Abstract
The transcription factor E2F-1 plays a crucial role in the control of cell proliferation. E2F-1 has tumor suppressive properties by inducing apoptosis and autophagy. In this study, E2F-1 and its truncated form (E2Ftr), lacking the transactivation domain (TAD), were compared for their ability to induce autophagy. In Gaussia luciferase-based assays, both E2F-1 and E2Ftr induced the proteolytic cleavage of the autophagic marker LC3. In addition, LC3 and autophagy protein 5 (Atg5) were upregulated by E2F-1 and E2Ftr. Likewise, both E2F proteins induced a punctate pattern of GFP-tagged LC3, indicating autophagosome formation. The presence of double-membrane autophagic vesicles induced by E2F-1 and E2Ftr was confirmed by transmission electron microscopy (TEM). The application of z-VAD-fmk, a caspase inhibitor, partially blocked both E2F-1 and E2Ftr-mediated cytotoxicity. Moreover, Atg5−/− cells were more resistant to the E2F-1 or E2Ftr-induced cell killing effect than Atg5 wt cells. The TAD of E2F-1 is not essential for induction of autophagy; apoptosis and autophagy cooperate for an efficient cancer cell killing effect induced by E2F-1 or E2Ftr. E2Ftr-induced autophagy is a promising approach to destroy tumors that are resistant to conventional treatments.
Introduction
Advanced cancer often presents itself as a metastatic disease highly resistant to available therapies.Citation1 Most chemotherapy and molecular therapies against cancer are based on induction of apoptosis, type I programmed cell death (PCD). However, tumors likely develop apoptosis-resistant mechanisms before or during treatment and become resistant to conventional therapy based on apoptosis-mediated cell death.Citation1-Citation3 For example, overexpression of anti-apoptotic genes, including members of the Bcl-2 family, the heat shock protein (HSP) family and the inhibitor of apoptosis protein (IAP) family, has been shown to play a critical role in decreasing apoptosis-therapeutic efficacy.Citation4-Citation6 Activation of other cell death mechanisms to kill apoptosis-resistant cancer cells seems to be a new therapeutic approach. Several studies indicate that autophagy (type II PCD) may play an important role in destroying cancer cells.Citation7-Citation10
Autophagy is mainly known for its survival role in cells during starvation.Citation11,Citation12 It is activated by several factors, such as hypoxia;Citation13 rapamycin and tamoxifen treatment;Citation14,Citation15 and DNA damage.Citation16-Citation18 Autophagy mediates the degradation of intracellular components, including organelles and long-lived proteins. It starts with the formation of double-membrane vesicles known as autophagosomes that contain cytoplasm and organelles. Ultimately, autophagosomes fuse with lysosomes where the bulk of cytoplasmic content undergoes degradation, resulting in the liberation of amino acids and fatty acids that can be reused by cells.Citation19,Citation20
Autophagy appears to have a dual role in cells by acting as a survival mechanism and as a caspase-independent form of PCD.Citation7-Citation10 For example, caspase-8 inhibition causes catalase degradation and cell death by autophagy in L929 cells,Citation8,Citation9 and the cardiac glycoside oleandrin induces autophagic cell death in human pancreatic cancer PANC-1 cells.Citation10
E2F-1 is a transcription factor that is capable of promoting both cell proliferation and apoptotic cell death.Citation21-Citation26 E2F-1 protein contains a DNA binding domain (DBD), a dimerization domain (DD) and a TAD. We have shown that adenoviral vectors expressing a truncated form of E2F-1 (E2Ftr; aa 1–375), which lacks the TAD, can still induce apoptotic cell death and inhibit tumor growth in vitro and in vivo.Citation27,Citation28
Recently, E2F-1 was shown to induce autophagy.Citation13,Citation17 Increased E2F-1 activity upregulated the expression of four autophagy genes: microtubule-associated protein-1 light chain-3 (LC3), autophagy-related gene-1 (ATG1), ATG5 and damage-regulated autophagy modulator (DRAM).Citation17 BNIP3 was also found to be an E2F target gene required for hypoxia-induced autophagy.Citation13 E2F-1 binding sites were identified in BNIP3,Citation13 ATG1 and LC3 promoters, as well as in the first exon of the human DRAM gene.Citation17 It is believed that the transcription activity of E2F-1 is associated with its capability of inducing autophagy.Citation13,Citation17
Herein, we report that the TAD in E2F-1 is not required for induction of autophagy. We found that the expression of E2Ftr lacking the wild-type TAD activates autophagy, upregulates the autophagic markers and causes autophagosome formation. We also observed that E2Ftr protein accumulates to high levels in cancer cells and leads to strong autophagy activity compared with wild-type E2F-1. Our results suggest that E2Ftr-induced autophagy might provide an alternative and potent approach to destroy tumors that are resistant to conventional treatments.
Results
E2Ftr activates both autophagy and apoptosis pathways
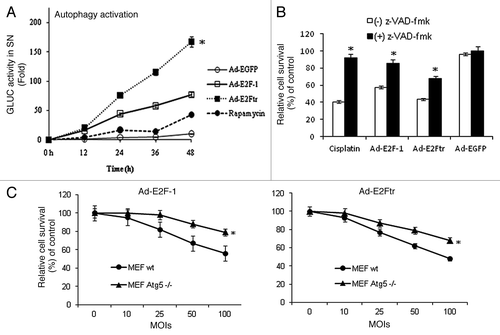
Polager et al. showed that E2F-1 regulates autophagy at the transcriptional level.Citation17 We examined whether the E2F-1 TAD is essential for autophagy activation. To study this, we performed Gaussia luciferase-based assays to monitor the activation of autophagy and apoptosis signaling pathways.Citation29,Citation30 The autophagy assay specifically measures the proteolytic cleavage of the autophagosome marker LC3 by the protease autophagy-related 4B (Atg4B). Atg4B cleaves at LC3 in the tripartite sensor protein (Actin-LC3-dNGLUC) encoded in the plasmid pEAK12-Actin-LC3-dNGLUC that was developed to monitor activation of the autophagic pathway.Citation29 The Atg4B-mediated cleavage of Actin-LC3-dNGLUC at LC3 results in release of GLUC that can be detected in supernatants. In this experiment, SK-MEL-2 cells were transfected with the autophagy sensor plasmid along with pCMV-E2F-1, pCMV-E2Ftr or empty control plasmid. Rapamycin, a well-known autophagy inducer, was used as a positive control. Supernatants were collected at 12, 24, 36 and 48 h for analysis of GLUC activity. We found that rapamycin (200 nM) increased GLUC activity 44-fold at 48 h, while pCMV-E2F-1 and pCMV-E2Ftr increased it by 24-fold and 20-fold respectively in comparison with empty vector, which was statistically significant (p < 0.05) ().
Figure 1. Quantification of autophagy and apoptosis activation by E2Ftr. A GLUC-based sensor to monitor autophagy or apoptosis was used. SK-MEL-2 cells were co-transfected with (A) pEAK12-Actin-LC3-dNGLUC (autophagy sensor) or (B) pEAK12-Actin-flagDEVDG2-dNGLUC (apoptosis sensor) and empty plasmid, pCMV-E2F-1 or pCMV-E2Ftr. Rapamycin (200 nM) was used as an autophagy inducer, and cisplatin (25 µM) was used as an apoptosis inducer. Supernatants were collected at different time points and analyzed for GLUC activity. Each point represents the mean of three independent experiments ± SD (bars) (*p < 0.05).
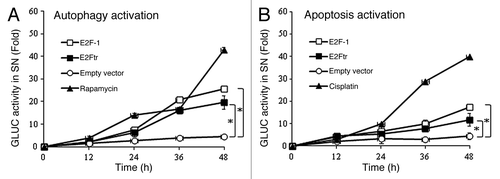
A method fundamentally similar to the one described above was used to detect apoptosis activation with the apoptosis sensor plasmid, pEAK12-Actin-flagDEVDG2-dNGLUC, where caspase-8 (extrinsic pathway) and caspase-9 (intrinsic pathway) cleaved at the conserved FDEVDG2 peptide.Citation29,Citation30 SK-MEL-2 cells were co-transfected with the apoptosis sensor plasmid and as described above. Supernatants were tested for apoptosis-related GLUC activity at different time points. Cisplatin, an apoptosis inducer, increased apoptosis activation 40-fold at 48 h. pCMV-E2F-1 and pCMV-E2Ftr increased release of GLUC 18- and 12-fold respectively in comparison with empty vector, which was statistically significant (p < 0.05) ().
The above experiments demonstrated that E2F-1 and E2Ftr can induce both autophagy and apoptosis activation, suggesting that the TAD deleted in E2Ftr is not required for activation of either of these pathways.
Upregulation of LC3-I and LC3-II by E2Ftr
There are two forms of LC3: LC3-I, a cytosolic protein and LC3-II, a lipidated form localized in the autophagosome membrane. LC3-II accumulation is a hallmark of autophagy.Citation31,Citation32 We analyzed the expression of LC3 and the conversion of LC3-I to LC3-II. LC3 expression was analyzed at 24 h after SK-MEL-2 melanoma cells were transfected with different concentrations of pCMV-E2F-1 or pCMV-E2Ftr. Untrasfected cells showed the endogenous E2F-1 expression levels. E2F-1 induced the highest levels of LC3-I and LC3-II at 2 µg of DNA (p < 0.05) (). In comparison, significant LC3-I and LC3-II expression was reached by E2Ftr when transfected with 0.25–0.5 μg of DNA (p < 0.05) (). We also observed that E2Ftr expression was detected at 0.1 µg of DNA, but no significant expression of LC3-I and LC3-II was observed.
Figure 2. E2Ftr induces autophagy-specific LC3-I and LC3-II protein expression. SK-MEL-2 cells were transfected at indicated concentrations of plasmids expressing (A) E2F-1 or (B) E2Ftr. Western blot and bar graphs of E2F-1, E2Ftr or LC3-II expression after transfection. Bars represent mean ± SEM expressed as fold change of E2F-1, E2Ftr or LC3-II vs α-actin (loading control) from 3 separate experiments, (*p < 0.05) increase in the level of expression.
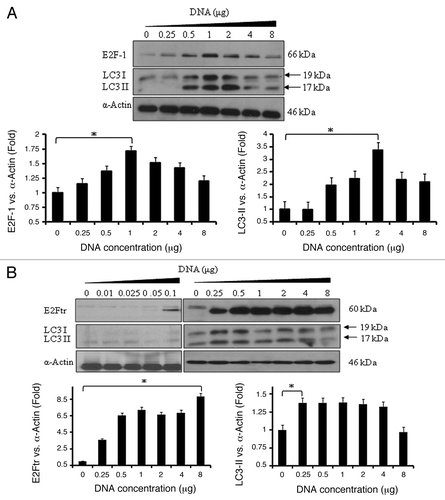
Expression of E2F-1 and LC3 started to decline at 4 µg of DNA. However, E2Ftr expression remained similar through increasing doses of DNA, but LC3 expression started to decline at 1 µg of DNA. These results show that E2F-1 and E2Ftr were both capable of upregulating LC3-I and LC3-II in a DNA dose-dependent manner and E2F-1 induced greater conversion of LC3-I to LC3-II than E2Ftr. Interestingly, lower concentrations of E2Ftr plasmid DNA (0.25–0.5 μg) are required for LC3 maximal induction compared with E2F-1 (1–2 μg).
E2Ftr induces the formation of autophagosomes
As mentioned above, the conversion of LC3-I to LC3-II is a useful and sensitive marker for distinguishing autophagy in mammalian cells. LC3-II is the only known mammalian protein identified that stably associates with the autophagosome membranes. To further validate the conversion of LC3-I to LC3-II, we used the GFP-tagged LC3 plasmid (pEGFP-LC3)Citation31 to monitor the localization of LC3 within cells. SK-MEL-2 cells were co-transfected with the monitory plasmid pEGFP-LC3 and empty plasmid or either pCMV-E2F-1 or pCMV-E2Ftr, rapamycin was used as positive control (). Co-transfection with pEGFP-LC3 and an empty plasmid showed a diffuse cytoplasmic pattern of fluorescence of GFP-LC3 (GFP-LC3-I form) within the cells. Conversion of diffuse cytosolic GFP-LC3 to membrane-associated GFP-LC3-II puncta were observed in cells transfected with pCMV-E2F-1 or pCMV-E2Ftr or treated with the positive control rapamycin ().
Figure 3. Autophagosome formation with GFP-LC3 incorporation is induced by E2Ftr. SK-MEL-2 cells were co-transfected with pEGFP-LC3 and empty plasmid, pCMV-E2F-1 or pCMV-E2Ftr. Incidence of punctate GFP-LC3 staining was analyzed under a fluorescence microscope. Rapamycin (200 nM) was used as the autophagy inducer. GFP diffuse expression was considered as GFP-LC3-I cytoplasmic localization. Incorporation of LC3 into the autophagosomes was depicted by GFP punctate pattern corresponding to GFP-LC3-II (arrows). chloroquine (10 µM) was used to analyze the autophagy flux. Images were obtained with Kodak MDS 290 software with the 40X objective. (B) Comparison of number of GFP dots per cell in absence or presence of chloroquine. A representative experiment is shown from three performed.
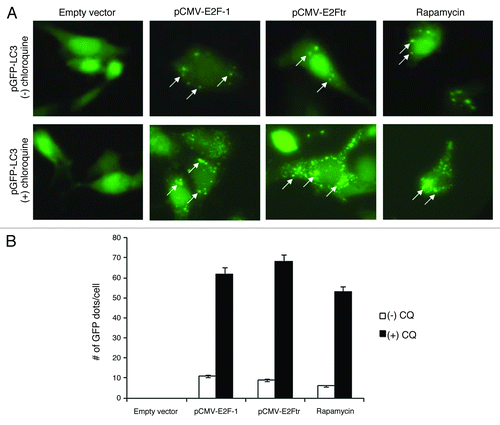
To verify that the fluorescence punctate pattern corresponds with autophagosomes, we used chloroquine (10 µM) to inhibit the fusion of autophagosomes with lysosomes, the later stage of autophagy. Chloroquine is a lysosomotropic agent that increases lysosomal pH and disrupts the autophagy flux, resulting in more autophagosomes accumulation. We observed that chloroquine treatment resulted in a higher accumulation of the fluorescent punctate pattern, from 11 to 62 GFP dots/cell in E2F-1 transfected-cells, from 9 to 68 GFP dots/cell in E2Ftr transfected-cells and from 6 to 53 GFP dots/cell in rapamycin-treated cells (). Therefore, these results verified that E2F-1 and E2Ftr are both capable of inducing autophagosomes formation.
Autophagy induced by E2Ftr is associated with increased Atg5 expression
Atg5 is essential for the formation of the phagophore during autophagosome assembly and conjugated with Atg12.Citation33,Citation34 We analyzed Atg5 and Atg12-Atg5 complex expression by transfecting SK-MEL-2 cells with 1 μg of either pCMV-E2F-1 or pCMV-E2Ftr. Atg5 and Atg12-Atg5 complex expression were monitored at different time points after transfection. Atg5 protein was induced at 12 h after transfection by both E2F-1 and E2Ftr and gradually increased up to 54 h (p < 0.05) (). However, no change was observed in the Atg12-Atg5 complex.
Figure 4. Effect of E2F-1 or E2Ftr expressions on Atg12-Atg5 complex or Atg5 protein levels and presence of Ad-wt in recombinant adenoviruses. (A) SK-MEL-2 cells were transfected with pCMV-E2F-1 or pCMV-E2Ftr. Expression of the Atg12-Atg5 complex and Atg5 expressions were analyzed at the indicated time points. (B) DM6 cells were infected with Ad-E2F-1 or Ad-E2Ftr at increasing MOI levels (0–100). Forty-eight hours after infection, the Atg12-Agt5 complex and Atg5 expressions were analyzed. Autophagy was associated with the appearance of a 32 kDa anti-Atg5-reactive protein. Bar graphs of Atg5 expression after transfection or infection. Bars represent mean ± SEM expressed as fold change of Atg5 vs α-actin (loading control) from 3 separate experiments, (*p < 0.05) increase in the level of expression. (C) Analysis of the possible presence of wild-type adenovirus in recombinant adenovirus vectors Ad-E2F-1, Ad-E2Ftr or Ad-EGFP. SK-MEL-2 cells were infected with Ad-wt, Ad-E2F-1, Ad-E2Ftr or Ad-EGFP at a MOI of 100. Thirty-six hours later, expression of E1A was analyzed with anti-E1A antibody. All immunoblots are representative of at least three independent experiments.
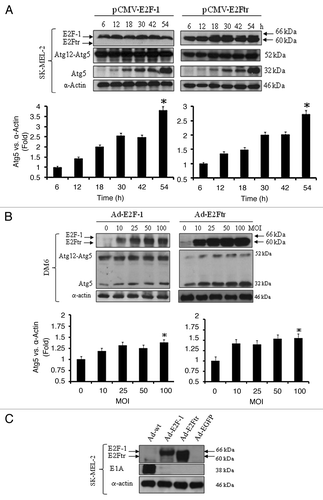
We have demonstrated so far that E2Ftr induces autophagy at similar levels to E2F-1 (–4A) and since we are interested in apply adenovirus E2Ftr gene transfer for gene therapy, we next infected DM6 melanoma cells with adenovirus expressing E2F-1(Ad-E2F-1) or E2Ftr (Ad-E2Ftr)Citation27 at increasing multiplicity of infection (MOI). Forty-eight hours after infection, a western blot was performed. We observed that, similar to SK-MEL-2 cells transfected with pCMV-E2F-1 or pCMV-E2Ftr, Atg5 expression increased in an Ad-E2F-1 or Ad-E2Ftr dose-dependent manner, whereas no change was observed in Atg12-Atg5 complex expression (). These results clearly indicate that E2Ftr, although lacking the TAD, was still able to upregulate autophagy gene Atg5 expression similar to wild-type E2F-1. These data also indicate that E2F-1- or E2Ftr-induced autophagic gene expression is reproducible in other melanoma cell type.
It has been previously reported that oncolytic adenoviruses can induce autophagy,Citation18,Citation35 and it is possible that recombinant adenoviruses may contain Ad-wt. To discard this possibility, SK-MEL-2 cells were infected with Ad-wt, Ad-E2F-1, Ad-E2Ftr or Ad-EGFP at a MOI of 100. Thirty-six hours later, expression of the E1A gene was analyzed. The adenoviral vector resulted in a significantly higher production of E2Ftr than E2F-1 in SK-MEL-2 cells (). We found that cells infected with Ad-E2F-1, Ad-E2Ftr or Ad-EGFP did not express E1A, whereas strong expression was detected only in cells infected with Ad-wt (). This result indicates that our Ad vectors are not contaminated with Ad-wt.
E2Ftr induces ultrastructural modifications in melanoma cells
The evident autophagy activation induced by E2F-1 and E2Ftr prompted us to examine the cell morphology closely. SK-MEL-2 cells were either not infected (mock) or were infected with Ad-EGFP, Ad-E2F-1 or Ad-E2Ftr at a MOI of 100. The cell morphology was first analyzed at 24 h after infection by light microscopy; cells were treated with Wright-Giemsa staining. The mock and cells infected with Ad-EGFP exhibited no difference in the cells’ morphology. Ad-E2F-1 and Ad-E2Ftr both induced changes in the cellular morphology, namely vesiculated-like cytoplasm (). Ad-E2Ftr caused a more flattened cell appearance.
Figure 5. Analysis of the cellular morphology induced by E2Ftr. (A) SK-MEL-2 cells were either not infected (mock) or were infected with Ad-EGFP, Ad-E2F-1 or Ad-E2Ftr at a MOI of 100. Wright-Giemsa staining was performed at 24 h, and cellular morphology was analyzed under a light microscope with the 40X objective. (B) SK-MEL-2 cells, infected as described above, were subjected to transmission electron microscope (TEM) at 24 h. Magnifcations are shown for mock [magnification x7000], Ad-EGFP [magnification, x7000], Ad-E2F-1, [magnification, x9800] or Ad-E2Ftr [magnification, x7000]) (Scale bar = 200 nm). Magnification of representative double-membrane vesicles is depicted at the bottom of the figure. Similar results were observed in two additional experiments.
![Figure 5. Analysis of the cellular morphology induced by E2Ftr. (A) SK-MEL-2 cells were either not infected (mock) or were infected with Ad-EGFP, Ad-E2F-1 or Ad-E2Ftr at a MOI of 100. Wright-Giemsa staining was performed at 24 h, and cellular morphology was analyzed under a light microscope with the 40X objective. (B) SK-MEL-2 cells, infected as described above, were subjected to transmission electron microscope (TEM) at 24 h. Magnifcations are shown for mock [magnification x7000], Ad-EGFP [magnification, x7000], Ad-E2F-1, [magnification, x9800] or Ad-E2Ftr [magnification, x7000]) (Scale bar = 200 nm). Magnification of representative double-membrane vesicles is depicted at the bottom of the figure. Similar results were observed in two additional experiments.](/cms/asset/dcb0d794-d8f9-4ec5-8acd-3044a3142032/kcbt_a_10921143_f0005.gif)
The presence of autophagic vesicles was further confirmed at the ultrastructural level by a detailed study through a transmission electron microscope (TEM), the gold standard method for autophagy morphology detection. As expected, cells infected with Ad-E2F-1 and Ad-E2Ftr showed evidence of double-membrane autophagic vesicles, a major characteristic of the autophagy mechanism (). In contrast, no evidence of autophagy was observed in neither mock nor Ad-EGFP-infected cells (). Collectively, these results demonstrate that E2Ftr, which lacks of the TAD, can induce autophagy as efficiently as E2F-1.
Adenovirus-mediated E2Ftr expression induces a strong activation of autophagy
We previously have shown that adenovirus Ad-E2Ftr can efficiently destroy cancer cells and inhibit tumor growth in vitro and in vivo.Citation27,Citation28 As E2Ftr can activate both the apoptosis and autophagy pathways, the Ad-E2Ftr may be a potent killer of apoptosis-resistant cancer cells. We investigated the Ad-E2Ftr capability in the promotion of autophagy activation in comparison with Ad-E2F-1. We tested autophagy activation in SK-MEL-2 cells transfected with pEAK12-Actin-LC3-dNGLUC and infected with Ad-EGFP, Ad-E2F-1 or Ad-E2Ftr at a MOI of 100. We observed that Ad-delivered E2Ftr increased GLUC activity 170-fold, significantly higher than the autophagic activities induced by Ad-E2F-1 (80-fold) and rapamycin (40-fold) (p < 0.05) ().
Figure 6. Autophagy activation and contribution of caspases pathway or autophagy in Ad-E2F-1 or E2Ftr-mediated killing effect. (A) SK-MEL-2 cells were transfected with pEAK12-Actin-LC3-dNGLUC followed by infection with Ad-EGFP, Ad-E2F-1 or Ad-E2Ftr at a MOI of 100 or treated with rapamycin (200 nM), which was used as autophagy inducer. Autophagy activation was monitored by Gaussia luciferase assay, as described in the Materials and Methods section. Each point represents the mean of three independent experiments ± SD (bars) (*p < 0.05). (B) DM6 cells were cultured in the absence or presence of general caspase inhibitor z-VAD-fmk at a concentration of 100 µM. Cells were then infected with Ad-E2F-1, Ad-E2Ftr or Ad-EGFP at a MOI of 100. Three days after infection, an MTT assay was performed. Each point represents the mean of three independent experiments ± SD (bars) (*p < 0.05). (C) Mouse embryonic fibroblast (MEF) wt or Atg5−/− were infected with Ad-E2F-1 or Ad-E2Ftr at increasing MOI levels. Three days after infection, an MTT assay was performed. Each point represents the mean of three independent experiments ± SD (bars) (*p < 0.05).
Apoptosis and autophagy cooperate for an efficient Ad-E2Ftr-cancer cell killing effect
We have demonstrated that adenovirus expressing E2F-1 or E2Ftr can induce apoptosisCitation28,Citation36 and autophagy. However, the contribution of apoptosis and autophagy to E2F-1 or E2Ftr-mediated cancer cell killing still remained unsolved. Therefore, we further investigated the contribution level of apoptosis and autophagy in E2F-1 or E2Ftr-mediated cancer cell death.
DM6 melanoma cells were cultured in the absence or presence of general caspase inhibitor z-VAD-fmk at a concentration of 100 µM. The cells then were treated with cisplatin (25 µM), which was used as a positive treatment, because it triggers typical caspase-associated cell death,Citation36 or infected with Ad-E2F-1, Ad-E2Ftr or Ad-EGFP at a MOI of 100. Three days after infection, an MTT assay revealed that z-VAD-fmk increased the relative survival rate from 43% to 90% of cells treated with cisplatin, indicating the effectiveness of z-VAD-fmk in inhibiting apoptosis in our experiments. In the absence of z-VAD-fmk, treatment with Ad-E2F-1 or Ad-E2Ftr resulted in 58% and 43% cell survival, respectively, whereas in presence of z-VAD-fmk, cell survival increased to 85% and 68%, respectively. This difference was statistically significant (p < 0.05). Ad-EGFP did not induce a significant cell killing effect (). The results suggest that Ad-E2F-1 and Ad-E2Ftr induces cancer cell killing, at least in part, by the caspase pathway.
We next assessed that ability of Ad-E2F-1 and Ad-E2Ftr to induce cell cytotoxicity independent of Atg5 autophagy gene. Mouse embryonic fibroblast (MEF) wt or Atg5−/−Citation37 were infected with Ad-E2F-1, Ad-E2Ftr or Ad-EGFP at increasing MOI levels. Three days after infection, an MTT assay revealed a dose-dependent cell killing effect with either Ad-E2F-1 or Ad-E2Ftr in wt or Atg5−/− cells; however, Atg5−/− cells were more resistant to Ad-E2F-1 or Ad-E2Ftr-mediated cytotoxicity than MEF cells wt for Atg5. This difference was statistically significant (p < 0.05) (). Ad-EGFP did not induce a significant cell killing effect (data not shown). The above results show that high levels of Ad-E2F-1 and E2Ftr strongly activate the autophagy pathway. It is therefore likely that autophagy and apoptosis cooperate in the Ad-E2F-1 or Ad-E2Ftr-mediated cancer cell killing effect. In addition, E2F-1 or E2Ftr-induced autophagy could be an early event that eventually results in apoptosis; or, autophagy and apoptosis are separate events, since both mechanisms were activated simultaneously ().
Discussion
The transcriptional activity of E2F-1 was previously reported to play an essential role in the induction of autophagy, upregulating the expression of autophagy genes LC3, ATG1, ATG5 and DRAM.Citation13,Citation17 In the present study, we investigated whether the E2F-1 TAD is required to induce autophagy. A GLUC-based assay revealed that E2Ftr was able to activate autophagy as efficiently as E2F-1 (); both E2F-1 and E2Ftr upregulated LC3 and Atg5 expression and caused a punctate distribution of GFP-tagged LC3 consistent with autophagosomes formation (–). Furthermore, analysis of the ultrastructural morphology of E2F-1 and E2Ftr-treated cells displayed the typical autophagic features: the presence of a highly vesiculated cytoplasm and double-membraned vesicles (). Our data demonstrated that the TAD deleted in E2Ftr is not required for activation of the autophagy pathway.
Previous studies have reported that Atg5 rarely exists as a monomer and is generally found forming a complex with Atg12.Citation33,Citation38 We found that Atg5 expression increases over the time, whereas no changes were observed in the Atg12-Atg5 complex in SK-MEL-2 cells transfected with E2F-1 or E2Ftr plasmids (). Similar results were found in DM6 cells infected with Ad-E2F-1 or Ad-E2Ftr (). We do not know why Atg5 was upregulated, but the Agt12-Atg5 complex remained unchanged. Other studies have demonstrated that autophagy is induced by virus or other agents and upregulation of Atg5 but not the Atg12-Atg5 complex.Citation39-Citation41 Free Atg5 is not functional in autophagy induction thus not an indicator of autophagy activity. It is possible that excessive Atg5 contributes to cell death. Indeed, it has recently been demonstrated that Atg5 plays a role in caspase-8 activation and apoptosis through intracellular death-inducing signaling complex (iDISC) formation.Citation42 The previous study of PolagerCitation17 showed that regulation of Atg5 by E2F-1 was indirect. Therefore, it is likely that an E2F-1/E2Ftr-targeted gene regulates Atg5. It would require an extensive screening to identify the gene targeted by E2F involved in Atg5 regulation and a more detailed study may determine the reason why Atg5, but not Atg12-Atg5, is upregulated by either E2F-1 or E2Ftr.
A possible mechanism for the activation of autophagy by E2F-1 may be through direct transcriptional activation of autophagy via E2F binding sites in the promoter of autophagy genes.Citation13,Citation17 Other mechanisms independent of the E2F-1 transactivation may also exist, as suggested by our study. A regulatory region within E2F-1 protein may directly induce autophagy without the transcriptional activity. However, E2Ftr still can increase the production of the autophagic proteins LC3 and Atg5 in a manner similar to E2F-1. E2Ftr may function as a dominant negative regulator by releasing the endogenous E2F-1 from the Rb/E2F-1 repression complex, with subsequent increase in LC3 and Atg5 transcription. It is also possible that E2Ftr activates autophagy by increasing the stability of the autophagy-related proteins or through other mechanisms involving protein-protein interactions. However, further investigation is required to elucidate the mechanism(s) by which E2Ftr induces autophagy.
Several reports have shown that induction of apoptosis is frequently associated with an increase of autophagy, which indicates that there is interplay between these two important cellular events.Citation43-Citation45 In this study, we showed that autophagy and apoptosis were activated simultaneously by E2F-1 or E2Ftr (). However, the reciprocal influence of the two pathways still remains to be clarified. We found that pretreatment of DM6 cells with caspase inhibitor z-VAD-fmk partially blocked the cytotoxicty induced by E2F-1 and E2Ftr (). On the other hand, MEF Atg5−/− were more resistant than MEF wt to E2F-1 or E2Ftr-mediated cell killing effect (). The effects of apoptosis inhibition (e.g., z-VAD) in cell death induced by Ad-E2F-1 and Ad-E2Ftr in WT vs Atg5−/− cells will be investigated in our further studies. It is possible that E2F-1 or E2Ftr-induced autophagy could be an early event in apoptosis, or that autophagy and apoptosis are separate events. However, it seems that autophagy and apoptosis cooperate for an efficient E2F-1 or E2Ftr-mediated cell killing effect. This close relationship has been reported previously when an Atg5 fragment produced by calpain cleavage had pro-apoptotic properties.Citation46,Citation47
Our efforts were to find the difference between E2F-1 and E2Ftr in regard to induction of autophagy. The only significant difference observed was the higher accumulation of E2Ftr in cells infected with the Ad vector. The high expression of E2Ftr may correspond to greater autophagy activity. Thus, our results suggest that E2Ftr can induce autophagy as efficiently as E2F-1, which may provide an alternative and potent approach to destroy tumors that are resistant to apoptosis-based treatments.
In summary, this study highlights a new mode of regulation of autophagy by E2F-1 that occurs independently of the TAD. We provide a new insight into the cell death mechanisms triggered by E2F-1 and E2Ftr. In addition to the classical apoptosis, we found that a caspase-independent death was also activated. Basically, what makes E2Ftr more promising for therapeutics is the absence of E2F-1 oncogenicity related to its transcriptional activity, and its high protein expression level, which is related to similar autophagy and cell death activity compared with E2F-1. Moreover, the activation of both apoptosis and autophagy pathways by E2Ftr might not only destroy apoptosis-sensitive cancer cells, but sensitize those tumors that are resistant to conventional apoptosis-based therapies.
Materials and Methods
Cell lines and culture conditions
The human melanoma SK-MEL-2 (ATCC # HTB-68) and human embryonic kidney HEK-293 (ATCC # CRL-1573) cell lines were purchased from the American Type Culture Collection. The human melanoma DM6 cell line was kindly provided to us by Dr. Douglas S. Tyler (Duke University Medical Center).Citation48 Wt and Atg5−/− MEF cells were kindly provided to us by Dr. Noboru Mizushima (Tokyo Medical and Dental University).Citation37
SK-MEL-2 and HEK-293 cell lines were cultured in α-minimal essential medium (α-MEM) (Cat # 32571). The DM6 cell line was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) (Cat # 12440). Wt and Atg5−/− MEF cells were cultured in Dulbecco’s Modification of Eagles Medium (DMEM) (Cat # 21063029). The media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Cat # 12664025) and penicillin/streptomycin (100 U/ml) (Cat # 15140122). All cell culture reagents were obtained from Gibco BRL. Cells were cultured in a humid incubator with 5% CO2 at 37°C. All experiments were repeated at least three times.
Plasmids
To construct the plasmid expressing the truncated E2F-1 (E2Ftr, 1–375), the 1.1 kb E2Ftr DNA fragment with EcoRI and XbaI ends was first ligated with the SV40 poly (A) fragment cleaved out with XbaI and HindIII from plasmid pUHD10–3.Citation49 The E2Ftr-poly (A) was then inserted into pUHD15–1Citation50 cleaved with EcoRI and HindIII, resulting in plasmid pHZ330 with the E2Ftr flanked with the CMV promoter and the SV40 poly (A). The E2Ftr expression cassette, cleaved from pHZ330 with XhoI and HindIII, was finally inserted into the plasmid pΔ1sp1A,Citation51 resulting in pHZ333 (pCMV-E2Ftr).
To construct the plasmid pCMV-E2F-1, the E2Ftr in pCMV-E2Ftr, digested with EcoRI and XbaI, was replaced with the 1.4 kb DNA fragment containing the E2F-1 coding sequence. The plasmid pΔ1sp1ACitation51 is used as an empty plasmid control in this study. The pEAK12-Actin-LC3-dNGLUC (autophagy sensor) and pEAK12-Actin-flagDEVDG2-dNGLUC (apoptosis sensor) were kindly provided by Dr. Brian Seed and were used for co-transfection with plasmids previously mentioned, as well as GFP-LC3 plasmid, which was generously donated by Dr. Tamotsu Yoshimori.
Recombinant adenoviral vectors
The three replication-defective recombinant adenoviral vectors used in this report were all deleted in the viral E1 gene. The Ad5CMV-E2F-1 (Ad-E2F-1) vector contained the transgene E2F-1 under the control of the CMV promoter as described.Citation36 AdTet-EGFP (Ad-EGFP) expressing enhanced green fluorescent protein and AdTet-E2Ftr3 (Ad-E2Ftr) expressing E2F-1 lacking the TAD were both recently reported by us.Citation27
Transient transfection
Cells (1 × 105) per well in 12-well plates were transfected with the indicated plasmids using Lipofectamine 2000 transfection reagent (Invitrogen, Cat # 11668019), according to the manufacturer’s protocol. For luciferase release assay, plasmids pCMV-E2F1 or pCMV-E2Ftr were used at 0.8 µg plus 0.8 µg pEAK12-Actin-LC3-dNGLUC or pEAK12-Actin-flagDEVDG2-dNGLUC. For GFP-LC3 puncta assay, pEGFP-LC3 was used at 0.8 µg. The conditions above mentioned yielded ~70% of transfection efficiency.
Luciferase release assay
Gaussia luciferase (GLUC) activity was determined using the Renilla Luciferase kit (Promega, Cat #E2810) as described.Citation29 To avoid harvesting luciferase activity from detached cells, supernatants were spun at 14,000 rpm for 5 min. Ten microliters of the supernatant were diluted 1:10 in 100 μL 1 × Renilla lysis buffer, and 20 μL of this mixture was added to 100 μL of Renilla substrate prior to analysis in a luminescence plate reader.
Western blot
Western blotting was performed by standard procedure using cell lysates and mouse anti-human E2F-1 monoclonal antibody (KH95) (Santa Cruz Biotechnology, sc-251), rabbit anti-LC3 polyclonal antibody (Novus Biologicals, NB100–2331), rabbit anti-Atg5 polyclonal antibody (C-terminal) (Sigma-Aldrich, A0713) and rabbit anti-human α-Actin polyclonal antibody (Sigma-Aldrich, A5060). Next, the membranes were incubated with anti-mouse immunoglobulin (Ig) or anti-rabbit Ig, peroxidase-linked, species-specific whole Abs (Amersham). Electrochemiluminescent (ECL) reagents were used to detect the signals, according to the manufacturer’s instructions (Amersham). All films were scanned with an optical scanner (HP Scan Jet 5550C) and quantified by measuring the density of each band using ImageJ 1.44p software (Wayne Rasband, NIH). To correct possible unequal loading, each band's density was normalized to its α-actin density. To allow multiple comparisons between gels, each sample was compared with its respective control on the same gel. Results are expressed as fold change.
GFP-LC3 puncta
Plasmid containing enhanced green fluorescent protein (EGFP) and microtubule-associated protein 1 light chain 3 (LC3) (pEGFP-LC3)Citation31 were used to detect autophagosome formation by co-transfecting with pE2F-1 or pE2Ftr into SK-MEL-2 cells. After transfection, chloroquine (10 µM) was or was not added. At the indicated times, cells were washed three times with 1xPBS and fixed with 4% paraformaldehyde (PFA). Then, cells were observed under a fluorescence microscope. Cells were classified as having a predominantly diffuse GFP stain or having numerous punctate structures representing autophagosomes. Images were obtained with Kodak MDS 290 software with the 40X objective.
Wright-Giemsa staining
Adherent cells were analyzed 24 h after infection. Cells were washed three times with 1xPBS and fixed with 4% paraformaldehyde. Then, cells were stained as described previouslyCitation52 and analyzed under a light microscope (Olympus Microsystems). Images were obtained in bright-field with the 40X objective.
Transmission electron microscopy
Electron microscopy was performed 24 h after infection. Briefly, melanoma cell samples were washed three times with 1x PBS, trypsinized, and collected by centrifuging at 1,000 x g for 5 min. The cell pellets were fixed with 4% paraformaldehyde overnight at 4°C, post-fixed with 2% OsO4 in 0.1M NaPO4 buffer for 1 h at room temperature, and dehydrated stepwise with ethanol. The pellets were embedded in Araldite 502 resin for sectioning. Images of thin sections were observed under a Philips CM 10 transmission electron microscope operated at 60 kv.
MTT assay
Cell viability was assessed at 72 h after infection by measuring the conversion of the tetrazolium salt 3-(4, 5-dimethylthiazol-2-)-2, 5-diphenyltetrazolium bromide (MTT) to formazan, according to the manufacturer’s instructions (Roche, Cat # 1465007).
Statistical analysis
The results of the in vitro assays were analyzed by the unpaired Student’s t test using a 1-way ordinary parametric analysis of variance. p < 0.05 was considered statistically significant.
| Abbreviations: | ||
| TAD | = | transactivation domain |
| PCD | = | programmed cell death |
| ATG | = | autophagy-related gene |
| LC3 | = | microtubule-associated protein-1 light chain-3 |
| E2Ftr | = | truncated E2F-1 |
| GFP | = | green fluorescent protein |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Lung Cancer Research Foundation (JGGG), award numbers R01CA129975 (HSZ) and R01CA90784 (KMM) from the National Cancer Institute and GMB081410 (KMM and HSZ) from the Kentucky Lung Cancer Research Program. AGG received a scholarship from the National Council of Science and Technology (CONACYT) of Mexico. We are grateful to Dr. Brian Seed (Harvard Medical School, Boston, MA) for kindly providing the pEAK12-Actin-LC3-dNGLUC and pEAK12-Actin-flagDEVDG2-dNGLUC plasmids and Dr. Tamotsu Yoshimori (Osaka University, Japan) for kindly providing the EGFP-LC3 plasmid. We thank Margaret A. Abby and Nancy Alsip for editing.
Notes
† These authors contributed equally to this work.
Related Research Data
References
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene 2003; 22:3138 - 51; http://dx.doi.org/10.1038/sj.onc.1206454; PMID: 12789290
- Hersey P, Zhang XD. Overcoming resistance of cancer cells to apoptosis. J Cell Physiol 2003; 196:9 - 18; http://dx.doi.org/10.1002/jcp.10256; PMID: 12767037
- Zhang XD, Wu JJ, Gillespie S, Borrow J, Hersey P. Human melanoma cells selected for resistance to apoptosis by prolonged exposure to tumor necrosis factor-related apoptosis-inducing ligand are more vulnerable to necrotic cell death induced by cisplatin. Clin Cancer Res 2006; 12:1355 - 64; http://dx.doi.org/10.1158/1078-0432.CCR-05-2084; PMID: 16489094
- Xanthoudakis S, Nicholson DW. Heat-shock proteins as death determinants. Nat Cell Biol 2000; 2:E163 - 5; http://dx.doi.org/10.1038/35023643; PMID: 10980714
- Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol 2001; 11:6 - 10; http://dx.doi.org/10.1016/S0962-8924(00)01874-2; PMID: 11146277
- Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev 1999; 13:239 - 52; http://dx.doi.org/10.1101/gad.13.3.239; PMID: 9990849
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol 2007; 78:217 - 45; http://dx.doi.org/10.1016/S0070-2153(06)78006-1; PMID: 17338918
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500 - 2; http://dx.doi.org/10.1126/science.1096645; PMID: 15131264
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 2006; 103:4952 - 7; http://dx.doi.org/10.1073/pnas.0511288103; PMID: 16547133
- Newman RA, Kondo Y, Yokoyama T, Dixon S, Cartwright C, Chan D, et al. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr Cancer Ther 2007; 6:354 - 64; http://dx.doi.org/10.1177/1534735407309623; PMID: 18048883
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211 - 6; http://dx.doi.org/10.1038/35056522; PMID: 11265251
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27:6229 - 42; http://dx.doi.org/10.1128/MCB.02246-06; PMID: 17576813
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 34; http://dx.doi.org/10.1038/nrc1692; PMID: 16148885
- Bursch W, Ellinger A, Kienzl H, Török L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996; 17:1595 - 607; http://dx.doi.org/10.1093/carcin/17.8.1595; PMID: 8761415
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860 - 4; http://dx.doi.org/10.1038/onc.2008.117; PMID: 18408756
- Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, Rao XM, Chen L, McMasters KM, et al. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011; 416:9 - 15; http://dx.doi.org/10.1016/j.virol.2011.04.017; PMID: 21575980
- Baehrecke EH. Autophagy: dual roles in life and death?. Nat Rev Mol Cell Biol 2005; 6:505 - 10; http://dx.doi.org/10.1038/nrm1666; PMID: 15928714
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Sala A, Nicolaides NC, Engelhard A, Bellon T, Lawe DC, Arnold A, et al. Correlation between E2F-1 requirement in the S phase and E2F-1 transactivation of cell cycle-related genes in human cells. Cancer Res 1994; 54:1402 - 6; PMID: 8137237
- Yang XH, Sladek TL. Overexpression of the E2F-1 transcription factor gene mediates cell transformation. Gene Expr 1995; 4:195 - 204; PMID: 7787412
- Xu G, Livingston DM, Krek W. Multiple members of the E2F transcription factor family are the products of oncogenes. Proc Natl Acad Sci U S A 1995; 92:1357 - 61; http://dx.doi.org/10.1073/pnas.92.5.1357; PMID: 7877982
- Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol 1994; 14:8166 - 73; PMID: 7969153
- Johnson DG, Cress WD, Jakoi L, Nevins JR. Oncogenic capacity of the E2F1 gene. Proc Natl Acad Sci U S A 1994; 91:12823 - 7; http://dx.doi.org/10.1073/pnas.91.26.12823; PMID: 7809128
- Bandara LR, Buck VM, Zamanian M, Johnston LH, La Thangue NB. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J 1993; 12:4317 - 24; PMID: 8223441
- Gomez-Gutierrez JG, Rao XM, Garcia-Garcia A, Hao H, McMasters KM, Zhou HS. Developing adenoviral vectors encoding therapeutic genes toxic to host cells: comparing binary and single-inducible vectors expressing truncated E2F-1. Virology 2010; 397:337 - 45; http://dx.doi.org/10.1016/j.virol.2009.11.021; PMID: 20003994
- Gomez-Gutierrez JG, Garcia-Garcia A, Hao H, Rao XM, Montes de Oca-Luna R, Zhou HS, et al. Adenovirus-mediated expression of truncated E2F-1 suppresses tumor growth in vitro and in vivo. Cancer 2010; 116:4420 - 32; http://dx.doi.org/10.1002/cncr.25322; PMID: 20549818
- Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy 2008; 4:801 - 6; PMID: 18641457
- Ketteler R, Sun Z, Kovacs KF, He WW, Seed B. A pathway sensor for genome-wide screens of intracellular proteolytic cleavage. Genome Biol 2008; 9:R64; http://dx.doi.org/10.1186/gb-2008-9-4-r64; PMID: 18387192
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84 - 91; http://dx.doi.org/10.4161/auto.1.2.1697; PMID: 16874052
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 2006; 22:463 - 75; http://dx.doi.org/10.1016/j.molcel.2006.04.014; PMID: 16713577
- Tyler MA, Ulasov IV, Lesniak MS. Cancer cell death by design: apoptosis, autophagy and glioma virotherapy. Autophagy 2009; 5:856 - 7; PMID: 19430207
- Dong YB, Yang HL, Elliott MJ, McMasters KM. Adenovirus-mediated E2F-1 gene transfer sensitizes melanoma cells to apoptosis induced by topoisomerase II inhibitors. Cancer Res 2002; 62:1776 - 83; PMID: 11912154
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2003; 35:553 - 61; http://dx.doi.org/10.1016/S1357-2725(02)00343-6; PMID: 12672448
- Alonso MM, Jiang H, Yokoyama T, Xu J, Bekele NB, Lang FF, et al. Delta-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol Ther 2008; 16:487 - 93; http://dx.doi.org/10.1038/sj.mt.6300400; PMID: 18253154
- Byun JY, Yoon CH, An S, Park IC, Kang CM, Kim MJ, et al. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 2009; 30:1880 - 8; http://dx.doi.org/10.1093/carcin/bgp235; PMID: 19783847
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 2011; 286:12924 - 32; http://dx.doi.org/10.1074/jbc.M110.138958; PMID: 21300795
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012; 287:12455 - 68; http://dx.doi.org/10.1074/jbc.M111.309104; PMID: 22362782
- Zhang JQ, Li YM, Liu T, He WT, Chen YT, Chen XH, et al. Antitumor effect of matrine in human hepatoma G2 cells by inducing apoptosis and autophagy. World J Gastroenterol 2010; 16:4281 - 90; http://dx.doi.org/10.3748/wjg.v16.i34.4281; PMID: 20818811
- Huang SW, Liu KT, Chang CC, Chen YJ, Wu CY, Tsai JJ, et al. Imiquimod simultaneously induces autophagy and apoptosis in human basal cell carcinoma cells. Br J Dermatol 2010; 163:310 - 20; http://dx.doi.org/10.1111/j.1365-2133.2010.09827.x; PMID: 20426785
- Archer CR, Koomoa DL, Mitsunaga EM, Clerc J, Shimizu M, Kaiser M, et al. Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochem Pharmacol 2010; 80:170 - 8; http://dx.doi.org/10.1016/j.bcp.2010.03.031; PMID: 20362557
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124 - 32; http://dx.doi.org/10.1038/ncb1482; PMID: 16998475
- Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol 2006; 8:1045 - 7; http://dx.doi.org/10.1038/ncb1006-1045; PMID: 17013414
- Ko SH, Ueno T, Yoshimoto Y, Yoo JS, Abdel-Wahab OI, Abdel-Wahab Z, et al. Optimizing a novel regional chemotherapeutic agent against melanoma: hyperthermia-induced enhancement of temozolomide cytotoxicity. Clin Cancer Res 2006; 12:289 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-05-0210; PMID: 16397054
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol 1994; 14:1669 - 79; PMID: 8114703
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 1992; 89:5547 - 51; http://dx.doi.org/10.1073/pnas.89.12.5547; PMID: 1319065
- Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci U S A 1994; 91:8802 - 6; http://dx.doi.org/10.1073/pnas.91.19.8802; PMID: 8090727
- Gomez-Gutierrez JG, Souza V, Hao HY, Montes de Oca-Luna R, Dong YB, Zhou HS, et al. Adenovirus-mediated gene transfer of FKHRL1 triple mutant efficiently induces apoptosis in melanoma cells. Cancer Biol Ther 2006; 5:875 - 83; http://dx.doi.org/10.4161/cbt.5.7.2911; PMID: 16861905