation of mitochondria-mediated apoptosis and autophagy. Eur J Pharm Sci 2011; 43:125 - 31; http://dx.doi.org/10.1016/j.ejps.2011.04.002; PMID: 21514382
- Martín-Cordero C, López-Lázaro M, Gálvez M, Ayuso MJ. Curcumin as a DNA topoisomerase II poison. J Enzyme Inhib Med Chem 2003; 18:505 - 9; http://dx.doi.org/10.1080/14756360310001613085; PMID: 15008515
- Snyder RD, Arnone MR. Putative identification of functional interactions between DNA intercalating agents and topoisomerase II using the V79 in vitro micronucleus assay. Mutat Res 2002; 503:21 - 35; http://dx.doi.org/10.1016/S0027-5107(02)00028-3; PMID: 12052500
- López-Lázaro M, Willmore E, Jobson A, Gilroy KL, Curtis H, Padget K, et al. Curcumin induces high levels of topoisomerase I- and II-DNA complexes in K562 leukemia cells. J Nat Prod 2007; 70:1884 - 8; http://dx.doi.org/10.1021/np070332i; PMID: 18076140
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739 - 45; http://dx.doi.org/10.1016/j.molcel.2007.11.015; PMID: 18082599
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547 - 51; http://dx.doi.org/10.1126/science.1074740; PMID: 12142523
- Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 2006; 40:209 - 35; http://dx.doi.org/10.1146/annurev.genet.40.051206.105231; PMID: 16805667
- Lu JJ, Cai YJ, Ding J. Curcumin induces DNA damage and caffeine-insensitive cell cycle arrest in colorectal carcinoma HCT116 cells. Mol Cell Biochem 2011; 354:247 - 52; http://dx.doi.org/10.1007/s11010-011-0824-3; PMID: 21526346
- Roy M, Sinha D, Mukherjee S, Biswas J. Curcumin prevents DNA damage and enhances the repair potential in a chronically arsenic-exposed human population in West Bengal, India. Eur J Cancer Prev 2011; 20:123 - 31; http://dx.doi.org/10.1097/CEJ.0b013e328341017a; PMID: 21332098
- Saleh EM, El-Awady RA, Abdel Alim MA, Abdel Wahab AH. Altered expression of proliferation-inducing and proliferation-inhibiting genes might contribute to acquired doxorubicin resistance in breast cancer cells. Cell Biochem Biophys 2009; 55:95 - 105; http://dx.doi.org/10.1007/s12013-009-9058-3; PMID: 19593673
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 8; http://dx.doi.org/10.1038/nature08467; PMID: 19847258
- Kepp O, Tesniere A, Schlemmer F, Michaud M, Senovilla L, Zitvogel L, et al. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis 2009; 14:364 - 75; http://dx.doi.org/10.1007/s10495-008-0303-9; PMID: 19145485
- Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al, Nomenclature Committee on Cell Death. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 2005; 12:Suppl 2 1463 - 7; http://dx.doi.org/10.1038/sj.cdd.4401724; PMID: 16247491
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14:1237 - 43; http://dx.doi.org/10.1038/sj.cdd.4402148; PMID: 17431418
- Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007; 72:29 - 39; http://dx.doi.org/10.1124/mol.106.033167; PMID: 17395690
- Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011; 7:176 - 87; http://dx.doi.org/10.4161/auto.7.2.14074; PMID: 21081844
- Ohno Y, Yagi H, Nakamura M, Masuko K, Hashimoto Y, Masuko T. Simultaneous induction of apoptotic, autophagic, and necrosis-like cell death by monoclonal antibodies recognizing chicken transferrin receptor. Biochem Biophys Res Commun 2008; 367:775 - 81; http://dx.doi.org/10.1016/j.bbrc.2008.01.030; PMID: 18201558