Abstract
The survival rates of patients with squamous cell carcinoma of the head and neck (HNSCC) have not improved significantly despite multi-modality therapy, including surgery, radiation therapy, and chemotherapy. Recently, molecular targeted agents have shown significant improvement in clinical outcomes; for example, in chronic myelogeneous leukemia with imatinib, breast cancer with trastuzumab, colon cancer with bevacizumab and cetuximab, and renal cell cancer with sorafenib and sunitinib. In HNSCC, the epidermal growth factor receptor antibody cetuximab has shown promising results in combination with radiation. Targeted agents including cetuximab induce stresses to activate prosurvival autophagy. Combining autophagy inhibitors with agents that induce autophagy as a prosurvival response may therefore increase their therapeutic efficacy. Whether autophagy contributes to the prosurvival response or to the antitumor effect of chemotherapeutic drugs is largely unknown. This review will discuss the possible role of autophagy as a novel target for anticancer therapy agents in HNSCC.
Introduction
Head and neck cancers, 90% of which are overwhelmingly squamous cell carcinomas, include cancers of the oral cavity, oropharynx, hypopharynx, larynx and, to a lesser degree, the nose and paranasal sinuses. Despite improvements in the treatment of head and neck cancer, many patients still succumb to their disease. Treatment for head and neck squamous cell carcinoma (HNSCC) has mainly consisted of a combination of surgery and radiation, or combined radiation and chemotherapy. Platinum-based agents provide the backbone of the standard chemotherapeutic regimens for HNSCC. Cisplatin is a widely used drug in the class of platinum-based chemotherapies, which also includes carboplatin and oxaliplatin. Platinum compounds work by the formation of DNA crosslinks within cells, leading to apoptosis and autophagy. The efficacy of cisplatin in HNSCC is greatly increased when combined with other chemotherapeutic agents, such as taxanes (paclitaxel and docetaxel) and 5-fluorouracil.Citation1 In HNSCC, new drugs targeting epidermal growth factor receptor (EGFR) are now approved and are entering clinical practice, either alone or in combination with conventional treatment approaches. Although treatment strategies continue to improve, HNSCC cells have been known to develop natural and acquired resistance to chemoradiotherapy, resulting in a marked decrease in the 5-year survival rate. Some evidence has indicated that autophagy facilitates the cancer cells’ resistance to chemotherapy and radiation treatment. The abrogation of autophagy through autophagy inhibitors or knockdown of autophagy-related molecules potentiates the resensitization of therapy-resistant cancer cells to anticancer treatment. This review delineates the possible role of autophagy as a novel target for anticancer therapy.
Autophagy in Anticancer Therapy
A number of anticancer therapies, including radiation therapy and chemotherapy, have been observed to induce autophagy in human cancer cell lines.Citation2 Autophagy is an evolutionarily conserved and highly regulated process of large-scale lysosomal degradation of long-lived proteins, macromolecules, ribosomes, and organelles, such as the endoplasmic reticulum (ER), Golgi apparatus, and mitochondria. Autophagy is morphologically characterized by the appearance of autophagosomes in the cytoplasm. In addition, LC3 has been found to be a specific biochemical marker for autophagy. Newly synthesized LC3, termed LC3- I, is evenly distributed throughout the cytoplasm. Upon induction of autophagy, some LC3-I is converted into LC3-II, which is tightly bound to the autophagosomal membranes, forming ring-shaped structures in the cytosol. The p62 protein is localized at the autophagosome formation site and directly interacts with LC3. Subsequently, p62 is incorporated into the autophagosome and then degraded. Therefore, impaired autophagy is accompanied by the accumulation of p62 followed by the formation of p62, and ubiquitinated protein aggregates because of both the self-oligomerization and ubiquitin binding of p62.Citation3 The signaling adaptor p62 is a scaffold protein for cell survival and death-signaling pathways such as the NF-κB pathway, Wnt signaling, and apoptosis. The current autophagy literature is often viewed as confusing because of its association with apparently contradictory roles, such as survival and cell death. Whether autophagy contributes to kill cancer cells or instead protects them from the cytotoxicity of drugs may depend on complex crosstalk between these pathways. As autophagy occupies the center of a complex network of cellular responses to stress, the relationship between autophagy and cancer cannot be recapitulated by a simple and uniform principle.
Autophagy as a protective cell survival mechanism
Autophagy seems to function as a protective cell survival mechanism against environmental and cellular stress, and perhaps causes resistance to anticancer therapies. Ionizing radiation, used to treat approximately 50% of all cancer patients, induces autophagy in human esophageal squamous carcinoma and malignant glioma cells. Pretreatment of breast cancer cell lines with 3-methyladenine (3-MA) antagonized ionizing radiation-induced autophagy. Likewise, pretreatment of radioresistant cells with autophagy inhibitors 3-MA or chloroquine (CQ) significantly reduced clonogenic survival of irradiated cells, suggesting that autophagy induced by radiation may be a protective survival mechanism.Citation4 Etoposide-induced autophagy promotes hepatoma cell adaptation and survival, and inhibition of autophagy by Beclin 1 siRNA improves the chemotherapeutic effect of etoposide on human hepatoma G2 cells.Citation5 In addition, acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy, because autophagy inhibitor 3-MA can overcome acquired cisplatin resistance through increasing the apoptotic effect of cisplatin.Citation6 These examples demonstrate that autophagy is activated in tumor cells as a prosurvival mechanism against cytotoxic agents and may therefore favor radioresistance and chemoresistance. Thus, inhibition of autophagy may be a strategy to sensitize cancer cells to radio- and chemotherapy-induced cell death.
Autophagy as a mechanism of cell death
Induction of autophagic cell death has been proposed as a mechanism of cell death, given that features of autophagy have been observed in dying cells. Autophagic cell death can be activated via different molecular pathways in cancer cell lines in response to various agents used in cancer treatment. Autophagic cell death is generally caspase-independent, does not involve classic DNA laddering, and is believed to be the result of extensive autophagic degradation of intracellular content. Studies show that cytotoxic signals can induce autophagy in cells that are resistant (or defective) to apoptosis, such as those expressing high Bcl-2 or Bcl-xL, those lacking Bax and Bak, or those being exposed to pancaspase inhibitors, such as zVAD-fmk ().Citation7 5-Fluorouracil induces autophagic cell death in Bax−/− or PUMA−/− human colon cancer cells, and etoposide induces autophagic cell death in Bax−/− and Bak−/− double knockout mouse embryonic fibroblasts that are resistant to apoptosis, indicating that autophagic cell death can be used as an alternative cell death pathway in apoptosis-defective cells.Citation8 Several new rapamycin derivatives may be used in combination with anticancer agents; however, in vivo evidence is limited, and there is still no concrete evidence showing that autophagy directly causes cell death.
Figure 1. Autophagy in cell survival and cell death. A variety of stressors can induce autophagy in HNSCC cells in order to avoid apoptosis. The process of autophagy can then go on to rescue cells and promote survival, enabling tumor growth and cell replication. In addition, autophagy has a more direct role in mediating cell death in specific contexts. The exact mechanisms regulating autophagic cell death remain to be determined. There is believed to be significant cross talk between the processes of autophagy and apoptosis, and if the latter process remains intact, some cells may ultimately undergo apoptosis following autophagy.
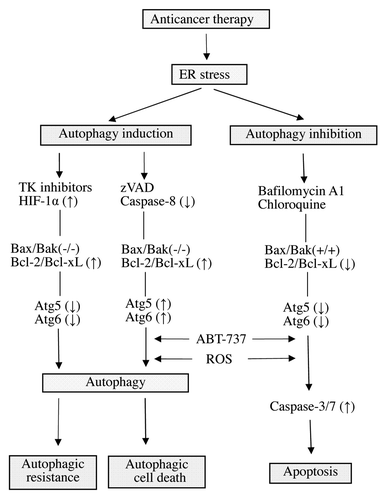
Autophagy and HNSCC
Receptor tyrosine kinase EGFR is frequently deregulated in HNSCC, which is highly expressed in 80–100% of head and neck cancers.Citation9 EGFR is a transmembrane tyrosine kinase receptor composed of an extracellular ligand-binding domain, a hydrophobic transmembrane region, and an intracellular domain with tyrosine kinase activity. EGFR is a member of the ErbB family of receptor tyrosine kinases that, once activated, can promote cell survival and proliferation. EGFR is a cell-surface protein and its variations are thought to contribute to the development of cancers of the head and neck. The dysregulation of EGFR is an early event in HNSCC that is associated with more aggressive disease, resistance to chemotherapy and poorer survival. Indeed, ligand activation of the EGFR by EGF, TGF-α or other ligands leads to the activation of several prosurvival signaling pathways, including Ras/MAPK, PI3K/Akt, and JAK/Stat.Citation8 Downregulation of EGFR inhibits cell growth of HNSCC in vitro. Two strategies targeting the receptor are available; they utilize mAbs, directed toward the extracellular domain of EGFR, and small molecule tyrosine kinase inhibitors, which bind the catalytic kinase domain of the receptor. Clinical trials using EGFR tyrosine kinase inhibitors in cancer therapy have been conducted, but blockage of tyrosine kinase activity alone does not seem to reach maximum therapeutic efficacy. Independent of its kinase activity, EGFR maintains the basal intracellular glucose level, thereby preventing cells from undergoing autophagic death.Citation10 This function of EGFR may endow tumor cells with increased survival capacity even in the presence of chemotherapeutic agents and tyrosine kinase inhibitors. Thus, the inhibition of this function and of the kinase activity of EGFR may both be necessary for eradication of HNSCC.
Akt/mTOR pathway in HNSCC
Inhibition of autophagy could sensitize tumor cells to many cytotoxic drugs or reverse resistance to chemotherapeutic drugs, representing a promising strategy to improve the efficacy of cancer treatment. The Akt/mTOR pathway is the most investigated as a potential target in autophagy. Accumulating evidence shows a prevalence of 47% of head and neck cancer with at least one of the PI3K/Akt/PTEN molecular alterations.Citation11 The defects accompanying the molecular aberrations contribute to the high prevalence of phosphorylated Akt (p-Akt) expression in head and neck cancer. Recent studies have focused on the disorder of this pathway in HNSCC with one of its key proteins, Akt, actively expressed in clinical tumor samples. Moreover, aberration of other cascade proteins has also been emphasized in HNSCC, including PTEN and mTOR.Citation12 In HNSCC, factors including PTEN dysregulation and point mutation in PIK3CA have both offered important contributions to the alteration of the PI3K/Akt/mTOR pathway. Dysregulation of these protein expressions is a frequent and early event during oral carcinogenesis. These findings highlight the signals leading to tumorigenesis, and specific targeting agents are designed for the treatment of the disease. Signaling pathways downstream of EGFR and other receptor tyrosine kinases, such as the PI3K/Akt pathway, are involved in the regulation of autophagy, indicating a potential link between receptor tyrosine kinase inhibition and autophagy. Indeed, a PI3K/mTOR inhibitor (NVP-BGT226) elicits autophagosome formation in cell lines of human head and neck cancer, and the depletion of p62 in treated cells suggests the induction of autophagic flux. In addition, BGT226 induces cancer cell death through activation of autophagy instead of apoptosis.Citation13 In the following section, it is demonstrated that valid inhibitors of HNSCC, which target EGFR and tyrosine kinase, have potential autophagic ability.
Small Molecule Tyrosine Kinase Inhibitors
Based on the essential role of EGFR-initiated signaling in tumor development and progression, this receptor tyrosine kinase has been recognized as a therapeutic target for HNSCC treatment.Citation14 Strategies have been developed to target EGFR, including mAbs, tyrosine kinase-specific inhibitors, ligand-linked immunotoxins, and antisense approaches. Tyrosine kinase inhibitors block the ATP binding pocket of the tyrosine kinase domain of EGFR, preventing activation of downstream targets. While mAbs are unable to cross the plasma membrane and target EGFR intracellular signaling apparatus, tyrosine kinase inhibitors have this potential; however, tumors overexpressing wild-type EGF receptor are far less sensitive to EGF receptor tyrosine kinase inhibitors. Unfortunately, cancer development interferes with multi-stage signal transduction pathways, and therefore blocking a single target only rarely results in disease regression.
Gefitinib (ZD1839; Iressa)
Gefitinib is an orally active, selective EGFR-tyrosine kinase inhibitor, which has principally been studied in non-small cell lung cancer. Gefitinib prevents the binding of ATP to the receptor and thereby inactivates EGFR. Cancer with certain activating mutations (point mutations or deletions of exons 18, 19 and 21) in EGFR is sensitive to gefitinib, although acquired resistance eventually develops. In vitro studies indicated that gefitinib potently inhibited EGFR tyrosine kinase activity at low concentrations that did not significantly affect other kinases tested.Citation15 In vivo studies showed that gefitinib had a favorable tolerability profile and antitumor activity in various xenograft models, and enhanced the antitumor activity of a variety of cytotoxic drugs, including platinum compounds.Citation16 Gefitinib has been tested in clinical trials in HNSCC, as a single agent, or in combination with other chemotherapies or radiation, but has shown limited clinical efficacy, with response rates of 10% to 15%. The mechanism of gefitinib resistance in HNSCC remains largely unknown. Gefitinib suppressed EGF-induced EGFR phosphorylation to basal levels at three phosphorylation sites, and it inhibited the activation-specific phosphorylation of the downstream signal pathway components Akt, ERK, Stat3, and NF-κB to various degrees in different HNSCC cell lines and tumors. Thus, gefitinib sensitivity is correlated with p-Akt and p-Stat3 activation in HNSCC cell lines and tumor specimens. p-Akt and p-Stat3 could serve as potentially useful biomarkers and drug targets for further development of novel therapeutic agents for HNSCC.Citation17
Gefitinib-induced autophagy
Gefitinib showed higher cytotoxicity against human tumor cell lines than against human normal oral cells. Gefitinib alone and combined with docetaxel induced internucleosomal DNA fragmentation and caspase-3 activation in human promyelocytic leukemia HL-60 cells, but not in HNSCC cell line HSC-2. It has been noted that sensitivity of tumor cells to gefitinib is closely correlated with their dependence on Akt activation for survival and proliferation, suggesting that inhibiting Akt may serve as an approach to improving the antitumor efficacy of EGFR inhibitors. Gefitinib simultaneously induced apoptosis and autophagy, and in the presence of the novel allosteric Akt inhibitor MK-2206, both apoptosis and autophagy were increased in tumor cells treated with gefitinib for 48 h. The synergism between MK-2206 and gefitinib appears to be associated with the modulation of autophagy and apoptosis in cells subjected to combination treatment.Citation18 Combination treatment with gefitinib and docetaxel induced the formation of acidic organelles and mitochondrial shrinkage, vacuolization and the production of autophagosomes and the loss of cell-surface microvilli, without destruction of the cell surface and nuclear membranes in HSC-2 and human glioblastoma T98G cells, suggesting the induction of autophagy in HSC-2 and T98G cells. Autophagy can be activated by gefitinib in lung cancer and promote cellular survival in the target therapy using EGFR-tyrosine kinase inhibitors. Blockage of autophagy by pharmacological or genetic approaches greatly enhanced the growth inhibitory effect of gefitinib.Citation19 Thus, inhibition of autophagy has the potential to improve the clinical efficacy of EGFR-tyrosine kinase inhibitors for cancer treatment. Interestingly, gefitinib-induced autophagy might be EGFR independent. Recent results indicated that gefitinib can have other targets, such as non-receptor tyrosine kinases that act also upstream of the PI3K/Akt/mTOR pathway.Citation20 Measurement of EGFR expression by immunohistochemistry was not useful for selecting patients to benefit from gefitinib therapy; however, it could not completely exclude the relevance of EGFR in gefitinib-induced autophagy.
Erlotinib (OSI-774; Tarceva)
A quinazoline derivative erlotinib is an orally active EGFR tyrosine kinase inhibitor. Erlotinib is metabolized through the cytochrome P450 system primarily by CYP3A4, which is mainly excreted in feces with a half-life of ~36 h. Erlotinib has shown clinical activity as well as the modulation of biomarkers related to EGFR inhibition properties in patients with HNSCC. Erlotinib binds competitively to the ATP-binding site at the kinase domain of EGFR and inhibits the activities of EGFR tyrosine kinases. Single-agent erlotinib in patients with locally advanced non-metastatic HNSCC administered before surgical resection results in a 29% objective response rate and inhibits activated EGFR and ERK.Citation21 In addition, full-dose erlotinib can be safely administered in combination with simultaneous cisplatin chemotherapy and radiation therapy in postsurgical patients with HNSCC. The ability of erlotinib to cross the plasma membrane and directly bind to and block EGFR tyrosine kinase, seems to be an advantageous therapy for tumors. Erlotinib-induced cytotoxicity is based on a specific mechanism of oxidative stress mediated by hydrogen peroxide production through NOX4 signaling.Citation22 These findings identify a novel mechanism of action to potentially increase the biological activity observed with the combination of EGFR inhibitors and conventional antineoplastic agents that increase oxidative stress, including cisplatin and ionizing radiation. This biochemical rationale also potentially represents a novel therapeutic strategy for reducing cancer cell resistance to therapy, commonly seen with EGFR inhibitors in the clinic.
Erlotinib-induced autophagy
Erlotinib produced a decline in the cell viability of A549 human lung cancer cells and induced cell apoptosis, coupled with rapid accumulation of ROS. Erlotinib induced the loss of mitochondrial membrane potential, the release of cytochrome c and AIF and activation of JNK. Thus, erlotinib has potent antitumor activity in A549 cells by activating ROS-dependent, JNK-driven cell apoptosis.Citation23 Individual clones with acquired resistance to cetuximab were treated with erlotinib, resulting in decreased activation of EGFR, HER2 and HER3, along with a decrease in the activation of the downstream signaling molecules Akt and MAPK. Erlotinib can induce a high level of autophagy in human lung cancer cells, which was accompanied by inhibition of the PI3K/Akt/mTOR signaling pathway. Moreover, cytotoxicity induced by erlotinib was greatly enhanced after autophagy inhibition by the pharmacological inhibitor CQ and siRNAs targeting Atg5 and Atg7, the most important components for the formation of autophagosome. At very high erlotinib concentrations (60 μM), the autophagic survival program ends in an autophagic cell death program due to the disposal of organelles in glioblastoma cell lines.Citation24 The inhibition of the rescue autophagic process with CQ made it possible to lower the toxic threshold of erlotinib to the therapeutic range.
Dasatinib (BMS-354825; Tykerb)
The second generation dasatinib is an ATP-competitive tyrosine kinase inhibitor that sensitively inhibits all members of the Src family of nonreceptor tyrosine kinases, including c-Src, Lck, Fyn, and Yes. Preclinical evaluations have provided a strong rationale for targeting c-Src in HNSCC. In human HNSCC, c-Src is overexpressed and correlated with lymph node metastases.Citation25 At higher concentrations (3–28 nM), dasatinib also inhibits the Src kinases Abl, c-Kit, PDGFR, and EphA2. Dasatinib is not completely specific, and it seems that not all of its biological and molecular effects are due to Src inhibition. Both cetuximab and radiation have been shown to induce the translocation of the EGFR to the nucleus, and nuclear EGFR has been clearly associated with resistance to both radiation and cetuximab treatment.Citation26 Although dasatinib as a single agent failed to demonstrate significant activity in patients with advanced, metastatic HNSCC, it could block cetuximab and radiation-induced nuclear translocation of the EGFR in HNSCC cell lines and this was correlated with decreased phosphorylation of EGFRY845, which may be necessary for the nuclear translocation of the EGFR. Despite evidence of the effective inhibition of p-Src, dasatinib did not appear to have an effect on tumor growth. It is possible that several signaling pathways are activated in patients with advanced HNSCC and therefore targeting p-Src is not sufficient. The complexities of cell signaling in advanced malignancies, including HNSCC, may result in upregulation of several feedback mechanisms when a single target is hit.
Dasatinib-induced autophagy
Treatment with dasatinib induced considerable levels of autophagy in glioma cells.Citation27 Cells expressing functional PTEN were more sensitive than PTEN-deficient cells. Nanomolar concentrations of dasatinib significantly increase autophagic cell death in glioblastoma. Dasatinib induced conversion of LC3 in p53 proficient B-cell chronic lymphocytic leukemia cells. In addition, a non-toxic concentration of p53 inhibitor pifithrin-α (25 μM) or 3-MA (0.3mM) decreased dasatinib-induced autophagy.Citation28 Dasatinib decreased LC3-I protein and increased LC3-II in both SKOv3 and HEY ovarian cancer cells. Dasatinib concurrently decreased p62 levels, consistently with its correlation with autophagy. Dasatinib inhibits ovarian tumor growth and induces autophagy and apoptosis in vivo. Beclin 1 and Atg12, but not cyclin-dependent kinase inhibitor p27Kip1, are critical for dasatinib-induced autophagy. Downstream targets of Akt, mTOR, and p70S6K, were significantly suppressed; therefore, dasatinib-induced autophagy partially depends on the Akt pathway and Bcl-2.Citation29 Autophagic cell death was magnified by combining dasatinib with low micromolar concentrations of DNA alkylating agent temozolomide, resulting in a significant augmentation of sensitivity to temozolomide. Less significant inhibition of cell proliferation was observed when dasatinib was combined with other cytotoxic chemotherapies such as carboplatin and irinotecan.Citation27 Temozolomide induces autophagy in glioma cells at doses > 100 μM. When combined with dasatinib, autophagy was induced at significantly lower doses of temozolomide. These two drugs were highly additive in combination. Dasatinib may also be successfully combined with radiation therapy or antiangiogenic therapy. These results may have implications for the clinical use of dasatinib.
Saracatinib (AZD0530)
Saracatinib is a novel, orally bioavailable, aniline-quinazoline that inhibits many cellular processes and signaling pathways in which Src kinases are involved. Saracatinib is highly selective for non-receptor tyrosine kinases including c-Src, c-Yes, Lck, and Bcr-Abl. Elevated expression and activation of Src family kinases have been implicated in a variety of cancers, including glioblastoma, colon, lung, breast, and prostate cancers.Citation30 Src is responsible for governing signaling pathways that regulate proliferation, angiogenesis, resistance to apoptosis, adhesion, motility and invasion.Citation31 High Src expression and/or activity is observed in metastases, supporting a role for Src in tumor progression by enhancing tumor invasion and metastatic potential. Elevated Src kinase activity is linked to the progression of solid tumors, including HNSCC. Src regulates HNSCC proliferation and tumor invasion, with the Src-targeted small molecule inhibitor saracatinib displaying potent anti-invasive effects in preclinical studies. Inhibition of Src kinase by saracatinib impairs the proinvasive activity of HNSCC by inhibiting Src substrate phosphorylation important for invadopodia formation and associated matrix metalloprotease activity.Citation32 Although the study was conducted to evaluate saracatinib in patients with recurrent or metastatic HNSCC, single-agent saracatinib does not merit further study in recurrent or metastatic HNSCC.
Saracatinib-induced autophagy
In the prostate cancer cell line PCa, saracatinib can inhibit cell proliferation and migration in vitro and lymph node metastasis in an orthotopic nude mouse model.Citation33 Flow cytometric analysis of the treated cells revealed significant growth arrest with only marginal apoptosis, a phenomenon also associated with other Src family kinase inhibitors. Saracatinib effectively induces autophagy in PCa cells, as does siRNA-targeted inhibition of Src expression.Citation34 These data suggest a role for Src activity in the suppression of autophagy. Importantly, inhibition of autophagy using either pharmacological inhibitors or RNA interference of essential autophagy genes promotes cell death induced by saracatinib. Notably, the combination of saracatinib with CQ, resulted in 64% tumor growth inhibition and enhanced apoptosis in a xenograft mouse model. This combination also led to at least a 2-fold increase in the number of apoptotic tumor cells in the group treated with saracatinib plus CQ, suggesting that suppression of autophagy drives cells into apoptosis. Taken together, inhibition of autophagy may enhance the therapeutic efficacy of saracatinib in the treatment of prostate cancer.
Emerging EGFR Antibody
As mentioned above, EGFR is an appealing target for molecular-targeted cancer therapy as it is expressed in HNSCC. EGFR is the first molecular target against which mAbs have been developed for cancer therapy.Citation35 Here the mechanisms underlying the effects of EGFR-specific mAb are reviewed in cancer therapy. Attention is focused on cetuximab and panitumumab, two mAbs introduced recently into clinical practice for treatment of head and neck cancer which target the external part of EGFR.
Cetuximab (IMC-225; Erbitux)
Recent advances in genetic engineering have made it possible to create chimera antibodies with sequences of murine and human proteins. Clinical application of fully murine mAbs is restricted, owing to the high incidence of serious immune-mediated side effects, particularly upon repeated exposure. Chimeric antibodies are formed of a variable murine region with antigenic activity, and a constant human region. Cetuximab is a recombinant human/mouse chimeric (65% human and 35% murine) IgG1 mAb that binds targets of an extracellular epitope in the EGFR ligand-binding domain, inhibits activation of the receptor tyrosine kinase, and stimulates receptor internalization and downregulation from the cell surface. Cetuximab has been used in clinical practice in combination with radiation in patients with locally advanced HNSCC and as monotherapy for recurrent and metastatic HNSCC;Citation36 however, overexpression of EGFR is a hallmark of head and neck cancers and confers increased resistance and inferior survival rates. Cetuximab induces unexpected EGFR phosphorylation in cetuximab-resistant HNSCC cells similarly to EGF, but this EGFR activation does not trigger EGFR internalization/degradation, the process currently implicated in the response to cetuximab. The level of Akt phosphorylation is unmodified in cetuximab-resistant cells. In addition, resistance to EGFR-targeted therapy is typically mediated though alternate means of ERK1/2 activation that bypass EGFR either via alternative receptors at the plasma membrane or constitutively active downstream components. Despite targeted agents against EGFR, almost half of treated patients fail this therapy, necessitating novel therapeutic strategies. By contrast, cetuximab is used in combination with chemotherapy to treat head and neck cancers. Cetuximab is effective in platinum-resistant recurrent or metastatic HNSCC. The addition of mAb to the standard first-line regimen cisplatin/5-fluorouracil not only increased the rate of objective responses but also improved progression-free and overall survival in patients with recurrent or metastatic HNSCC.Citation37
Cetuximab-induced autophagy
The therapeutic effects of cetuximab on cancer cells include cell cycle arrest and/or the induction of apoptotic cell death. In addition, the treatment of human head and neck cancer cell lines with cetuximab triggered autophagosome formation, conversion of LC3 from its cytoplasmic to membrane-associated autophagosome form, and increased acidic vesicular organelle formation.Citation38 Furthermore, autophagy-lysosomal inhibition blocked cetuximab-induced autophagic flux, leading to the accumulation of LC3-II,Citation39 strongly indicating that autophagic flux was activated by cetuximab treatment. The role of autophagy induced by cetuximab is to protect or limit cancer cell death caused by cetuximab-induced apoptosis, suggesting that autophagy is a protective cellular response to cetuximab treatment. The autophagy induced after cetuximab treatment occurs as a response to cetuximab-induced apoptosis. Autophagy was detected only in cancer cells in which cetuximab induced either strong or weak apoptosis; inhibition of apoptosis by a caspase inhibitor prevented the induction of autophagy. Autophagy occurred when cetuximab inhibited the class I PI3K/Akt/mTOR pathway and it was accompanied by decreased levels of HIF-1α and Bcl-2. Under hypoxic conditions, Bcl-2 overexpression determines an increase of HIF-1α protein expression and HIF-1 transcriptional activity.Citation40 Decrease in HIF-1α through inhibition of protein synthesis by cetuximab plays a major role in cetuximab-mediated antitumor activity. Cetuximab acts by promoting an association between Beclin 1 and its major physiological partner, Vps34, which is inhibited by overexpression of Bcl-2.
Panitumumab (ABX-EGF; Vectibix)
Panitumumab is the first fully human IgG2 mAb targeting EGFR, which binds the ectodomain of EGFR, blocks binding of EGF and TGF-α to the receptor, and inhibits receptor autophosphorylation. Panitumumab has a longer half-life and higher affinity (Kd = 1.33 ± 0.29 nM) for EGFR on the HNSCC cell line (UM-SCC-22B) than other mAbs and inhibits EGF-dependent tumor cell activation and proliferation.Citation41 Panitumumab is potentially less immunogenic than chimeric mAbs, such as cetuximab, and rarely causes a severe infusion reaction. As cetuximab is an IgG1 antibody, it is potentially able to fix complement and initiate ADCC, whereas panitumumab, as an IgG2 antibody, would not have significant ability to do so. Preclinical studies indicate that panitumumab can improve HNSCC radiation sensitization by augmenting DNA damage and suppressing EGFR activation induced by radiation. Panitumumab can prevent radiation-induced phosphorylation of EGFR and downstream signaling pathways, and PCNA expression, which appears mostly in the S-phase of the cell cycle and is a good marker of cellular proliferation, is reduced in xenografts treated with the combination of panitumumab and radiation compared with either modality alone.Citation42
Panitumumab-induced autophagy
Although the cellular targets of panitumumab are known, the mechanisms through which panitumumab exerts its antitumor activity are not as yet well documented. Panitumumab causes cell cycle arrest at the G0/G1 interphase in vitro and inhibits tumor colony formation with a similar mechanism of action to cetuximab. Augmentation of radiation-induced DNA damage by panitumumab could enhance the apoptotic response to irradiation.Citation42 The augmentation of radiation-induced apoptosis by panitumumab could also be mediated by blockade of the radiation-induced EGFR-p-Stat3 signaling well characterized in HNSCC.Citation43 Activated Stat3 plays a central role in protecting cells against apoptosis through the transcriptional modulation of survival genes, such as Bcl-xL, Bcl-2, and survivin. Immunoblotting analysis revealed that panitumumab increased protein levels of Beclin 1, a marker of autophagy. In addition, an increase in the GSH level was noted following panitumumab treatment, reflecting an imbalance in the redox status of the cells. Redox imbalance within the cytosol may be directly responsible for the induction of autophagy. Panitumumab reduces colon cancer cell proliferation through autophagy and possibly via protein destabilization, regardless of EGFR levels and K-ras mutations.Citation44
Multi-Targeted Inhibitors
Targeted molecular therapy is a new paradigm in cancer treatment, in which drugs selectively interfere with molecules considered important in oncogenesis. Whereas conventional chemotherapy aims to kill all proliferating cells including tumors, targeted molecular therapy aims to disrupt cancer-specific signaling pathways involved in tumor growth and proliferation. Compared with the toxicity of chemotherapy, targeted molecular therapies seem to be relatively tolerable. There are multiple types of targeted molecular therapies but, among them, multi-target tyrosine kinase inhibitors have received particular attention. Tyrosine kinases regulate important cell functions, including survival, differentiation, and proliferation. When mutated or overexpressed, they have key roles in many cancers: increasing tumor cell growth and proliferation, inducing resistance to apoptosis, and promoting angiogenesis and metastasis.
Sunitinib (SU01148; Sutent)
Sunitinib is an oral tyrosine kinase inhibitor that targets multiple tyrosine kinase receptors, including PDGFR, VEGFR, c-Kit, Flt3, GDNFR, and CSF-1R.Citation45 Sunitinib showed potent antitumor effects in many tumor cell lines, including human leukemia cells (HL-60), human breast cancer cells (SKBr-3), human lung cancer cells (A549), human gastric cancer cells (SGC-7901) and human colon cancer cells (HCT116), with IC50 values approximately between 2.2 and 5.5 μM, respectively. Recent research has explored the potential antiangiogenic effects of sunitinib, given the critical role of many of these receptor kinases in tumor angiogenesis. Sunitinib inhibited the activation of VEGFR2/3 signaling and the downstream molecules MEK, ERK, and Akt induced by VEGF-C and VEGF-D in lymphatic endothelial cells; however, sunitinib has limited activity in unselected patients with recurrent or metastatic HNSCC. The prognosis of patients with palliative HNSCC unsuitable for chemotherapy or with progression after platinum-based therapy is poor, with a median overall survival ranging from 3–6.7 mo. The severity of some of the complications highlights the importance of improved patient selection for future studies of sunitinib in head and neck cancer. Sunitinib should not be used outside clinical trials in HNSCC.Citation46
Sunitinib-induced autophagy
Sunitinib markedly induced the apoptosis of rat pheochromocytoma cells (PC12) in a dose- and time-dependent manner. Sunitinib induced both a reduction in the expression of the antiapoptotic molecule Bcl-2 as well as dephosphorylation of the proapoptotic molecule Bad, which resulted in the activation of Bad in these cells. Sunitinib inhibited Stat3 and induced direct renal cell carcinoma cell apoptosis, independent of tumor vasculature destruction. Sunitinib inhibited the phosphorylation of Akt and mTOR followed by a reduction of p70S6K, which is a well-known target of mTOR.Citation47 Sunitinib-treated cells have increased levels of LC3-II in H9c2 cardiac muscle cells, which contribute to the formation of autophagosomes and remain attached to matured autophagosomes, indicating that exposure of cells to sunitinib resulted in the formation of autophagosomes. Sunitinib markedly increased autophagic flux in cells, which was associated with elevated cell death. Inhibition of autophagy by Beclin 1 knockdown resulted in significant attenuation of cell death. This result confirms that autophagy plays an important role in sunitinib-mediated cell cytotoxicity;Citation48 however, sunitinib-induced cytotoxicity in cells does not proceed markedly through apoptosis, as measured by cellular morphology, PARP cleavage or caspase-3 cleavage. Caspase inhibitor zVAD-fmk had little effect on the survival of cells. These results suggest that autophagy might be involved in sunitinib-induced cytotoxicity. Thus, pretreatment with autophagy inhibitors might constitute a potentially therapeutic option to prevent sunitinib-induced cytotoxicity.
Sorafenib (BAY 43–9006; Nexavar)
Sorafenib is a multi-kinase inhibitor that was originally developed as an inhibitor of Raf-1, a component of the ERK1/2 pathway, but which was subsequently shown to inhibit multiple other kinases, including class III tyrosine kinase receptors, such as PDGF, VEGFR1/2, c-Kit, and Flt3. Sorafenib simultaneously inhibits molecular components of the Raf/MEK/ERK signaling pathway, abrogating tumor growth, and inhibiting angiogenesis in head and neck cancers.Citation49 Sorafenib mediates its antitumor effects independent of p53 status. This is important as more than 50% of head and neck tumors have mutant p53, and some of the targeted inhibitors selectively inhibit cell growth in cancer cells with wild-type p53 only. In the clinic, sorafenib is believed to act by suppressing tumor angiogenesis through inhibition of VEGFR family receptors. In head and neck cancers, sorafenib seems to act mainly by inhibiting EGFR/Ras/Raf/MEK/ERK signaling, tumor proliferation, and apoptosis. Despite these findings, the lack of meaningful clinical activity suggests that advanced head and neck cancers may not entirely depend on these pathways. Sorafenib is reasonably well tolerated in patients with advanced or metastatic HNSCC, as it has a toxicity profile similar to that observed in other trials with this agent.Citation50 Sorafenib has also demonstrated good tolerability and promising antitumor activity in combination studies, including combinations with oxaliplatin, 5-fluorouracil, paclitaxel, gemcitabine, doxorubicin, docetaxel, and irinotecan. Full clinical activity of sorafenib may be achieved by combining it with chemoradiation or other signal transduction inhibitors.
Sorafenib-induced autophagy
Sorafenib kills human leukemia cells at concentrations below the maximum achievable dose of 15–20 μM through a mechanism involving downregulation of the antiapoptotic Bcl-2 family member Mcl-1.Citation51 Sorafenib-mediated Mcl-1 downregulation occurred through a translational rather than a transcriptional or posttranslational process that was mediated by ER stress signaling and the regulation of autophagy. Sorafenib induced both apoptosis and autophagy in human hepatocellular carcinoma cells through a mechanism that involved ER stress and was independent of the MEK1/2-ERK1/2 pathway,Citation52 whereas recent findings have also revealed that sorafenib generated a protective form of autophagy in gastrointestinal and pancreatic tumor cells. The mTORC1 inhibition by sorafenib treatment in carcinoma cells induced morphological and biochemical hallmarks of autophagy, for example, the generation of autophagosomes, GFP-LC3 redistribution and LC3-II accumulation. Upregulation of IRE1 (the protective component of ER stress) signals from sorafenib-induced ER stress was critical for the induction of autophagy. Moreover, autophagy activation alleviated ER stress-induced cell death. Inhibition of autophagy using either pharmacological inhibitors or essential autophagy gene knockdown (Atg7 gene) enhanced cell death in sorafenib-treated hepatocellular carcinoma cell lines. The combination of sorafenib with the autophagy inhibitor CQ produced more pronounced tumor suppression in both in vivo and in vitro. In vivo, sorafenib/CQ treatment resulted in a marked increase in TUNEL-positive tumor cells compared with sorafenib-treated tumors. Interestingly, combination of sorafenib with vorinostat interacts in a synergistic fashion to kill carcinoma cells by activating CD95. Exposure to sorafenib plus HDAC inhibitor generated a CD95- and Beclin 1-dependent protective form of autophagy. Sorafenib (3 or 6 μM) and vorinostat (500 nM) treatment promoted higher levels of CD95 phosphorylation, which correlated with DISC formation, receptor surface localization, and autophagy. Thus, sorafenib contributes to CD95 activation by promoting receptor tyrosine phosphorylation, whereas vorinostat contributes to CD95 activation via the initial facilitation of ROS generation and subsequently of CD95 ligand expression.Citation53
Lapatinib (GW572016; Tykerb)
Lapatinib is a novel, selective, and highly potent (9–10 nM) dual inhibitor that targets the tyrosine kinase domains of both EGFR and human HER2 (ErbB2) by interfering with ATP binding, thus blocking autophosphorylation and resultant downstream signaling activities, including cellular proliferation and survival.Citation54 ErbB2 heterodimerization with EGFR may mediate disease progression. Lapatinib in turn inhibits the activation of downstream signaling pathways such as ERK1/2 and Akt in cell lines and xenografts. Lapatinib elicits cytostatic or cytotoxic antitumor effects, depending on the cell type, and has demonstrated clinical activity in several solid tumors. Lapatinib has shown single-agent activity in in vitro and in vivo xenograft studies in HNSCC cell lines. Lapatinib may be more effective in combination with cisplatin or paclitaxel, and lapatinib might provide useful clinical benefits to HNSCC patients. Although paclitaxel shows antiangiogenic activity, a synergistic effect with lapatinib was not observed. Since the therapy efficacy of lapatinib was subjected to intrinsic and acquired resistance, it is an urgent issue to uncover the potential mechanisms conferring resistance to lapatinib.
Lapatinib-induced autophagy
Although apoptotic cells detected by TUNEL were frequent with lapatinib, the change in lapatinib treatment was not statistically significant in the caspase-3 assay. The lack of apoptotic changes despite the observation of tumor shrinkage in some patients suggests that caspases may not be involved in lapatinib-induced cell death or that lapatinib does not induce apoptosis, and its mechanism of action is through some other pathway in locally advanced HNSCC.Citation55 In breast cancer cells with HER2 amplification, lapatinib induced apoptosis in association with upregulation of the proapoptotic protein Bcl-2 interacting mediator of cell death (BIM) through inhibition of the MEK/ERK signaling pathway;Citation56 however, the clinical response of lapatinib was seriously limited by drug resistance. The preliminary data demonstrated increased autophagosome formation in stable resistant cells. Autophagy inhibitors such as hydroxychloroquine (HCQ) and 3-MA might be helpful to restore the sensitivity of the resistant cells to lapatinib via abolishing the formation process and degradation ability of autophagosome. Alternatively, the abrogation of key molecules to form autophagosomes, including at least 30 Atg genes, also might be useful for resensitization to lapatinib treatment. Autophagy could have facilitated ErbB2-positive cancer cell resistance to lapatinib and promoted the survival of resistant cells. The abrogation of autophagy might restore drug sensitivity and autophagy might be one of the targets to overcome lapatinib resistance.Citation57 In contrast, co-administration of lapatinib with Bcl-2/Bcl-xL/Mcl-1 antagonist obatoclax caused synergistic cell killing by eliciting autophagic cell death that was dependent upstream on mitochondrial ROS generation, increased p62 levels, and downstream upon activation of p38 MAPK and inactivation of mTOR.
Other Promising Agents
Effective therapies for patients with HNSCC are urgently needed. Targeted therapies are directed at the unique molecular signature of cancer cells to produce greater efficacy with less toxicity. The development and use of such therapeutics will enable us to improve cancer care. Molecular targeted therapy (other than those above) based on signal transduction pathway alterations detected in cancer offers the hope of reaching this goal.
Bortezomib (PS-341; Velcade)
The dipeptide boronic acid bortezomib is a reversible inhibitor of the chymotryptic activity of the 26S proteasome. Although the molecular mechanism of bortezomib has not yet been fully understood, this agent became the first proteasome inhibitor to enter clinical use. Clinical trials with bortezomib have shown promising results for some types of cancers, but not for others. Its antitumor activity has been attributed to an effect on several pathways, including the inhibition of NF-κB.Citation58 NF-κB is overexpressed in HNSCC and its expression is associated with its aggressive nature and poor prognosis. HNSCC cell lines with increased NF-κB expression demonstrated a lower apoptosis rate and increased radioresistance. Tumors with a high risk of recurrence harbored a higher expression of genes that are associated with the activation of NF-κB signaling. Bortezomib was shown to inhibit NF-κB activation and has cytotoxic, anti-angiogenic, and radiosensitizing activity in HNSCC and other tumors. In HNSCC cell lines, the combination of bortezomib with docetaxel showed significantly increased growth inhibition compared with either alone. Although bortezomib has been shown to inhibit NF-κB activation and to enhance the cytotoxicity of docetaxel with known efficacy in HNSCC,Citation59 the response rate was lower than anticipated. It is unclear why bortezomib did not improve patient outcome when added to docetaxel. In addition, bortezomib attenuated the effects of cetuximab- and radiation-induced EGFR degradation and inhibition of prosurvival signaling in the HNSCC cell line UMSCC-1. Because EGFR is degraded by the ubiqutin-proteasome system, it seems likely that proteasome inhibition by bortezomib could attenuate the cytotoxic effects of cetuximab and radiation by protecting EGFR from degradation.Citation60 Consequently, proteasome inhibitor-induced activation of EGFR can induce MAPK, Akt, and Stat3 prosurvival pathways. Together, the results of these studies show that bortezomib in combination with cetuximab or reirradiation results in incomplete clinical and molecular responses in HNSCC. Other studies suggested that bortezomib induced apoptosis of HNSCC cells through inhibition of protein phosphatase 2A-dependent Akt activation.
Bortezomib-induced autophagy
Bortezomib potently activated the caspase cascade and induced apoptosis in human HNSCC cell lines. Proteasome inhibition by bortezomib induced ER stress and ROS in HNSCC cells. The inhibition of ROS significantly suppressed caspase activation and apoptosis induced by bortezomib.Citation61 Bortezomib potently induces autophagy in HNSCC cells, as demonstrated by the upregulation of LC3-II and Beclin 1, and relocalization of GFP-LC3 to punctate distribution in the cytoplasm. Although the induction of autophagy following proteasome inhibition has been observed in other cell types, with autophagy serving a prosurvival role in colon, prostate, and ovarian cancer cells, whether bortezomib-induced autophagy will play a prosurvival or prodeath role in a particular cell type remains to be determined. Interestingly, targeting HDAC6 by RNA interference abolishes autophagy and enhances cell death induced by bortezomib in breast cancer cells. HDAC6 regulates the recruitment of the autophagic mechanism to destroy the aggregates. Bortezomib treatment of three human HNSCC cell lines led to the phosphorylation/activation of JNK1 enzyme, but not JNK2, accompanied by JNK-dependent phosphorylation of Bcl-2 on Ser,Citation70 which causes disruption of Bcl-2/Beclin 1 complexes, liberating Beclin 1 to promote autophagy.Citation62 Bcl-xL is also known to be overexpressed in a majority of HNSCC cell lines and primary specimens. DNA-damaging agent oxaliplatin induces Bcl-xL phosphorylation at the SerCitation62 residue in a JNK-dependent manner. Bcl-xL antiapoptotic function may be more critically related to its phosphorylation status than to its expression level. Eventually, bortezomib activates two different, while interconnected pathways of cell death, autophagy and apoptosis.
Rapamycin (Sirolimus; Rapamune)
Among the multiple molecular mechanisms dysregulated in HNSCC, the experimental findings support the importance of the Akt/mTOR signaling route in HNSCC progression. Indeed, activation of mTOR and Akt has been observed in more than 80% of all HNSCC lesions, often correlating with poor prognosis.Citation63 mTOR is a serine-threonine protein kinase that belongs to the PI3K-related family, and has been recognized as an important and attractive therapeutic target for cancer therapy. Rapamycin was found to display potent antitumor activity in a variety of solid tumors and was discovered as the first mTOR inhibitor; however, it has poor aqueous solubility and chemical stability and therefore its utilization at doses able to produce an effect as an anticancer agent is limited. Rapamycin binds to intracellular FKBP12 to generate a drug-receptor complex that then binds to and inhibits the kinase activity of mTORC1, which regulates cell growth through effectors such as p70S6K and 4E-BP1. Rapamycin used as a single agent displays anti-proliferative effects and induces apoptosis in HNSCC cell lines, and promotes the rapid regression of HNSCC-tumor xenografts in mice.Citation64 On the other hand, mTOR controls the accumulation of HIF-1α and the consequent expression of VEGF in HNSCC cells. HNSCC cells are the primary target of rapamycin and provide evidence that its anti-angiogenic effects may represent a downstream consequence of mTOR inhibition in HNSCC cells.Citation65 In addition, rapamycin may increase the antitumor activity of a variety of cytotoxic agents, including cisplatin, carboplatin, doxorubicin, paclitaxel, topotecan and mitoxantrone in HNSCC cell lines;Citation64 however, it increasingly clear that rapamycin does not have universal antitumor effects in people and that only a fraction of patients respond to the drug.
Rapamycin-induced autophagy
Rapamycin induced autophagy, and the inhibition of autophagy by siRNA directed against Beclin 1 attenuated the cytotoxicity of rapamycin in rapamycin-sensitive tumor cells, indicating that autophagy was a primary mediator of rapamycin’s antitumor effect rather than a protective response. In general, mTOR acts as a molecular link between apoptosis and autophagy. Indeed, mTOR inhibitors induce apoptosis in some types of tumor cells, whereas they trigger autophagy in other settings.Citation66 Because the incidence of autophagy induced by rapamycin was enhanced by the suppression of kinase activity, rapamycin treatment did not sufficiently inhibit mTOR kinase activity.Citation67 Rapamycin does not inhibit all mTOR functions because mTOR exists in cells at least two distinct multiprotein complexes, only one of which binds to FKBP12-rapamycin. The rapamycin-sensitive complex is defined by the interaction of mTOR with the accessory protein raptor (mTORC1), and the rapamycin-insensitive complex interacts with rector (mTORC2). Rapamycin is only a partial inhibitor of mTORC1 kinase activity. Although autophagy is generally considered a prosurvival mechanism that preserves viability, there is evidence that it could drive an alternative programmed cell death pathway in cells with defects in apoptosis. Autophagy is an alternative cell death pathway that is induced by mTOR inhibitors and upregulated when apoptosis is defective.
Everolimus (RAD001; Afinitor)
Everolimus is an orally administered inhibitor of mTOR, a component of an intracellular signaling pathway that regulates cellular metabolism, growth, proliferation, and angiogenesis. Everolimus, a derivative of rapamycin, binds to an intracellular protein FKBP12, forming a complex that inhibits the mTOR serine/threonine kinase in its complex mTORC1. In this regard, everolimus also reduced p-AktS473 levels in primary tongue lesions and their metastases, suggesting that everolimus can also reduce mTORC2 activity in HNSCC, likely indirectly. There is a paucity of data concerning the preclinical activity of everolimus in head and neck cancer. Everolimus can markedly reduce tumor burden and even recurrence in HNSCC tumor xenografts and in chemically-induced and genetically-defined animal models demonstrating HNSCC initiation and progression.Citation68 In addition, treatment with everolimus caused a marked decrease in the number of invaded lymph nodes, which was reflected in a significant increase in the overall survival of all everolimus-treated animals.Citation69 The combination of everolimus and cisplatin may be a useful therapeutic strategy in HNSCC. Inhibition of mTOR, a downstream signaling mediator of Akt, has been shown to enhance sensitivity to cisplatin in some cancer cell lines. At the concentrations required to inhibit cell growth, everolimus could induce apoptosis rather than cell cycle arrest and exert an additive effect on HNSCC cell growth when combined with cisplatin. Unfortunately, paradoxical upregulation of Akt following everolimus treatment suggests the presence of a feedback loop on Akt signaling in some malignant cells.
Everolimus-induced autophagy
Everolimus increased Beclin 1 expression, the conversion of the soluble form of LC3 to the autophagic vesicle-associated form LC3-II and the occurence of lysosomes/autophagosomes. Everolimus was confirmed to act on the target by analyzing the phosphorylation status of the mTOR target proteins 4E-BP1 and p70S6K. The effects of everolimus were abrogated by RNA interference knockdown of Atg5. Everolimus synergistically enhances both chemosensitivity to doxorubicin and radiosensitivity, and seems to do so through autophagy. Autophagic activation resulted in Src activation and MET inactivation, and the anticancer effects of autophagic activation are mediated largely through MET. Activated c-MET is responsible for triggering a number of intracellular signaling cascades and plays important roles in cell proliferation, survival, migration, and angiogenesis. Everolimus might promote autophagy-mediated programmed cell death, a non-apoptotic escape pathway important in cells that are defective or deficient in apoptosis.Citation70
Temsirolimus (CCI-779; Torisel)
Temsirolimus is a water-soluble synthetic rapamycin ester that has been developed in both oral and intravenous formulations. Temsirolimus has only modest activity when used alone and may induce acquired resistance by activating upstream mTORC2 and Akt. Tumors that do not depend upon PI3K/Akt/mTOR signaling for survival are primarily resistant. The limited clinical efficacy of temsirolimus is due to a compensatory increase in survival signaling pathways downstream of Akt as well as incomplete block of 4E-BP1-controlled proliferative processes downstream of mTOR. Temsirolimus effectively abrogated the downstream effects of mTOR activation in an HNSCC cell line, decreasing expression of the translational effectors p70S6K and 4E-BP1 and resulting in decreased expression of b-FGF and VEGF. When administered in combination with irradiation, anti-EGFR and anti-angiogenic therapies in head and neck cancer patients, temsirolimus exhibits additive antiproliferative effects.Citation71 Although irradiation enhances the prosurvival PI3K/Akt/mTOR pathway, which is already upregulated in many types of cancer, including HNSCC, the combination of temsirolimus and radiotherapy was significantly more effective in reducing tumor burden than conventional chemoradiotherapy with cisplatin. Doses of temsirolimus that significantly inhibit mTOR signaling have a growth-inhibitory effect, and importantly cause significant inhibition of VEGF production in the cells.Citation72 HNSCC may be an excellent tumor type for the use of mTOR inhibitors as single agent adjuvant therapy since temsirolimus inhibited mTOR and did not cause an increase in p-Akt but, on the contrary, a decrease.
Temsirolimus-induced autophagy
Temsirolimus increased the number of acidic vesicular organelles and the amount of LC3 processing, which are characteristic of autophagy, without the induction of apoptosis.Citation73 Temsirolimus acts primarily by promoting G1 cell-cycle arrest and autophagy in mantle cell lymphoma cells. The downregulation of p21 was shown to be critical for the induction of autophagy, especially in the presence of Beclin 1. Downregulation of p21 was associated with sustained ERK activation, thus favoring autophagy. Cell death can be achieved in vivo through more prolonged exposure to the drug as a result of sustained autophagy. It is also possible that temsirolimus may indirectly kill tumor cells in vivo by inhibiting angiogenesis.Citation74 Primarily resistant cells to temsirolimus lack robust Akt signaling, are unable to phosphorylate Akt at baseline, and express PTEN. Dual treatment with temsirolimus and the PI3K inhibitor ZSTK474 or the PI3K/mTOR inhibitor BEZ235 overcame temsirolimus-induced Akt hyper-phosphorylation, which is a marker for developing acquired resistance. The mechanism of cell death involves massive autophagy which proceeds to caspase-independent apoptosis. The combination of vorinostat and temsirolimus demonstrated synergy, suggesting that this combination may have a clinical value in patients with lymphoma. This synergistic effect seems to be related to a variety of molecular events, including enhancement of caspase-3 activation and possibly the inhibition of ERK phosphorylation. Temsirolimus induced autophagy, while vorinostat induced caspase-mediated apoptosis. The effect of vorinostat was associated with inhibition of ERK phosphorylation, but without altering Bcl-2 or Beclin 1 levels.
Hsp90 in HNSCC
Small molecule kinase-targeted drugs have been at the forefront of a new class of cancer therapeutics; however, cancer cells routinely develop drug resistance to these targeted therapies. Therefore, there is a need to be able to identify novel targets that regulate multiple proteins and signal transduction pathways. Hsp90 is a ubiquitously-expressed molecular chaperone that is required for the conformational maturation and stability of a range of client proteins. Hsp90 clients include key mediators of signaling cascades that regulate cellular proliferation as well as block apoptotic signaling.Citation75 Many of the clients are tyrosine kinases (Bcr-Abl, EGFR family members), serine/threonine kinases (Akt, Raf-1), and transcription factors (p53, Stat3) that are involved in multiple signal transduction pathways.Citation76 Furthermore, Hsp90 is overexpressed in HNSCC, and inhibition of Hsp90 allows for the development of small molecules that exhibit high differential selectivity;Citation77 therefore, inhibition of Hsp90 disrupts multiple signaling pathways that contribute to malignancy.
Hsp90 inhibitors (17-AAG, KU363, EC5)
Inhibitors of Hsp90 against HNSCC have been studied previously. Multiple studies have demonstrated that Hsp90 inhibition leads to the degradation of client proteins and enhances tumor cell death. Hsp90 inhibitor 17-AAG, a geldanamycin-derived Hsp90 inhibitor, has entered clinical trials in cancer patients and shows evidence of biological and clinical activity. Recent clinical trials, however, have demonstrated that Hsp90 inhibitors were not therapeutically effective and that 17-AAG displayed dose-limited toxicity and was somewhat difficult to formulate. KU363, a novobiocin-derived Hsp90 inhibitor, functions by binding at the C terminus of Hsp90, thus resulting in inhibition of its chaperone activity. In vitro, it was observed to reduce cell viability and proliferation in four different HNSCC cell lines. It also triggered significant induction of apoptosis, which was seen with flow cytometry and confirmed by caspase-3 activation. Western data also revealed the downregulation of Hsp90 client proteins ErbB2/HER2 and Raf-1.Citation78 Low-dose KU363 displayed comparable levels of efficacy with the high dose but without the overt evidence of systemic toxicity that was observed on gross pathology in some high-dose mice. EC5 is a novel ansamycin-based compound, a dimer of geldanamycin, designed to engage both N-terminal binding sites on the Hsp90 dimer simultaneously, thus stabilizing the drug-target interaction. Although 17-AAG has some limitations as a therapeutic agent due to poor solubility, EC5 was designed to stabilize the drug-target interaction for increased effectiveness.Citation77 Both 17-AAG and EC5 inhibited tumor cell proliferation effectively, but EC5 was more potent, with IC50 below 200 nM in most cell lines tested. EC5 induced anti-proliferative effects in all the HNSCC cell lines. The anti-proliferative effect by EC5 in HNSCC can be explained by G1 arrest followed by apoptotic cell death with the degradation of client proteins including EGFR, c-Raf-1, Akt, and Cdk4 and inhibition of Akt phosphorylation. In vivo, EC5 markedly reduced the growth rate of established HNSCC xenografts in nude mice and decreased the expression of EGFR and Akt within the xenografts.Citation79 These results suggest that ansamycin-based Hsp90 inhibitors affect multiple pathways involved in tumor development and progression, and may represent a new strategy for the treatment of HNSCC patients.
Hsp90 inhibitor-induced autophagy
In most cancer cells, the hyperactivation of Hsp90 stabilizes its client proteins and protects cells from stress conditions. In contrast, the specific inhibition of Hsp90 by some chemicals can lead to the degradation of its clients via either the ubiquitin proteasome system or autophagy.Citation80 17-AAG not only causes the upregulation of heat shock proteins, but is also an effective inducer of the autophagic pathway, which may provide a means to modulate autophagy in cells. Hsp90 inhibitors induce cytoprotective autophagy in myeloma cells. Inhibiting the autophagic process could be a valuable strategy to facilitate the death of multiple myeloma cells by other agents targeting the intrinsic apoptotic pathway. In this respect, Hsp90 inhibitors represent an emerging class of compounds with potent anti-myeloma actions whose activity could be further enhanced by preventing autophagy. Hsp90 inhibitor-induced autophagy is associated with Akt protein degradation in a mechanism dependent on Hsp90 inhibition and Akt-mediated inhibition of mTOR activity in human melanoma A-375 cells. Recent studies have reported that IκB kinase, an essential activator of NF-κB, is a client protein of Hsp90 that can be selectively degraded by autophagy when Hsp90 is inhibited. Consequently, the inhibited nuclear translocation of NF-κB leads to various changes in cellular programs. For example, Hsp90 forms a complex with Beclin 1 through an evolutionarily conserved domain to maintain the stability of Beclin 1. There are three NF-κB binding sites inside the first intron of the Becn1 promoter.Citation81 Paradoxically, geldanamycin effectively promoted proteasomal degradation of autophagy protein Beclin 1 in a concentration- and time-dependent manner. NF-κB can regulate the autophagic process in a positive or negative manner. Taken together, these observations provide an Hsp90-NF-κB-Beclin1 link as a potential biological regulation for autophagic signaling.Citation82 Many drug candidates which target Hsp90 are currently undergoing clinical trials for multiple indications, either as a single agent or in combination therapy.
HDAC in HNSCC
HDACs play a key role in the epigenetic regulation of genes by catalyzing the removal of acetyl groups, stimulating chromatin condensation and promoting transcriptional repression. Modulation of expression levels of genes encoding HDACs has been reported for different types of cancer. For example, overexpression of HDAC1 has been reported in gastric and HDAC2 and HDAC3 in colorectal cancer. Decreased transcription of the HDAC5 gene has been observed in colorectal cancer. Increased expression of HDAC6 is linked to better survival in breast cancer. SIRT8 was found to be overexpressed in thyroid cancer, while SIRT2 gene expression is downregulated in human gliomas. The inhibition of HDAC can reverse epigenetic silencing that is commonly observed in cancer, and various HDAC inhibitors have been developed for cancer therapy. The altered activities of both histone acetyl transferases and HDACs have been reported in HNSCC,Citation83 and recently, HDAC2 expressions in HNSCC had been implicated in advanced stage and poor prognosis, indicating they may represent a potential therapeutic target in this disease.Citation84 HDAC2 maintains HIF-1α stability, probably at the level of protein modification, which in turn leads to an increase in cell invasion⁄migration ability in oral cancer progression. These findings implicate the potential of HDAC inhibitors for oral cancer therapy. AP-2α is involved in a variety of processes, including adipogenesis, neuronal development, and is suspected in cancer progression, in which AP-2α plays a role not only in epigenetic silencing, but also in genomic instability. The potential importance of AP-2α’s transcriptional regulation of target genes is revealed in the pathogenesis of HNSCC.
HDAC inhibitors (Vorinostat, Apicidin)
HDAC inhibitors have been shown to induce growth arrest and differentiation and to promote apoptosis in HNSCC, and some of them are in advanced clinical development either as single agents or in combination with conventional chemotherapeutics or targeted agents.Citation85 Vorinostat (SAHA; Zolinza) induces synergistic antiproliferative and proapoptotic effects in combination with gefitinib in HNSCC cells, including cells strongly resistant to gefitinib. The differential modulation of ErbB receptors by vorinostat may represent a mechanistic basis for the observed synergism. In HNSCC (CAL27) cancer cells, which have a prevalent epithelial phenotype and express EGFR, ErbB2, and ErbB3, vorinostat downregulates the expression and signaling of all three receptors. In HNSCC cells, gefitinib resistance was linked to ErbB2- and ErbB3-mediated signaling, and synergistic antiproliferative activity was demonstrated by combining gefitinib and agents targeting ErbB2.Citation86 Apicidin is a novel cyclic tetrapeptide with a broad spectrum of antiproliferative activity against a variety of cancer cell lines. Apicidin significantly inhibited the proliferation of HNSCC cells in a dose-dependent manner, and markedly upregulated p21WAF1 led to G2/M phase arrest in a p53-independent manner. In addition, apicidin treatment leads to the release of cytochrome c to the cytosol and activation of caspase-9, -7 and -3, which was confirmed by PARP cleavage in HNSCC cells.Citation87
HDAC inhibitor-induced autophagy
There are several reports of HDAC inhibitor-induced autophagy. Valproic acid (Depakene) can induce autophagy, which constituted the main cause of cell death instead of apoptosis in glioma cell lines, whereas FK228 mediated autophagy in rhabdomyosarcoma cells. Vorinostat induced autophagy in hepatocellular carcinoma cells as revealed by LC3-II accumulation. Vorinostat induced autophagy through downregulation of Akt/mTOR signaling and induction of the ER stress response. Inhibition of autophagy by 3-MA or Atg5 knockout reduced vorinostat-induced cytotoxicity, indicating that vorinostat-induced autophagy led to cell death.Citation88 Vorinostat decreased the expression of p62 in a dose-dependent manner. The combination of autophagy inducers with vorinostat might be attractive for the treatment of hepatocellular carcinoma, and pharmacological targeting of autophagy provides promise for the management of cancer therapy. Apicidin induced not only apoptosis but also autophagy in HNSCC (YD-8 and YD-10B) cells. Apicidin markedly increased the levels of LC3-II expression, Atg5 protein expression and the accumulation of acidic vesicular organelles. CQ treatment with apicidin resulted in marked increases in the levels of LC3-II. Apicidin treatment in the presence of CQ for 48 h significantly reduced cell viability as compared with apicidin treatment alone, indicating that inhibition of autophagy enhanced apicidin-mediated cytotoxicity through an increase in apoptosis.Citation89 These results indicated that autophagy plays a protective role in apicidin-mediated cell killing and its inhibition enhances apicidin-induced apoptosis in HNSCC cells. Although little data are available about the possible impact of the different HDAC isoforms on autophagy, the current experiments revealed that HDAC1 and HDAC2 are required for the autophagic response, and HDAC6 is a novel component that controls the fusion of autophagosomes and lysosomes associated with autophagy. Refer to for autophagy induction of targeted agents available for HNSCC.
Table 1. Autophagy induction of targeted agents available for HNSCC
Conclusions and Future Perspectives
One of the most daunting clinical issues is frequent tumor progression after standard treatment, mainly due to therapeutic resistance. Conventional treatments are not adequate for the majority of advanced or recurrent HNSCC patients because of the marked resistance of tumors to chemotherapy and radiation, and the situation is especially devastating for first-time treatment failure. It is an urgent issue to elucidate the mechanisms that induce anticancer drug resistance. Most, if not all, anticancer treatments including novel targeted therapies stimulate autophagy, and this enhanced autophagy is involved in regulating tumor cell resistance to anticancer agents. Cancer cells may take advantage of autophagy to survive despite treatment with anticancer therapy regimens, such as chemotherapy or radiation. Currently, there are several potential strategies to inhibit autophagy, including pharmacological and genetic approaches, to resensitize resistant cancer cells to anticancer therapy. For example, repression of autophagy with siRNA targeted to Beclin 1 and Atg7 enhances the therapeutic efficacy of cisplatin and 5-fluorouracil in esophageal cancer, respectively. A key contributor to drug resistance in autophagic cancer cells is undoubtedly their failure to engage in apoptosis. The overall hypothesis in these trials is that autophagy is a survival mechanism of therapeutic resistance and that blocking autophagy might increase the effect of anticancer treatment. Pharmacological clinical trials are underway for evaluating the potential benefit of CQ in combination with conventional therapy for a variety of malignancies, although the verification that the effects of CQ in these settings were through modulation of autophagy is yet to be established. 3-MA was shown to enhance cell death induced by 5-fluorouracil and cytotoxicity induced by the tyrosine kinase inhibitor.
It must be remembered that the anticancer effect of these different molecules might not be solely due to their inhibition of autophagy. Although there are several pharmacological inhibitors of autophagy, none of the available autophagy inhibitors is specific for autophagy, and hence, affects other pathways. For instance, CQ has been reported to induce apoptosis due to lysosomal disruption, mitochondrial membrane permeabilization and decreased protein degradation. In mouse breast cancer cell lines, CQ may act through mechanisms other than by inhibition of autophagy. CQ can induce p53-independent death in gliomas that do not require caspase-mediated apoptosis. 3-MA suppresses autophagy via the inhibition of class III PI3K; however, 3-MA can suppress proteolysis even in Atg5-deficient cells, suggesting that its effects on protein degradation extend beyond its role in autophagy inhibition. More selective inhibitors of autophagy need to be developed. Current knowledge leaves no doubt that autophagy has great potential for the development of novel therapeutics.
Acknowledgments
The author apologizes to all colleagues whose work may not have been cited for space reasons. He is grateful to D. Mrozek for editing the manuscript.
Related Research Data
References
- Fury MG, Pfister DG. Current recommendations for systemic therapy of recurrent and/or metastatic head and neck squamous cell cancer. J Natl Compr Canc Netw 2011; 9:681 - 9; PMID: 21636539
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 34; http://dx.doi.org/10.1038/nrc1692; PMID: 16148885
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001 - 4; http://dx.doi.org/10.1016/j.cell.2009.05.023; PMID: 19524504
- Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol 2011; 99:287 - 92; http://dx.doi.org/10.1016/j.radonc.2011.06.002; PMID: 21722986
- Xie BS, Zhao HC, Yao SK, Zhuo DX, Jin B, Lv DC, et al. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int J Mol Med 2011; 27:599 - 606; PMID: 21274505
- Ren JH, He WS, Nong L, Zhu QY, Hu K, Zhang RG, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm 2010; 25:75 - 80; http://dx.doi.org/10.1089/cbr.2009.0701; PMID: 20187799
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221 - 8; http://dx.doi.org/10.1038/ncb1192; PMID: 15558033
- Henson ES, Gibson SB. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: implications for cancer therapy. Cell Signal 2006; 18:2089 - 97; http://dx.doi.org/10.1016/j.cellsig.2006.05.015; PMID: 16815674
- Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer 2011; 11:9 - 22; http://dx.doi.org/10.1038/nrc2982; PMID: 21160525
- Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008; 13:385 - 93; http://dx.doi.org/10.1016/j.ccr.2008.03.015; PMID: 18455122
- Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer 2005; 114:242 - 8; http://dx.doi.org/10.1002/ijc.20711; PMID: 15543611
- Nathan CO, Amirghahari N, Rong X, Giordano T, Sibley D, Nordberg M, et al. Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Res 2007; 67:2160 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-2449; PMID: 17332346
- Chang KY, Tsai SY, Wu CM, Yen CJ, Chuang BF, Chang JY. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res 2011; 17:7116 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-11-0796; PMID: 21976531
- Hegymegi-Barakonyi B, Eros D, Szántai-Kis C, Breza N, Bánhegyi P, Szabó GV, et al. Tyrosine kinase inhibitors - small molecular weight compounds inhibiting EGFR. Curr Opin Mol Ther 2009; 11:308 - 21; PMID: 19479664
- Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res 2002; 62:5749 - 54; PMID: 12384534
- Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res 2000; 6:4885 - 92; PMID: 11156248
- Pernas FG, Allen CT, Winters ME, Yan B, Friedman J, Dabir B, et al. Proteomic signatures of epidermal growth factor receptor and survival signal pathways correspond to gefitinib sensitivity in head and neck cancer. Clin Cancer Res 2009; 15:2361 - 72; http://dx.doi.org/10.1158/1078-0432.CCR-08-1011; PMID: 19318490
- Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 2012; 11:154 - 64; http://dx.doi.org/10.1158/1535-7163.MCT-11-0606; PMID: 22057914
- Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 2011; 6:e18691; http://dx.doi.org/10.1371/journal.pone.0018691; PMID: 21655094
- Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, et al. Cellular targets of gefitinib. Cancer Res 2005; 65:379 - 82; PMID: 15695376
- Vergez S, Delord JP, Thomas F, Rochaix P, Caselles O, Filleron T, et al. Preclinical and clinical evidence that Deoxy-2-[18F]fluoro-D-glucose positron emission tomography with computed tomography is a reliable tool for the detection of early molecular responses to erlotinib in head and neck cancer. Clin Cancer Res 2010; 16:4434 - 45; http://dx.doi.org/10.1158/1078-0432.CCR-09-2795; PMID: 20660574
- Orcutt KP, Parsons AD, Sibenaller ZA, Scarbrough PM, Zhu Y, Sobhakumari A, et al. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res 2011; 71:3932 - 40; http://dx.doi.org/10.1158/0008-5472.CAN-10-3425; PMID: 21482679
- Qian X, Li J, Ding J, Wang Z, Zhang W, Hu G. Erlotinib activates mitochondrial death pathways related to the production of reactive oxygen species in the human non-small cell lung cancer cell line A549. Clin Exp Pharmacol Physiol 2009; 36:487 - 94; http://dx.doi.org/10.1111/j.1440-1681.2008.05091.x; PMID: 19673930
- Eimer S, Belaud-Rotureau MA, Airiau K, Jeanneteau M, Laharanne E, Véron N, et al. Autophagy inhibition cooperates with erlotinib to induce glioblastoma cell death. Cancer Biol Ther 2011; 11:1017 - 27; http://dx.doi.org/10.4161/cbt.11.12.15693; PMID: 21508666
- Canel M, Secades P, Rodrigo JP, Cabanillas R, Herrero A, Suarez C, et al. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res 2006; 12:3272 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-05-1583; PMID: 16740747
- Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol 2010; 97:330 - 7; http://dx.doi.org/10.1016/j.radonc.2010.06.010; PMID: 20667610
- Milano V, Piao Y, LaFortune T, de Groot J. Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther 2009; 8:394 - 406; http://dx.doi.org/10.1158/1535-7163.MCT-08-0669; PMID: 19190119
- Amrein L, Soulières D, Johnston JB, Aloyz R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk Res 2011; 35:99 - 102; http://dx.doi.org/10.1016/j.leukres.2010.05.029; PMID: 20573397
- Le XF, Mao W, Lu Z, Carter BZ, Bast RC Jr.. Dasatinib induces autophagic cell death in human ovarian cancer. Cancer 2010; 116:4980 - 90; http://dx.doi.org/10.1002/cncr.25426; PMID: 20629079
- Yeatman TJ. A renaissance for SRC. Nat Rev Cancer 2004; 4:470 - 80; http://dx.doi.org/10.1038/nrc1366; PMID: 15170449
- Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res 2006; 12:1398 - 401; http://dx.doi.org/10.1158/1078-0432.CCR-05-2692; PMID: 16533761
- Ammer AG, Kelley LC, Hayes KE, Evans JV, Lopez-Skinner LA, Martin KH, et al. Saracatinib impairs head and neck squamous cell carcinoma invasion by disrupting invadopodia function. J Cancer Sci Ther 2009; 1:52 - 61; http://dx.doi.org/10.4172/1948-5956.1000009; PMID: 20505783
- Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene 2008; 27:6365 - 75; http://dx.doi.org/10.1038/onc.2008.250; PMID: 18679417
- Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer 2010; 1:40 - 9; http://dx.doi.org/10.1177/1947601909358324; PMID: 20811583
- Mehra R, Serebriiskii IG, Dunbrack RL Jr., Robinson MK, Burtness B, Golemis EA. Protein-intrinsic and signaling network-based sources of resistance to EGFR- and ErbB family-targeted therapies in head and neck cancer. Drug Resist Updat 2011; 14:260 - 79; http://dx.doi.org/10.1016/j.drup.2011.08.002; PMID: 21920801
- Tejani MA, Cohen RB, Mehra R. The contribution of cetuximab in the treatment of recurrent and/or metastatic head and neck cancer. Biologics 2010; 4:173 - 85; PMID: 20714355
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 27; http://dx.doi.org/10.1056/NEJMoa0802656; PMID: 18784101
- Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res 2010; 70:5942 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-10-0157; PMID: 20634405
- Li X, Lu Y, Pan T, Fan Z. Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy 2010; 6:1066 - 77; http://dx.doi.org/10.4161/auto.6.8.13366; PMID: 20864811
- Trisciuoglio D, Gabellini C, Desideri M, Ziparo E, Zupi G, Del Bufalo D. Bcl-2 regulates HIF-1α protein stabilization in hypoxic melanoma cells via the molecular chaperone HSP90. PLoS One 2010; 5:e11772; http://dx.doi.org/10.1371/journal.pone.0011772; PMID: 20668552
- Foon KA, Yang XD, Weiner LM, Belldegrun AS, Figlin RA, Crawford J, et al. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys 2004; 58:984 - 90; http://dx.doi.org/10.1016/j.ijrobp.2003.09.098; PMID: 14967460
- Kruser TJ, Armstrong EA, Ghia AJ, Huang S, Wheeler DL, Radinsky R, et al. Augmentation of radiation response by panitumumab in models of upper aerodigestive tract cancer. Int J Radiat Oncol Biol Phys 2008; 72:534 - 42; http://dx.doi.org/10.1016/j.ijrobp.2008.06.1490; PMID: 18793955
- Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene 2000; 19:2489 - 95; http://dx.doi.org/10.1038/sj.onc.1203483; PMID: 10851047
- Giannopoulou E, Antonacopoulou A, Matsouka P, Kalofonos HP. Autophagy: novel action of panitumumab in colon cancer. Anticancer Res 2009; 29:5077 - 82; PMID: 20044619
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007; 25:884 - 96; http://dx.doi.org/10.1200/JCO.2006.06.3602; PMID: 17327610
- Machiels JP, Henry S, Zanetta S, Kaminsky MC, Michoux N, Rommel D, et al. Phase II study of sunitinib in recurrent or metastatic squamous cell carcinoma of the head and neck: GORTEC 2006-01. J Clin Oncol 2010; 28:21 - 8; http://dx.doi.org/10.1200/JCO.2009.23.8584; PMID: 19917865
- Saito Y, Tanaka Y, Aita Y, Ishii KA, Ikeda T, Isobe K, et al. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am J Physiol Endocrinol Metab 2012; 302:E615 - 25; http://dx.doi.org/10.1152/ajpendo.00035.2011; PMID: 21878661
- Zhao Y, Xue T, Yang X, Zhu H, Ding X, Lou L, et al. Autophagy plays an important role in sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol Appl Pharmacol 2010; 248:20 - 7; http://dx.doi.org/10.1016/j.taap.2010.07.007; PMID: 20637791
- Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol 2006; 24:2659 - 65; http://dx.doi.org/10.1200/JCO.2005.05.4577; PMID: 16763280
- Williamson SK, Moon J, Huang CH, Guaglianone PP, LeBlanc M, Wolf GT, et al. Phase II evaluation of sorafenib in advanced and metastatic squamous cell carcinoma of the head and neck: Southwest Oncology Group Study S0420. J Clin Oncol 2010; 28:3330 - 5; http://dx.doi.org/10.1200/JCO.2009.25.6834; PMID: 20498388
- Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res 2011; 71:4955 - 67; http://dx.doi.org/10.1158/0008-5472.CAN-11-0898; PMID: 21622715
- Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7:1159 - 72; http://dx.doi.org/10.4161/auto.7.10.16818; PMID: 21691147
- Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, et al. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther 2010; 9:2220 - 31; http://dx.doi.org/10.1158/1535-7163.MCT-10-0274; PMID: 20682655
- Shewchuk L, Hassell A, Wisely B, Rocque W, Holmes W, Veal J, et al. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J Med Chem 2000; 43:133 - 8; http://dx.doi.org/10.1021/jm990401t; PMID: 10633045
- Del Campo JM, Hitt R, Sebastian P, Carracedo C, Lokanatha D, Bourhis J, et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer 2011; 105:618 - 27; http://dx.doi.org/10.1038/bjc.2011.237; PMID: 21829197
- Tanizaki J, Okamoto I, Fumita S, Okamoto W, Nishio K, Nakagawa K. Roles of BIM induction and survivin downregulation in lapatinib-induced apoptosis in breast cancer cells with HER2 amplification. Oncogene 2011; 30:4097 - 106; http://dx.doi.org/10.1038/onc.2011.111; PMID: 21499301
- Chen S, Li X, Feng J, Chang Y, Wang Z, Wen A. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med Hypotheses 2011; 77:206 - 8; http://dx.doi.org/10.1016/j.mehy.2011.04.013; PMID: 21570197
- Ishii Y, Waxman S, Germain D. Targeting the ubiquitin-proteasome pathway in cancer therapy. Anticancer Agents Med Chem 2007; 7:359 - 65; http://dx.doi.org/10.2174/187152007780618180; PMID: 17504161
- Chung CH, Aulino J, Muldowney NJ, Hatakeyama H, Baumann J, Burkey B, et al. Nuclear factor-kappa B pathway and response in a phase II trial of bortezomib and docetaxel in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol 2010; 21:864 - 70; http://dx.doi.org/10.1093/annonc/mdp390; PMID: 19850643
- Argiris A, Duffy AG, Kummar S, Simone NL, Arai Y, Kim SW, et al. Early tumor progression associated with enhanced EGFR signaling with bortezomib, cetuximab, and radiotherapy for head and neck cancer. Clin Cancer Res 2011; 17:5755 - 64; http://dx.doi.org/10.1158/1078-0432.CCR-11-0861; PMID: 21750205
- Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 2004; 24:9695 - 704; http://dx.doi.org/10.1128/MCB.24.22.9695-9704.2004; PMID: 15509775
- Li C, Johnson DE. Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via JNK activation. Cancer Lett 2012; 314:102 - 7; http://dx.doi.org/10.1016/j.canlet.2011.09.020; PMID: 21993018
- Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses García A, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res 2007; 13:4964 - 73; http://dx.doi.org/10.1158/1078-0432.CCR-07-1041; PMID: 17785546
- Aissat N, Le Tourneau C, Ghoul A, Serova M, Bieche I, Lokiec F, et al. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother Pharmacol 2008; 62:305 - 13; http://dx.doi.org/10.1007/s00280-007-0609-2; PMID: 17912526
- Amornphimoltham P, Patel V, Leelahavanichkul K, Abraham RT, Gutkind JS. A retroinhibition approach reveals a tumor cell-autonomous response to rapamycin in head and neck cancer. Cancer Res 2008; 68:1144 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-07-1756; PMID: 18281490
- Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ 2002; 9:99 - 100; http://dx.doi.org/10.1038/sj.cdd.4400978; PMID: 11840159
- Iwamaru A, Kondo Y, Iwado E, Aoki H, Fujiwara K, Yokoyama T, et al. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 2007; 26:1840 - 51; http://dx.doi.org/10.1038/sj.onc.1209992; PMID: 17001313
- Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res 2005; 65:9953 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-05-0921; PMID: 16267020
- Patel V, Marsh CA, Dorsam RT, Mikelis CM, Masedunskas A, Amornphimoltham P, et al. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res 2011; 71:7103 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-10-3192; PMID: 21975930
- Lin CI, Whang EE, Donner DB, Du J, Lorch J, He F, et al. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol Cancer Res 2010; 8:1217 - 26; http://dx.doi.org/10.1158/1541-7786.MCR-10-0162; PMID: 20736296
- Bozec A, Etienne-Grimaldi MC, Fischel JL, Sudaka A, Toussan N, Formento P, et al. The mTOR-targeting drug temsirolimus enhances the growth-inhibiting effects of the cetuximab-bevacizumab-irradiation combination on head and neck cancer xenografts. Oral Oncol 2011; 47:340 - 4; http://dx.doi.org/10.1016/j.oraloncology.2011.02.020; PMID: 21421338
- Ekshyyan O, Rong Y, Rong X, Pattani KM, Abreo F, Caldito G, et al. Comparison of radiosensitizing effects of the mammalian target of rapamycin inhibitor CCI-779 to cisplatin in experimental models of head and neck squamous cell carcinoma. Mol Cancer Ther 2009; 8:2255 - 65; http://dx.doi.org/10.1158/1535-7163.MCT-08-1184; PMID: 19625495
- Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 2008; 36:443 - 50; http://dx.doi.org/10.1016/j.exphem.2007.12.008; PMID: 18343280
- Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P, et al. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res 2006; 66:2028 - 37; http://dx.doi.org/10.1158/0008-5472.CAN-04-4559; PMID: 16489002
- Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med 2004; 10:283 - 90; http://dx.doi.org/10.1016/j.molmed.2004.04.006; PMID: 15177193
- Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 2004; 82:488 - 99; http://dx.doi.org/10.1007/s00109-004-0549-9; PMID: 15168026
- Brandt GE, Blagg BS. Alternate strategies of Hsp90 modulation for the treatment of cancer and other diseases. Curr Top Med Chem 2009; 9:1447 - 61; http://dx.doi.org/10.2174/156802609789895683; PMID: 19860731
- Cohen SM, Mukerji R, Samadi AK, Zhao H, Blagg BS, Cohen MS. Novel C-terminal Hsp90 inhibitor for head and neck squamous cell cancer (HNSCC) with in vivo efficacy and improved toxicity profiles compared with standard agents. Ann Surg Oncol 2011; http://dx.doi.org/10.1245/s10434-011-1971-1; PMID: 21837531
- Yin X, Zhang H, Burrows F, Zhang L, Shores CG. Potent activity of a novel dimeric heat shock protein 90 inhibitor against head and neck squamous cell carcinoma in vitro and in vivo. Clin Cancer Res 2005; 11:3889 - 96; http://dx.doi.org/10.1158/1078-0432.CCR-04-2272; PMID: 15897590
- Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-κ B2 p100 involving protection of NF-κ B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res 2007; 17:520 - 30; http://dx.doi.org/10.1038/cr.2007.47; PMID: 17563756
- Copetti T, Demarchi F, Schneider C. p65/RelA binds and activates the beclin 1 promoter. Autophagy 2009; 5:858 - 9; PMID: 19458474
- Jiang Q, Wang Y, Li T, Shi K, Li Z, Ma Y, et al. Heat shock protein 90-mediated inactivation of nuclear factor-κB switches autophagy to apoptosis through becn1 transcriptional inhibition in selenite-induced NB4 cells. Mol Biol Cell 2011; 22:1167 - 80; http://dx.doi.org/10.1091/mbc.E10-10-0860; PMID: 21346199
- Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett 2009; 280:168 - 76; http://dx.doi.org/10.1016/j.canlet.2008.10.047; PMID: 19103471
- Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol 2009; 45:610 - 4; http://dx.doi.org/10.1016/j.oraloncology.2008.08.011; PMID: 18951835
- Iglesias-Linares A, Yañez-Vico RM, González-Moles MA. Potential role of HDAC inhibitors in cancer therapy: insights into oral squamous cell carcinoma. Oral Oncol 2010; 46:323 - 9; http://dx.doi.org/10.1016/j.oraloncology.2010.01.009; PMID: 20207580
- Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res 2006; 12:4103 - 11; http://dx.doi.org/10.1158/1078-0432.CCR-05-2404; PMID: 16818711
- Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW, et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res 2000; 60:6068 - 74; PMID: 11085529
- Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6:1057 - 65; http://dx.doi.org/10.4161/auto.6.8.13365; PMID: 20962572
- Ahn MY, Ahn SG, Yoon JH. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol 2011; 47:1032 - 8; http://dx.doi.org/10.1016/j.oraloncology.2011.07.027; PMID: 21856210