Abstract
One of the hallmarks of cancer is metabolic deregulation. Many tumors display increased glucose uptake and breakdown through the process of aerobic glycolysis, also known as the Warburg effect. Less studied in cancer development and progression is the importance of the glutamine (Gln) pathway, which provides cells with a variety of essential products to sustain cell proliferation, such as ATP and macromolecules for biosynthesis. To this end Gln dependency was assessed in a panel of non-small cell lung cancer lines (NSCLC). Gln was found to be essential for the growth of cells with high rates of glutaminolysis, and after exploring multiple genes in the Gln pathway, GLS1 was found to be the key enzyme associated with this dependence. This dependence was confirmed by observing the rescue of decreased growth by exogenous addition of downstream metabolites of glutaminolysis. Expression of the GLS1 splice variant KGA was found to be decreased in tumors compared with normal lung tissue. Transient knock down of GLS1 splice variants indicated that loss of GAC had the most detrimental effect on cancer cell growth. In conclusion, NSCLC cell lines depend on Gln for glutaminolysis to a varying degree, in which the GLS1 splice variant GAC plays an essential role and is a potential target for cancer metabolism-directed therapy.
Introduction
A great variety of extracellular and intracellular changes take place during the initiation and development of tumors. A hallmark of cancer is altered energy balance. For sustained expansion of cancer cells, both energy in the form of ATP, and macromolecules such as nucleotides and lipids need to be generated at increased rates.Citation1
It is generally accepted that cancer cells can depend on both glucose (Glc) and the amino acid glutamine (Gln) for viability and growth. As such, they can show increased uptake of both to accommodate this dependence.Citation2-Citation4 Through the process of glycolysis, Glc can be metabolized to lactate and secreted or enter the trichloric acid (TCA) cycle in the mitochondria. Citrate efflux for de-novo fatty acid synthesis will leave a void in the cycle, which can be supplemented by the process of glutaminolysis. In the process of glutaminolysis, Gln enters the cell and is then converted by glutaminase (GLS) into glutamate and subsequently α ketoglutarate, which then enters the TCA cycle. It can exit the cycle as malate through malic enzyme and be processed into lactate. Glutamine also provides a nitrogen source necessary for biosynthesis in proliferating cells.Citation5 Finally, glutamate can be converted into glutathione, a well-known anti-oxidant that can help defend cancer cells against oxidative stress and result in chemoresistance.Citation6,Citation7
GLS is the initial enzyme in the glutaminolysis pathway, and catalyzes the hydrolytic deamidation of glutamine to glutamate and ammonia. GLS can be expressed as two isoforms in human cells: GLS1, which is also known as kidney glutaminase (KGA), and GLS2, also known as liver glutaminase (LGA). GLS1 can exist as several splice variants through alternative splicing, and KGA is considered the full length GLS1. KGA is derived from exons 1–14 and 16–19, whereas a second GLS1 splice variant, GAC has an alternative carboxy-terminus (from exon 15 only), resulting in a lower molecular weight protein.Citation8 Both variants have intact GLS enzyme domains, and only KGA contains ankyrin repeats in its unique carboxy terminus. After localizing to the mitochondria, the N-terminus of both GLS1 variants can be cleaved to give rise to the final, active enzyme.Citation9
Recently, regulation of GLS1 and glutaminolysis in cancer has been shown to be under oncogenic control. Oncogenic levels of c-Myc can facilitate glutamine addiction of cancer cells by suppressing miR-23a/b and subsequently increasing GLS1 expression and glutamine metabolism.Citation10,Citation11 Glutamine metabolism is also found essential for K-ras-mediated anchorage-independent growth as depletion of Gln or transient knockdown of GLS1 inhibits colony formation observed in K-ras mutant cells.Citation12 Inhibition of GLS1 inhibits oncogenic transformation by Rho GTPase in fibroblasts, and also the growth of breast and lymphoma cancer cells.Citation13 Finally, in glioma cells expressing mutant IDH1, inhibition of GLS inhibits cell growth.Citation14 Beyond oncogene-mediated glutamine addiction, other studies describe that stable loss of GLS1 expression in Ehrlich ascites cells decreases growth, tumorigenicity, and also anti-oxidant capacity.Citation15,Citation16
Of all tumor types, lung cancer is the most common cause of mortality, with 159,390 deaths each year in the USA alone.Citation17 The two main classifications of lung cancer are small cell lung carcinoma (SCLC) and non-small cell lung carcinoma (NSCLC). There are three main sub-types of NSCLC: squamous cell lung carcinoma, adenocarcinoma and large cell lung carcinoma. Patients with SCLC respond generally better to initial chemotherapy and radiation, whereas patients with NSCLC are less responsive. For this reason we chose to study the potential importance of the glutamine pathway in NSCLC.
Although the importance of glycolysis has been studied extensively in tumor development, glutamine dependence and glutaminolysis is less well understood. In this study, the role of glutaminolysis, and GLS1 in particular, in cancer cell growth was studied in NSCLC cell lines.
Results
NSCLC cell lines show a range of glutamine dependency
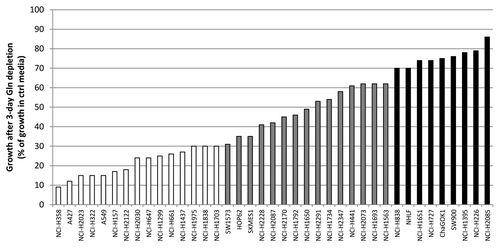
The role of glutaminolysis in cancer cell growth is not well understood. We therefore sought to investigate the relative dependency on Gln in non-small cell lung cancer (NSCLC). A panel of 39 cell lines of which 38 NSCLC lines and 1 non-cancerous lung cell line was subjected to growth in the presence or absence of Glc or Gln for 72 h. The effect of metabolite dependency on cell growth was assayed by measuring total ATP levels. The NSCLC lines showed a range of dependency on Gln, as demonstrated by reduced ATP levels (; Table S1). A total of 15 out of 39 lines showed Gln dependency as these lines had a 70% or more reduced growth in Gln free media. Fifteen lines showed reduced growth between 30–70% and were considered to show intermediate Gln dependence. Finally, 9 out of 39 lines showed a relative insensitivity to Gln depletion, and displayed reduced cell growth of 30% or less. The non-cancerous line NHLF displayed a relative insensitivity to Gln depletion.
Figure 1. Gln dependency of NSCLC lines. Cells were grown in the presence or absence of Gln after which cell growth was assessed. Gln-dependency was rated arbitrarily: white bars represent Gln-dependent lines with growth in absence of Gln being equal or less than 30% of control; gray bars represent intermediate lines (growth between 30 and 70%), and black bars Gln-independent lines (growth equal or more than 70%).
Glutamine-dependent cell lines consume more glutamine
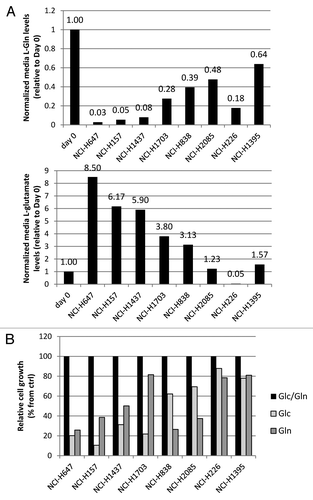
We next sought to understand if the cause of Gln dependency observed among the cell lines tested was due to their ability to uptake exogenous Gln or if there were differences in intracellular mechanisms of Gln processing. As Gln and its direct metabolite glutamate are amino acids, a panel of cell lines, both Gln-dependent and independent, were grown in media containing both Glc and Gln, and subsequently levels of amino acids were measured as an indication of consumption or production (). Of the four Gln-dependent lines (NCI-H1703,NCI-H647, NCI-H157 and NCI-H1437), three lines displayed high Gln uptake (92–97%), which was accompanied with high glutamate secretion (5.9- to 8.5-fold increase over time 0) under normal growth conditions, and NCI-H1703 cells showed Gln uptake of 72% and 3.8-fold glutamate secretion. In contrast, the four Gln-independent lines NCI-838, NCI-H2085, NCI-H226 and NCI-H1395 showed lower Gln uptake of 61, 52, 82 and 36%, and glutamate secretion of 3.1-, 1.2-, 0.05- and 1.6-fold respectively. Qualitatively, Gln-dependent cell lines generally display high basal glutamine consumption compared with Gln-independent lines. To confirm that these glutamine consumption correlate with Gln dependency only and not with general nutrient dependency, these cell lines were grown in normal media, or in media depleted of Glc or Gln, and subsequently cell growth was assessed (). Results of this assay suggest that the correlation of glutaminolysis rates (from ) with Gln dependency is not observed as compared with cell line Glc dependency.
Figure 2. Amino acid analysis of NSCLC lines. (A) Relative free glutamine and glutamate levels in conditioned media. (B) Effect of Gln or Glc depletion of these lines on cell growth. Values were calculated from triplicate samples.
GLS 1 is the rate-limiting enzyme in glutaminolysis in Gln-dependent NCI-H1703 cells
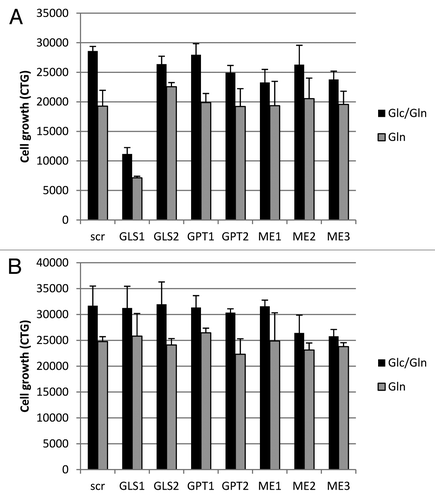
Our results indicate a preference for basal glutamine consumption in Gln-dependent NSCLC lines compared with independent lines. We therefore investigated which enzyme in the glutaminolysis pathway is rate-limiting for glutamine consumption. To this end, the Gln-dependent line NCI-H1703 and Gln-independent line NCI-H226 were transiently transfected with SMARTpool siRNA directed against enzymes with key roles in glutaminolysis and are not part of the essential Krebs cycle. These were GLS1, GLS2, glutamate pyruvate transaminase (GPT) isoforms, which convert glutamate into α ketoglutarate, and malic enzyme (ME) isoforms that are responsible for the conversion of malate into pyruvate which subsequently is released from cells after conversion to lactate. Following siRNA transfection, cells were grown in Glc-containing or depleted media for an additional 72 h and cell growth was assessed. Knockdown of GLS 1 (GLS1) showed a decrease in cell growth of NCI-H1703 cells (61% decrease in glucose containing media and 63% decrease in the absence of glucose; ). In contrast, GLS1 knockdown caused no negative effect on growth in the Gln-independent NCI-H226 cells (). Knockdown of the other enzymes had no effect on cell growth. These results suggest that GLS1 is an essential enzyme in NCI-H1703 cells for glutaminolysis, while other cells rely on alternative mechanisms for Gln processing.
Figure 3. Knockdown of glutaminolysis targets in Gln-dependent and independent cell lines. Cell growth was assayed in NCI-H1703 (A) and NCI-H226 cells (B), after being transiently transfected with siRNA for a variety of enzymes involved in glutaminolysis, and subsequently grown in media with or without Glc for 4 d. The data points shown represent the mean ± SD of triplicate samples.
Glutamine depletion and GLS1 inhibition have similar effects on cell viability
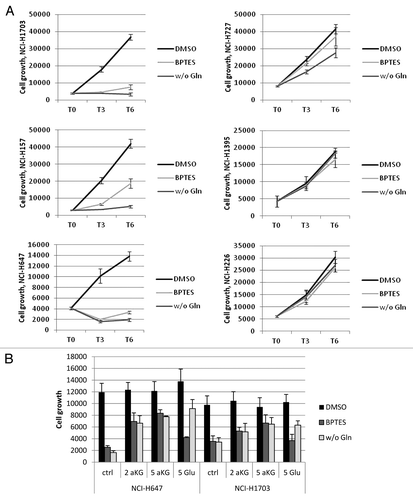
As transient knockdown of GLS1 in the Gln-dependent line NCI-H1703 decreased cell growth, which was not observed in the Gln-independent line NCI-H226, it is plausible that either withdrawal of exogenous Gln or loss of GLS1 function would affect growth through decreased glutaminolysis. However, a variety of enzymes other than GLS1 process Gln, and the withdrawal of exogenous Gln could affect growth through these alternate pathways.Citation20 To study the importance of GLS1 in the observed effects of Gln withdrawal on cell growth, a panel of cell lines, both Gln-dependent and independent, were incubated with the GLS1-specific inhibitor bis-2[5-phenylacetamido-1,2,4-thiadiazol-2-yl]ethylsulfide (BPTES).Citation21 Alternatively, cells were grown either in the presence or absence of Gln, after which cell growth was assessed. Gln-dependent lines show similar decreases in growth with both Gln depletion and BPTES treatment, varying from 86–91% decrease at day 6 with Gln depletion and 55–79% with BPTES treatment (, left panel). In contrast, little decrease was observed under either condition in the Gln-independent lines, (2–33% without Gln, and 11–14% with BPTES) treatment (, right panel). These results indicate that GLS1 action, rather than other Gln utilizing enzymes, is accountable for effects observed with Gln withdrawal. As glutamate is formed by GLS1, we next examined whether glutamate or the downstream product of glutaminolysis, α ketoglutarate (aKG) could rescue either Gln depletion or GLS1 inhibition (). Both Gln-dependent lines NCI-H647 and NCI-H1703 show similar decreases in growth by Gln withdrawal and BPTES treatment for 2 d (86 and 79% growth reduction respectively in NCI-H647, and 65 and 64% growth reduction in NCI-H1703 cells). The addition of both glutamate and aKG to glutamine free media allowed cells to grow up to ~70% of their normal growth potential in both cell lines. In contrast to Gln withdrawal, where both products are able to rescue cell growth, with BPTES treatment, only aKG and not glutamate, is able to rescue growth. In conclusion, effects of Gln depletion on cell growth are caused largely by decreased glutaminolysis as GLS1 inhibition has similar negative effects on growth and downstream metabolites are able to partially rescue growth.
Figure 4. Importance of GLS1 function in Gln-dependent and independent lines. The data points shown represent the mean ± SD of triplicate samples. (A) Cells were grown for indicated times in the absence or presence of Gln, or treated with 10uM BPTES, after which cell growth was assessed. (B) Cells grown in the absence of Gln or treated with BPTES were co-treated with 2 mM or 5mM DM-aKG or DM-Glu for 2 d to rescue growth. Cell growth numbers were ATP values measured by CTG.
GAC is the predominant GLS1 isoform in NSCLC
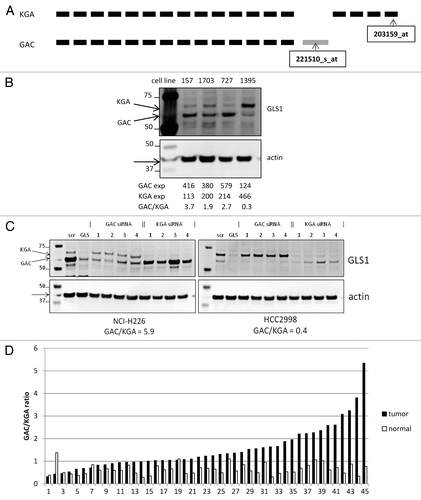
Having established that glutaminolysis, and in particular GLS1 function, is essential for the growth of a subset of NSCLC cell lines, the expression of GLS1 was studied in greater detail. It has been described that the expression is not limited to full length GLS1, which is also known as KGA (669 amino acids of length), but a shortened splice variant named GAC (which is 598 amino acids long) can also be expressed.Citation8 GAC is derived from a transcript with an alternative exon in the 3′ end, replacing the last four exons in the KGA transcript, resulting in lack of ankyrin repeats that are present in the C-terminus of KGA. Expression of these two GSL1 splice variants is not well studied, but it is suggested that GAC is overexpressed in tumors.Citation22 We analyzed Affymetrics U133plus2 gene array probe sequences and discovered that each GLS1 splice variant has a specific probe on the chip (). By analyzing these specific probes for either GAC or KGA, expression in NSCLC lines was estimated (Table S1). To confirm these mRNA ratios, and also study the expression of GAC and KGA protein, cell lines with specific GAC:KGA ratios, based on mRNA levels, were analyzed by western blot for GLS1 protein expression using an antibody directed against the common region of both variants (). The two bands observed were of predicted protein sizes for GAC and KGA after post-translation cleavage of the N-terminus (58 and 66 kDa respectively), and the ratio observed reflected the more quantitative mRNA ratio for each cell line. This suggests that both splice variants can be detected at the protein level, and that protein and transcript ratios are similar. To further study GLS1 splice variant expression in NSCLC cell lines, specific siRNA oligos targeting either GAC or KGA or both were designed and introduced in cancer cell lines NCI-H226 and HCC2998, and subsequent knockdown was analyzed by western blot analysis (). GLS1 SMARTpool siRNA treatment resulted in decreased expression of both bands, whereas GAC siRNA treatment only targeted the lower GLS band, and conversely KGA siRNA treatment targeted the upper band. As analysis of GLS1 splice variant expression was now possible on both the mRNA and protein levels, we next assessed the relative transcript expression of the two GLS1 splice variants in primary NSCLC tumors compared with matched normal lung tissues (). The GAC:KGA ratio was found to be significantly increased in lung tumors compared with matched normal lung tissue, resulting in GAC being the predominant GLS splice variant in tumors (p value = 2E-7). This increased GAC:KGA ratio in primary NSCLC tumors was caused by a significant decrease of KGA expression, from median value of 416 in normal lung to 164 in tumor (p value = 0.043; Fig. S1A). GAC expression, however, was not significantly different between tumor and normal tissue (p value = 0.67; Fig. S1B). The same predominance of GAC over KGA was also observed in NSCLC cell lines (Table S1).
Figure 5. Analysis of GLS1 splice variant expression/ratio. (A) Schematic representation of GLS1 splice variant transcripts, and the location of the variant specific probes. (B) NSCLC cell lysates were analyzed for GLS1 expression. Transcript expression levels are depicted as Affymetrics MAS5 values. (C) Cell lines with varying GAC:KGA ratios were transiently transfected with siRNA oligos for either GAC or KGA after which GLS1 splice variant expression was analyzed by western blot analysis. (D) NSCLC tumors and matched normal lung tissues were analyzed for GAC:KGA ratio.
Loss of GAC affects NSCLC cell growth more than loss of KGA
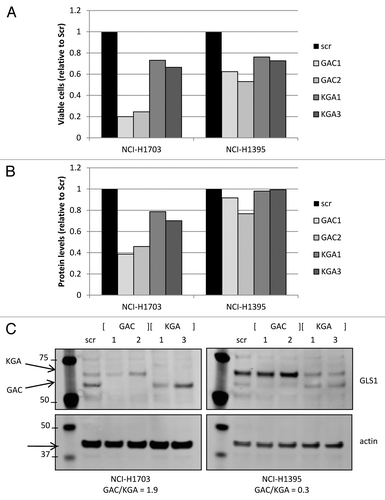
As specific transient downregulation of individual GLS1 splice variants, either GAC and/or KGA, can be achieved through siRNA oligos directed against their unique 3′-end transcript sequences, we next wanted to address the requirement of each individual GLS1 splice variant for cell growth. To this end, cell lines with different GAC:KGA ratios were transiently transfected with one of two siRNA oligos targeting either GAC or KGA, and viable cells were counted, or total protein levels were measured (). GAC knockdown resulted in 75–80% and 38–47% reduced cell growth in NCI-H1703 cells and NCI-H1395 cells respectively, whereas KGA knockdown only reduced growth by 27–34% and 24–27% respectively. Similar trends were observed with total protein levels as growth readout. Importantly, these observations were made in both Gln-dependent NCI-H1703 cells with predominant GAC expression, and in Gln-independent NCI-H1395 cells in which KGA is the predominant splice variant. As can be observed in , knockdown of the individual GLS1 splice variants was achieved, when analyzing the protein expression of equalized cell lysates from the growth assay. These data suggest that GAC is the more essential GLS1 splice variant in these cancer cells.
Figure 6. Transient GLS1 splice variant knockdown. Cell growth was assessed by measuring viable cell count (A) or total protein levels (B; BCA analysis) in cell lines that had GLS1 splice variant transiently knocked down by specific siRNA oligos directed against either GAC or KGA. Equalized protein lysates of experimental samples (from A) were analyzed for GLS1 splice variant and actin expression (C).
Discussion
NSCLC cell lines were analyzed for their dependency on metabolites, particularly glutamine. Lines that are sensitive to Gln depletion do have relatively high basal glutamine consumption, and inhibitor and knockdown studies show that GLS1 is a key component in Gln utilization. As GLS1 can be expressed as two splice variants, namely GAC and KGA, the expression of these variants was investigated. GAC:KGA ratios were found to be higher in tumors compared with normal tissue, and transient knockdown of GAC had a greater effect on growth than knockdown of KGA.
We showed that Gln-dependent lines have a higher glutaminolysis rate compared with independent lines. Further studies will be needed to determine the cause for this relative Gln dependency. Expression levels of the rate-limiting enzyme GLS1, or specifically GAC levels, could play a determining role in establishing Gln deprivation sensitivity. However, neither absolute GLS1 expression levels nor GLS1 splice variant ratio correlate with sensitivity (Table S1). Also, expression levels of glutamine synthase, which is responsible for the reverse reaction (glutamate to Gln), do not inversely correlate with Gln dependency (data not shown). Certain oncogenes have been linked to increased dependency of cell lines to Gln. For instance, Myc regulates GLS1 expression, and Gln is found essential for K-ras, IDH1 mutant and Rho GTPase-induced cancer cell growth.Citation10-Citation14 However, no correlation between Gln dependency and selected oncogene status was observed (Table S1). Likely, a combination of various factors determines the sensitivity to Gln in NSCLC cancer cells.
We demonstrated that in primary lung tumors the ratio of GAC over KGA increases, and showed the essential role for GAC in tumor cell growth, even in the presence of glucose. Increased glutaminolysis and GLS1 expression has been demonstrated previously in cancer.Citation23 The increased GAC:KGA ratio is consistent between previous studies and our results.Citation8,Citation24 In the tissue culture environment, the GAC:KGA ratio did not influence glutamine dependency. However, more work is needed to explore the consequences of altering this ratio in the metabolic microenvironment of primary NSCLC. What is the reason for increasing the GAC:KGA by increasing GAC expression, or by decreasing KGA expression? One explanation for one mechanism vs. the other could be the difference of net Gln influx between tumors. It has been shown that expression of Gln transporters such as ASCT2 and SN2 is found increased by oncogenic Myc.Citation11 Myc can also increase GLS1 expression.Citation10 In NSCLC tumors, this increased Gln transporter expression is not observed, or observed to a lesser degree, requiring the cells to downregulate KGA rather than upregulate GAC.
It is plausible that the GAC:KGA ratio is not only altered in NSCLC by downregulation of KGA transcription, but also by increased GAC protein expression. It is hypothesized that pH-response elements in the 3′-untranslated region (3′-UTR) of GAC can stabilize this transcript under acidic conditions, which can occur in tumors, and thus resulting in increased protein expression.Citation25 Although an attractive explanation, no experimental evidence has been described to date, and preliminary in-house data with cancer cells grown under various pH conditions showed no effect on absolute GAC protein expression (data not shown). Further studies are needed to elucidate if this is a mechanism of regulating protein expression. Structurally, both GAC and KGA have identical GLS domains, while only the carboxy termini differ between them. Kinetic studies and the X-ray crystallographic structures of GAC suggest that GAC is the better adapted GLS variant to exert its function at increasing levels of its activator phosphate.Citation26
As tumors display an altered GAC:KGA ratio, and the fact that these cells are fast proliferating cells with increased need for nucleotides, it is plausible to assume that the changed ratio in these cells is to shift the Gln pool in part toward nucleotide synthesis. Compatible with this idea, is the observation that KGA can be localized in the cytosol, whereas GAC is mitochondrial.Citation26 Also, the observation that downstream metabolites glutamate and α ketoglutarate were not able to fully rescue growth with GLS1 inhibition or Gln depletion, suggests that glutamine can be used for other purposes such as nucleotide synthesis. An interesting link can be drawn in this respect with the alternative splicing occurring with the PKM gene, which displays a shift from the PKM1 to the PKM2 splice variant in cancer, resulting in a shift from glucose feeding into the TCA cycle toward glucose providing biosynthesis of nucleotides, amino acids and phospholipids.Citation27
Previous studies have been performed with either transient or stable GLS1 knockdown.Citation10,Citation12,Citation15,Citation16 These studies are in agreement with our observed importance of GLS1 for tumor cell growth, but do not address the specific contribution of individual splice variants GAC and KGA. Small molecules, such as Gln mimetic 6-diazo-5-oxo-L-norleucine (DON), have been shown to reduce tumor growth in combination with modified diet in animal models.Citation28 In cell systems, DON inhibits cell proliferation by disruption of mitochondrial function, and premature senescence.Citation29,Citation30 However, being a Gln mimetic, DON can inhibit a variety of Gln-utilizing enzymes, and not exclusively GLS.Citation20 BPTES is an allosteric specific inhibitor of GLS1, presumably affecting both KGA and GAC.Citation21 As a tool compound it has been exploited in studies with mutant IDH1 to inhibit cancer cell growth.Citation14 Recently, a novel GLS inhibitor was identified in an inhibitory screen with Rho GTPase-transformed cells.Citation13 The target identified was GAC, although KGA targeting was not tested. In conclusion, given the increased GAC:KGA ratio observed in tumors compared with normal tissues, targeting GAC specifically may maximize anti-tumor effects while minimizing effects on normal tissues.
Materials and methods
Cell lines
Cell lines, free of Mycoplasma, were grown in RPMI (Gibco 72400) containing 10% fetal bovine serum (Hyclone), trypsinized using TrypLE (Gibco) and counted on Vi-Cell XR counter (Beckman Coulter).
Metabolic screening
NSCLC lines were seeded in a 96-well plate format in RPMI containing Glc and glutamax, and supplemented with 10% FBS at a growth rate-dependent density. After 24 h, media was carefully removed and cells were placed in 100ul RPMI containing Glc and Gln (Gibco 11875), or without Gln (Gibco 21700), or without Glc (Gibco 11879), and supplemented with 10% FBS. Cells were immediately assayed for cell growth, or grown for 72 or 144 h before assaying. The media of cells was refreshed after 72 h.
Knockdown assay
Cells were transiently transfected in a 6- or 96-well format using a reverse transfection protocol with lipofectamine RNAiMAX according to the manufacturer’s protocol (Invitrogen). Cells were introduced to either 10nM non-specific scrambled, or GLS1, or other glutaminolysis target targeting SMARTpool siRNA (Dharmacon). For knockdown of GAC or KGA, specific siRNA oligos were used (GAC: GGAAAGUCUGGGAGAGAAAUU, CUAUGAAAGUCUCCAACAAUU, CCUUUGGACCAUUGGACUAUU, AAAAGAGACAGUAUGGAAAUU; KGA: CCCAAGGACAGGUGGAAUAUU, CUGGAAGCCUGCAAAGUAAUU, GGACUAUGAUUCUAGAACAUU, GUACACACCUCAAGGAGAUUU). After 24 h, media was replaced with RPMI containing Gln with or without Glc, and supplemented with 10% FBS, and cells were incubated for an additional 72 h period, unless indicated otherwise, after which they were assayed.
Inhibitor/rescue studies
Cells were seeded in a 96-well plate format in RPMI containing Glc and glutamax, and supplemented with 10% FBS at a growth rate-dependent density. After 24 h, media was carefully removed and cells were placed in 100 µl RPMI, which was supplemented with 10% FBS, and contained Gln with or without 10 µM of BPTES, and in presence of absence of dimethyl-α ketoglutarate (DM-aKG) and dimethyl-glutamate (DM-Glu) (Sigma). Cells were subsequently grown for the indicated periods of time after which they were assayed.
Cell growth assay
Cell growth was assessed by measuring total ATP levels using CellTiterGlo (Promega), according to the manufacturer’s protocol.
Amino Acid analysis
Cells were seeded in a 6-well plate format in at a growth rate-dependent density. After 24 h, media was replaced by RPMI containing Glc and Gln, and supplemented with 10% FBS. After 3 d, conditioned media was collected and analyzed on Hitachi High-Technologies Model L-8900 dedicated amino acid analyzer (AminoAcids.com). Amino acid levels were normalized to total media amino acid levels minus Gln and glutamate, and subsequently calculated relative to hour 0.
Gene expression analysis
Gene expression was analyzed as described.Citation18 For NSCLC tumors and normal lung tissues, data was obtained from NCBI’s GEO database using data set GSE18842.Citation19 The raw data files from GSE18842 were similarly processed by normalizing all probe signal intensities to a value of 100 using the mas5 algorithm in the Affymetrix Microarray Analysis Suite 5.0. The expression values were obtained from the following probes: 221510_s_at (GAC), 203159_at (KGA), 205531_s_at (GLS2), 202431_s_at (c-Myc), 214058_at (l-Myc) and 209757_s_at (n-Myc). NSCLC cell line expression data was generated internally and available through National Cancer Institute caBIG Website: https://cabig.nci.nih.gov/caArray_GSKdata/.
Western blot analysis
Protein samples were prepared by cell lysis in RIPA buffer (Sigma) complemented with protease and phosphatase inhibitors. Protein content was measured by BCA assay (Pierce), after which samples were equalized for protein concentration. Generally, 10–20 µg protein was subjected to western blot, and subsequently analyzed by Li-Cor. GLS1 antibody was obtained from Abcam (ab93434), and actin antibody from Sigma.
| Abbreviations: | ||
| NSCLC | = | non-small cell lung cancer |
| Glc | = | glucose |
| Gln | = | glutamine |
| Glu | = | glutamate |
| aKG | = | alpha-etoglutarate |
| GLS | = | glutaminase |
| GAC | = | glutaminase C |
| KGA | = | kidney glutaminase |
| GPT | = | glutamate pyruvate transaminase |
| ME | = | malic enzyme |
| BPTES | = | bis-2[5-phenylacetamido-1,2,4-thiadiazol-2-yl]ethylsulfide |
Additional material
Download Zip (227.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank members of the Cancer Metabolism Discovery Performance Unit at GlaxoSmithKline for helpful discussions.
Supplemental Material
Supplemental material may be found here:
Related Research Data
References
- Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 2010; 10:267 - 77; http://dx.doi.org/10.1038/nrc2817; PMID: 20300106
- Fischer CP, Bode BP, Souba WW. Adaptive alterations in cellular metabolism with malignant transformation. Ann Surg 1998; 227:627 - 34, discussion 634-6; http://dx.doi.org/10.1097/00000658-199805000-00003; PMID: 9605654
- Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, et al. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer 2011; 117:2926 - 38; http://dx.doi.org/10.1002/cncr.25868; PMID: 21692052
- DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313 - 24; http://dx.doi.org/10.1038/onc.2009.358; PMID: 19881548
- Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle 2010; 9:3921 - 32; http://dx.doi.org/10.4161/cc.9.19.13139; PMID: 20935507
- Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 2002; 34:439 - 58; http://dx.doi.org/10.1016/S1357-2725(01)00143-1; PMID: 11906817
- Backos DS, Franklin CC, Reigan P. The role of glutathione in brain tumor drug resistance. Biochem Pharmacol.
- Elgadi KM, Meguid RA, Qian M, Souba WW, Abcouwer SF. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics 1999; 1:51 - 62; PMID: 11015561
- Perera SY, Chen TC, Curthoys NP. Biosynthesis and processing of renal mitochondrial glutaminase in cultured proximal tubular epithelial cells and in isolated mitochondria. J Biol Chem 1990; 265:17764 - 70; PMID: 2211660
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762 - 5; http://dx.doi.org/10.1038/nature07823; PMID: 19219026
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105:18782 - 7; http://dx.doi.org/10.1073/pnas.0810199105; PMID: 19033189
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010; 107:8788 - 93; http://dx.doi.org/10.1073/pnas.1003428107; PMID: 20421486
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18:207 - 19; http://dx.doi.org/10.1016/j.ccr.2010.08.009; PMID: 20832749
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 2010; 70:8981 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-10-1666; PMID: 21045145
- Lobo C, Ruiz-Bellido MA, Aledo JC, Márquez J, Núñez De Castro I, Alonso FJ. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 2000; 348:257 - 61; http://dx.doi.org/10.1042/0264-6021:3480257; PMID: 10816417
- Lora J, Alonso FJ, Segura JA, Lobo C, Márquez J, Matés JM. Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem 2004; 271:4298 - 306; http://dx.doi.org/10.1111/j.1432-1033.2004.04370.x; PMID: 15511236
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225 - 49; http://dx.doi.org/10.3322/caac.20006; PMID: 19474385
- Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, et al. Molecular target class is predictive of in vitro response profile. Cancer Res 2010; 70:3677 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-09-3788; PMID: 20406975
- Sanchez-Palencia A, Gomez-Morales M, Gomez-Capilla JA, Pedraza V, Boyero L, Rosell R, et al. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int J Cancer 2011; 129:355 - 64; http://dx.doi.org/10.1002/ijc.25704; PMID: 20878980
- Pinkus LM. Glutamine binding sites. Methods Enzymol 1977; 46:414 - 27; http://dx.doi.org/10.1016/S0076-6879(77)46049-X; PMID: 909432
- Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 2007; 406:407 - 14; http://dx.doi.org/10.1042/BJ20070039; PMID: 17581113
- Roberg BA, Torgner IA, Kvamme E. Kinetics of a novel isoform of phosphate activated glutaminase (PAG) in SH-SY5Y neuroblastoma cells. Neurochem Res 2010; 35:875 - 80; http://dx.doi.org/10.1007/s11064-009-0077-7; PMID: 19894115
- Szeliga M, Obara-Michlewska M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009; 55:71 - 5; http://dx.doi.org/10.1016/j.neuint.2009.01.008; PMID: 19428809
- Szeliga M, Matyja E, Obara M, Grajkowska W, Czernicki T, Albrecht J. Relative expression of mRNAS coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res 2008; 33:808 - 13; http://dx.doi.org/10.1007/s11064-007-9507-6; PMID: 17940881
- Porter LD, Ibrahim H, Taylor L, Curthoys NP. Complexity and species variation of the kidney-type glutaminase gene. Physiol Genomics 2002; 9:157 - 66; PMID: 12045296
- Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 2012; 109:1092 - 7; http://dx.doi.org/10.1073/pnas.1112495109; PMID: 22228304
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 2011; 43:969 - 80; http://dx.doi.org/10.1016/j.biocel.2010.02.005; PMID: 20156581
- Yoshioka K, Takehara H, Okada A, Komi N. Glutamine antagonist with diet deficient in glutamine and aspartate reduce tumor growth. Tokushima J Exp Med 1992; 39:69 - 76; PMID: 1412455
- Unterluggauer H, Mazurek S, Lener B, Hütter E, Eigenbrodt E, Zwerschke W, et al. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 2008; 9:247 - 59; http://dx.doi.org/10.1007/s10522-008-9134-x; PMID: 18317946
- Wu F, Lukinius A, Bergström M, Eriksson B, Watanabe Y, Långström B. A mechanism behind the antitumour effect of 6-diazo-5-oxo-L-norleucine (DON): disruption of mitochondria. Eur J Cancer 1999; 35:1155 - 61; http://dx.doi.org/10.1016/S0959-8049(99)00099-4; PMID: 10533463