Abstract
Dihydrofolate reductase (DHFR) is an essential enzyme involved in de novo purine and thymidine biosynthesis. For several decades, selective inhibition of DHFR has proven to be a potent therapeutic approach in the treatment of various cancers including acute lymphoblastic leukemia, non-Hodgkin’s lymphoma, osteogenic sarcoma, carcinoma of the breast, and head and neck cancer. Therapeutic success with DHFR inhibitor methotrexate (MTX) has been compromised in the clinic, which limits the success of MTX treatment by both acquired and intrinsic resistance mechanisms. We report that benzamide riboside (BR), via anabolism to benzamide adenine dinucleotide (BAD) known to potently inhibit inosine monophosphate dehydrogenase (IMPDH), also inhibits cell growth through a mechanism involving downregulation of DHFR protein. Evidence to support this second site of action of BR includes the finding that CCRF-CEM/R human T-cell lymphoblasic leukemia cells, resistant to MTX as a consequence of gene amplification and overexpression of DHFR, are more resistant to BR than are parental cells. Studies of the mechanism by which BR lowers DHFR showed that BR, through its metabolite BAD, reduced NADP and NADPH cellular levels by inhibiting nicotinamide adenine dinucleotide kinase (NADK). As consequence of the lack of NADPH, DHFR was shown to be destabilized. We suggest that, inhibition of NADK is a new approach to downregulate DHFR and to inhibit cell growth.
Introduction
Dihydrofolate Reductase (DHFR) catalyzes the reduction of folic acid and dihydrofolate to tetrahydrofolate, which after further modification, participates as an essential cofactor in the transfer of single carbon moieties in the synthesis of purines and thymidylate and some amino acids. Given its participation in nucleotide biosynthesis, DHFR has been exploited as a therapeutic target in the treatment of various malignancies as well as for the treatment of infectious diseases.Citation1 Lower doses of methotrexate (MTX) are commonly used for the treatment of rheumatoid arthritis and psoriasis and as an immunosuppressant.
Despite its prevalent use, resistance to MTX still hinders its success in treating many cancers. Gene amplification represents one mechanism by which cancer cells increase DHFR levels and attain resistance to MTX treatment.Citation2 It is thought that even low level gene amplification in ALL is enough to elicit resistance to MTX and thus permit a relapse in this disease.Citation3 While less common, mutations in DHFR affecting the binding affinity of MTX may also elicit resistance.Citation4 The most prevalent mechanisms of MTX resistance are those involving drug uptake. MTX is actively transported into the cell via the ubiquitous high capacity, low-affinity reduced folate carrier (RFC) and the low capacity high affinity folate receptor (FR) system,Citation5 or the recently described acid pH proton coupled transporter.Citation6 Mutations in RFC have been implicated in MTX resistance in both leukemia and osteosarcoma.Citation7 Lack of retention of MTX in cells may also lead to resistance to high dose pulse treatment. Upon entering the cell, several glutamate residues are added to MTX via polyglutamyl synthetase (FPGS). This polyglutamylation process is a critical step in assuring intracellular retention and accumulation of MTX. The status of FPGS and its ability to facilitate polyglutamation is considered an important predictor of MTX efficacy in patients.Citation1,Citation8 A novel mechanism of MTX resistance recently described is a polymorphism in the DHFR 3′ UTR that inhibits binding of microRNA-24 to DHFR transcripts that leads to mRNA and DHFR overexpression.Citation9
NAD(P) analogs have been of growing interest in recent years. Initially, investigators were reluctant to pursue developing NAD analogs due to lack of specificity. Studies have shown that it is possible to target enzymes with NAD analogs in a highly specific manner, and NAD analogs are used in the clinic today.Citation10 Examples include the inosine monophosphate dehydrogenase (IMPDH) inhibitor tiazofurin, which is used for treatment of chronic myelogenous leukemia, and mycophenoloic acid (MPA), which binds at the nicotinamide subdomain of IMPDH, and as a result is used in the clinic as an immunosuppressant. In addition, inhibitors of poly-ADP-ribose polymerase (PARP), which plays an important role in the DNA damage sensor pathway, are used alone or in combination with DNA damaging agents in the treatment of BRCA deficient breast and ovarian cancers.Citation11
Benzamide derivatives were initially recognized for significant PARP inhibition.Citation12 In an effort to reduce toxicity, the conjugated benzamide, benzamide riboside (3-β-D-ribofuranosyl) benzamide(BR), was synthesized.Citation13 While BR showed minimal activity against PARP, its primary metabolite, benzamide adenine dinucleotide (BAD), acts as a potent inhibitor of IMPDH.Citation14 BR is converted intracellularly to its metabolite BAD via NMN adenylyltransferase, the rate-limiting enzyme in NAD biosynthesis.Citation10 It is also thought that BAD may be further modified with the addition of a phosphate group via NAD kinase, however few studies have examined the activity of BADP in cells.Citation15 Cytotoxicity studies demonstrated high sensitivity to BR in several tumor cell lines.Citation16 Although BR as a nucleoside penetrates cell membranes well, BAD as a pyrophosphate does not. BAD is unstable in vitro being cleaved to BR by the action of phosphoesterases and phosphatases. Therefore in our studies we used methylenebis(phosphonate)-BAD (β-BAD), and other NAD analogs. We found earlier that that replacement of the pyrophosphate oxygen of BAD by a –CH2- group afforded methylene biphosphonate BAD (β-BAD), which is a metabolically stable mimic of BAD.Citation10 Over the years numerous methylene bis (phosphonate) NAD analogs have been synthesized and these have been found to be close analogs of their corresponding pyrophosphates.Citation10
We previously identified an analog of NAD(P), thionicotinamide adenine dinucleotide, in a search for novel compounds that would inhibit dihydrofolate reductase (DHFR) via binding to the NADPH site.Citation17 NADS and NADPS were equally cytotoxic, indicating that NADS was converted to NADPS intracellularly. Although NADPS was only a weak inhibitor of DHFR, NADPS caused a marked decrease in the levels of DHFR. We subsequently showed that NADPS is an inhibitor of NAD kinase, and as a result lowered NADPH levels.Citation17 Earlier studies from this and other laboratories had shown a critical role for NADPH in protecting DHFR from proteolysis.Citation18,Citation19 Further, lowered levels of intracellular levels of DHFR increase the susceptibility of cells to methotrexate, a powerful stoichiometric inhibitor of this enzyme. BR is converted to benzamide adenine dinucleotide (BAD) in cells and has been shown to be a potent inhibitor of NAD Kinase.Citation20 Herein we show that BR has a second site of action: its metabolite, BAD, by inhibiting NAD kinase decreased NADPH levels, and destabilized DHFR leading to effective cell killing.
Results
NAD analogs exhibit weak inhibition of DHFR
A library of putative NAD(P) analogs, were initially tested as potential inhibitors of IMPDH.Citation10 We hypothesized that given the structural similarity of the molecules in this library to that of NAD, if converted to NADP(H) analogs in cells, may also bind to the NADP and NADPH binding site in DHFR, and inhibit its activity. In order to investigate whether or not these molecules inhibited DHFR activity we employed a spectrophotometric assay (see methods). In the presence of the NAD analogs little or only weak DHFR inhibition was observed (). All of the compounds tested did not achieve 50% enzyme inhibition (IC50) at concentrations of 1 mM, with the exception of compound 1277, which had an IC50 of 0.47 mM. The data do not rule out the possibility that these molecules could operate as pro-drugs and converted to metabolites with greater inhibitory activity against DHFR in vivo.
Table 1. Evaluation of DHFR activity was accomplished by measuring absorbance of NADPH using spectrophotometric analysis at 340 nm
BR inhibits cell growth through a mechanism involving DHFR
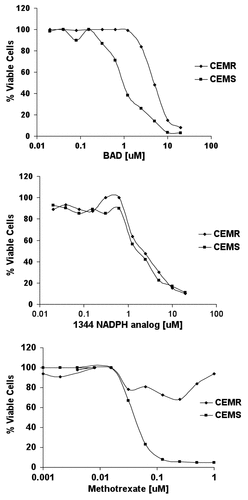
To determine if the NAD analogs were anabolized in cells and inhibited DHFR, CCRF-CEM and CCRF-CEM/R cells were treated with varying concentrations of drug (0.01 μM- 10 μM) for 96 h. CCRF-CEM/R cells are a sub-line of human CCRF-CEM T-cell lymphoblasts possessing 5 times the DHFR gene copy number and enzyme levels as CCRF-CEM cells, and are resistant to MTX treatment (9). We reasoned that if a drug were acting via inhibition of DHFR, it would be less effective against cells with increased DHFR activity. Following a 96-h treatment, all CCRF-CEM experimental groups showed a decrease in cell viability (). The concentration at which 50% cell kill was achieved (IC50) was 1 μM for BR, 1.2 μM for 1344, an NADPH analog specifically inhibiting IMPDH and 0.06 μM for MTX (). While the IC50 remained the same for CCRF-CEM/R and CCRF-CEM cells treated with 1344(), the IC50 values for CCRF-CEM/R cells treated with BR were increased 7-fold (). As a control, CCRF-CEM/R cells treated with MTX did not reach 50% viability even at high drug concentrations (). While CCRF-CEM/R cells were not as resistant to BR treatment as compared with MTX, these data suggested that BR mediates inhibition of cell growth, in part, through a mechanism involving DHFR.
Figure 1. Three thousand CCRF-CEMR or CCRF-CEMS were plated in RPMI media containing varying concentrations of drug, highest concentration of 20 μM, and incubated at 37ºC for 96 h. Cytotoxicity was measured using the trypan blue exclusion assay and cell viability was evaluated using the Vi-CELL Series Cell Viability Analyzer.
Human NAD kinase catalyzes the formation of BADP from BAD in vitro
BAD is the major metabolite of BR, and is a potent inhibitor of IMPDH and human NAD kinase.Citation16,Citation20 However, few studies investigated any further cellular modification of BAD that could possibly contribute to, or alter its activity. One study suggested that while an inhibitor of NAD kinase, BAD may also be a substrate for phosphorylation.Citation15 In its phosphorylated state we predicted that the added negative charge on BAD may increase its affinity for the NADP binding pocket of DHFR and thus increase its potency for inhibition. We sought to investigate if β-BAD is a substrate for NAD kinase and if so, whether or not β-BADP inhibited DHFR activity in vitro.
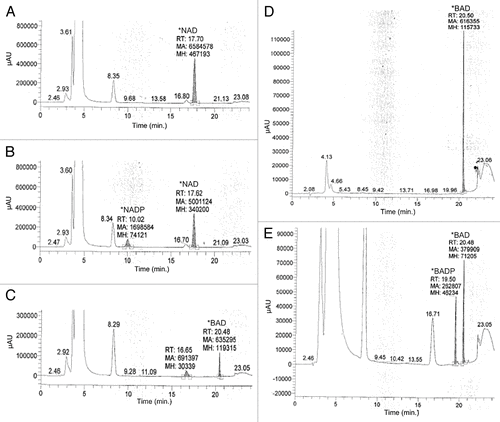
β-BAD and NAD were incubated for one hour in the presence of recombinant human NAD kinase. Following incubation, samples were analyzed by HPLC. When NAD was incubated with NAD kinase, a second peak appeared along with the NAD peak with retention time (RT) of 10 min, corresponding to the RT seen with the NADP standard (data not shown), thus validating the activity of NAD kinase (). Similarly, when β-BAD was incubated with NAD kinase, a small second peak, in addition to the β-BAD peak with a retention time of 20.5 min, appeared at a RT of 19.5 min (). This second peak is likely β-BADP. When β-BAD was incubated with increasing concentrations of NAD kinase, the peak at the retention time of 19.5 min increased (data not shown) indicating that phosphorylation of β-BAD increased with higher concentrations of NAD kinase. While it is clear that β-BAD can be phosphorylated by NAD kinase, we found in preliminary work that by generating μM concentrations of β-BADP using the technique described above and then adding this reaction mixture to the DHFR spectrophotometric assay, inhibition of DHFR by β-BADP was not observed (data not shown).
Figure 2. In order to evaluate the potential for BAD to act as a substrate of phosphorylation via human recombinant NAD kinase, 10 μM BAD was incubated for 1 h in a solution containing 50 mM Tris/HCL, pH 8.0, 20 mM MgCl2, 5 mM ATP, and 1–4 μg of human recombinant NAD kinase. To validate the activity of NAD kinase, 10 μM NAD was also incubated in the aforementioned solution for 1 h. Following incubation, samples were evaluated via HP LC using a Prtisil-10 SA X column at an absorbance of 254 nm as described by Saunders et al.Citation14 Data shows samples in which NAD was incubated with reaction solution lacking NAD kinase (A), NAD incubated in the reaction solution with 2 μg NAD kinase (B), BAD incubated with reaction solution lacking NAD kinase (C), 10 μM BAD standard (D), and 10 μM BAD incubated in reaction solution containing 4 μg NAD kinase (E).
BADP is not generated in significant quantities in cells
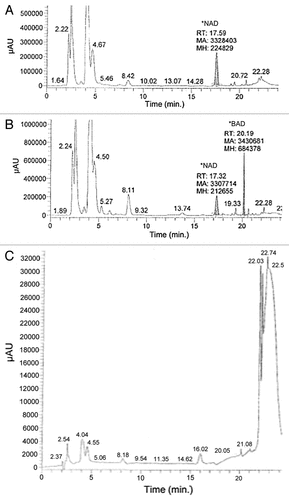
While β-BADP can be formed in vitro using high concentrations of recombinant NADK, we sought to determine if BAD, is phosphorylated to BADP in cells. CCRF-CEM/R cells were incubated in media containing 50 μM BR for three hours. Following incubation, cells were treated with perchloric acid and the solution was neutralized. All precipitate was removed and the supernatant was analyzed by HPLC. Cells were treated with BR and not β-BAD because BR readily crosses cell membranes via a passive mechanism whereas BAD, in contrast to β-BAD, is unable to enter the cell intact.Citation10
Lysate from cells treated with BR show a peak with a RT of 20.19 min, which is close to the retention time of β-BAD (20.5 min) and is expected for BAD (). This peak is absent in those cells not treated with BR. Interestingly, there is a small peak with a RT of 19.33 min also seen in . Although small, this peak has a retention time similar to what is expected for BADP (19.5 min), as shown in . Thus BADP is not formed in significant quantities via the phosphorylation of BAD in CCRF-CEM/R cells and is unlikely to mediate DHFR inhibition.
Figure 3. Sixty million CCRF-CEMR cells were plated in RPMI media with or without 50 μM BR and incubated at 37ºC for 3 h. Following incubation, cells were lysed with 140 μL of ice cold HCl04, kept at room temperature for 10 min and then spun down for 1 min at 16,000 RPM. An amount of 112 μL of ice cold K2CO3 was added to each sample, and samples were then kept at -20ºC for 15 min. The samples were centrifuged and the supernatant was collected and analyzed via HP LC at conditions described by Saunders et al.Citation14 (A) HP LC data of cell lysate from those cells not treated with BR, and (B) shows HP LC data of cells lysate from those cells treated with 50 μM BR. Finally, (C) is the HP LC data of 50 μM BR in water.
NADP and NADPH levels are decreased in cells treated with BR
NADPH is a critical coenzyme in many biosynthetic pathways. As BAD has been shown to inhibit NAD kinase,Citation20 we measured levels of NADP and NADPH in cells treated with BR. Cells were treated with BR, MTX, or β-BAD for up to 24 h. At various time points cells were lysed and the lysate analyzed for levels of NADP and NADPH. At 8 h post-treatment, NADPH levels were markedly decreased in the BR group as compared with the negative control group and the β-BAD group (). At the 24-h time point, all groups showed further reductions in NADPH, with the greatest reduction in the BR group (). Similar results were seen in all groups for levels of NADP ().
Figure 4. Five million CCRF-CEMR cells were plated in RPMI media containing 10 μM BR, BAD or no drug. Cells were allowed to incubate for up to 24 h harvested and then analyzed for NADP+/NADPH levels as described in Methods. (A) Levels of NADPH for the three experimental groups at 0, 8 and 24 h. (B) Levels of NADP+ at the same conditions. 132, BR; 106, BAD.
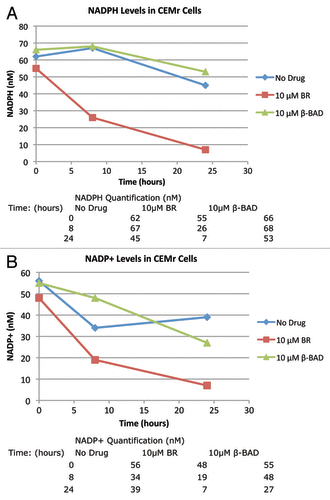
Interestingly, NADP and NADPH levels were decreased in cells treated with BR but were not significantly decreased in those cells treated with the metabolite, β-BAD. A decrease in NADPH levels is significant for the function of DHFR as this coenzyme protects DHFR from proteolysis.Citation18,Citation19 Inhibition of NADK contributes to decreased levels of NADPH and NADP, however whether or not BR inhibits other pathways involving NADPH/NAD+ should be considered. The cytotoxicity of BR on CEM/S cells and its effect on cell cycle distribution as a function of BR concentration at 24 and 48 h showed that at higher concentrations an increase in the percent of cells in the S-Phase occurred with increasing numbers of subG-1 cells (data not shown).
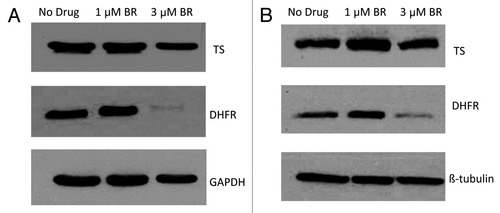
Cellular levels of DHFR protein decrease following BR treatment
DHFR is important for nucleotide biosynthesis and overall cell viability. Given that NADPH stabilizes DHFR, and BR decreases cellular levels of NADPH, we hypothesized that levels of DHFR protein would decrease following treatment with BR. Cells were treated with BR for a 24 or 48-h period. Following treatment, cells were lysed and DHFR protein levels were quantified by western blot. As controls we also measured levels of tubulin and thymidylate synthase (TS), not requiring NADPH for activity.
Following the 24-h treatment, DHFR levels in the control and the experimental group treated with 1 μM BR were similar (). However, cells treated with 3 μM BR at 24 h, showed a marked decrease in DHFR levels. Controls of tubulin and TS did not show this change. We conclude that a decrease in NADP/NADPH levels due to inhibition of NAD kinase at levels of BR slightly higher than the IC50 concentration causes DHFR instability and lowered levels of DHFR.
Figure 5. The effect of BR treatment on DHFR levels. CCRF-CEM/S cells were pelleted in RPMI media with BR (1 or 10 μM ) for 24 h (A) or 48 h (B). Following treatment cells were lysed and protein levels analysed by western blotting.
Discussion
An earlier study showed that BR toxicity is only partially reversed by guanosine, and the authors suggested that BR inhibits growth not only by inhibition of IMPDH, but also by another mechanismCitation15. In this study we show that BR also targets DHFR. While it is clear that BR, or its metabolites BAD and BADP do not target DHFR directly, BR via conversion to BAD and inhibition of NAD kinase, leads to a decrease in levels of NADP and NADPH, and as a result instability and lower levels of DHFR. Further studies are required to understand the detailed mechanism(s) by which DHFR is degraded by BR.
Specific inhibitors of NADK may possess synergistic potential with MTX and should be further investigated in combination therapy studies, or as a single agent in patients whose tumors posses either intrinsic or acquired resistance to DHFR inhibitors. The pharmacologic attributes of BR allow it to circumvent many of the resistance mechanisms encountered with MTX treatment. BR is passively taken into the cell and, once in the cell, BR is quickly metabolized to its active form, BAD, which is unable to cross the cell membrane and is retained in the cell. One study reported that the DHFR amplified, MTX-resistant human adenocarcinoma cell line, HT29, is highly sensitive to BR and MTX combination treatment, but not sensitive to either BR or MTX alone.Citation21 It was suggested that concomitant inhibition of DHFR and IMPDH may account for the increased sensitivity observed. However our data suggests that downregulation of DHFR by BR may also contribute to drug synergism. These findings are encouraging and lay the foundation for further MTX/NADK inhibitor combination studies in both sensitive and other MTX-resistant cancer cell lines.
Mutations have been described in enzymes catalyzing the conversion of BR to BAD, which decrease cell sensitivity to BR treatment. Mutations in NMN adenylyltransferase (NMNAT), the rate-limiting step in the conversion of BR to BAD have a BR IC50 100 fold greater than that observed in sensitive cells.Citation22 Myelosuppression, a side effect of traditional chemotherapy, should be considered when this drug is co-administered with MTX and other antifolates. Furthermore, toxicity studies in mice showed skeletal muscle necrosis following BR treatment suggesting that muscle toxicity may also present as a BR related toxicity.Citation23 To the best of our knowledge, BR has not yet been evaluated in a human clinical trial.
This study presents the first indication that BR, or its metabolites affect levels of DHFR, via inhibiting NAD kinase and decreasing levels of NADPH. These results provide a foundation toward future efforts in the development of drugs specifically targeting NADK.
Materials and Methods
Cell Culture and Materials
CCRF-CEM and CCRF-CEM/R cells were plated in RPMI-1640 media supplemented with 10% dialysed fetal bovine serum (FBS) and 1% streptomycin and penicillin and incubated at 37°C. and 5% CO2. All materials used in cell culture media were purchased from Invitrogen (Carlsbad, CA). CCRF-CEM/R have 5-times the copy number of DHFR than CCRF-CEM, and are resistant to MTX treatment.Citation24
Cell Toxicity Assay
Three thousand cells were plated in RPMI-1640 media supplemented with 10% FBS and 1% streptomycin and penicillin, and incubated with NAD analogs for 96 h. Cytotoxicity was measured using the trypan blue exclusion assay and cell viability was evaluated using the Vi-CELL Series Cell Viability Analyzer (Beckman Coulter, CA). Cytotoxicity data was analyzed and plotted using Excel 2007 (Microsoft Inc.) and IC50 values (drug concentration eliciting 50% decrease in total cell viability) were obtained using a regression curve formulated by Excel. Experimental groups were run in triplicate.
NADPH/NADP quantification assay
CCRF-CEM/R cells were plated in RPMI-1640 media supplemented with 10% FBS and 1% streptomycin and penicillin, and incubated overnight to achieve a concentration of 2 million cells/ml. (Five million were taken out of culture and plated in 15mm x 60mm Falcon tissue culture dishes. NAD analog was added to the appropriate tissue culture dishes and cells were incubated at 37°C for varying amounts of time. Four million cells were harvested from the experimental groups and washed in 5 mL 1% PBS, and then split in two equal groups for the purpose of quantifying NADPH or NADP levels. NADPH/NAD+ levels were quantified using the Fluorescent NADP/NADPH Detection Kit (Cell Technology, CA). Samples were kept overnight at -80°C. NADP+/NADPH fluorescence was measured using the Victor3 1420 Multilabel Counter (Perkin Elmer Life Sciences, CA) with fluorescence excitation at 535 nm and emission at 590 nm. Fluorescence intensity was analyzed using Wallac 1420 Manager Software (Perkin Elmer Life Sciences, CA). NADPH standard (provided by Cell Technology) was analyzed using the plate reader and data points were plotted and a regression line was generated using Excel (Microsoft, WA). From the NADPH standard data, levels of NADPH/NADP+ were determined and plotted using Excel (Microsoft, WA).
DHFR Enzyme Activity Assay
Measurement of DHFR enzyme activity in the presence of NAD analog was done with conditions described in.Citation25 Final cuvette concentrations were: 100 μM NADPH, and 50 μM dihydrofolate. Background absorbance was measured in the absence of dihydrofolate. Activity was determined by measuring the decrease in absorbance at 340 nm due to NADPH oxidation and conversion of dihydrofolate to terahydrfolate, using a Beckman DU 640 spectrophotometer (Brea, CA).
High-pressure Liquid Chromatography
High-pressure liquid chromatography (HPLC) was used in order to determine the phosphorylation of β-BAD using NADK (Enzo Life Sciences, PA). Samples were prepared and analyzed using conditions described by Saunders et al.Citation15
Western blot analysis
Total protein was extracted from CCRF-CEMS cells and total protein concentration was quantified via the Bio-RAD protein assay die reagent (Hercules, CA). After blocking with 5% powdered milk in PBS, the nitrocellulose membrane was incubated with primary antibody over night at 4°C. Anti-GFP primary antibody (Roche Diagnostics Corporation, Indianapolis, IN) was used at concentrations suggested by the manufacturer. Purified mouse anti-DHFR antibody was from BD Bioscience and human polyclonal anti-thymidylate synthase antibody was a gift from Dr. Frank Maley (Albany, NY).
NAD Kinase Assay
The total assay volume of 150 μL consisted of 50 mM Tris/HCL, pH 8.0, 20 mM MgCl2, 5 mM ATP, and 1–4 μg of human recombinant NAD kinase (Enzo Lifesciences, Farmingdale, NY). Ten μM β-BAD or 10 μM NAD (Sigma-Aldrich, St. Louis, MO) were added to the reaction mixture and the sample was incubated at 37°C for one hour. Samples were then analyzed for NADP formation using HPLC. NAD kinase activity was determined according to Saunders et al. with minor modificationsCitation15. An Ultrasphere C-18 (4.6 x 250 mm 5μ) column was used instead of Partisil-10 SAX column and the wavelength set at 260 nm instead of 254 nm.
NAD Kinase Whole Cell Assay
120 million CCRF-CEM/R cells were incubated in a 600mL flask in RPMI-1640 media supplemented with 10% FBS and 1% streptomycin and penicillin. Cells were split into two 250 mL flasks and re-suspended in fresh media. Benzamide riboside was added to one of the flasks and cells were incubated for three hours at 37°C. Following incubation, cells were washed in PBS and lysed with 140 μL of ice cold perchloric acid, kept at room temperature for 10 min, and then spun down for one minute at 16,000 RPM. Ice cold K2CO3 (112 μL) was added to each sample and samples were then kept at -20°C for 15 min. The samples were then centrifuged and the supernatant was collected and analyzed via HPLC.
NAD/NADP Analogs
All NAD analogs were provided by Krzysztof W. Pankiewicz. Methylene bis (phosphonate)-BAD (β-BAD) was obtained by an improved synthetic method.Citation26
| Abbreviations: | ||
| DHFR | = | dihydrofolate reductase |
| MTX | = | methotrexate |
| BR | = | benzamide riboside |
| BAD | = | benzamide adenine dinucleotide |
| β-BAD | = | methylenebis(phosphonate)-BAD |
| NADK | = | nicotinamide adenine dinucleotide kinase |
| IMPDH | = | inosine monophosphate dehydrogenase |
Acknowledgments
This work was supported by the National Cancer Institute [RO1-CA08010].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kamen BACP, Bertino JR. Folate antagonists. Cancer medicine: BC Decker 2003:727-38.
- Alt FW, Kellems RE, Bertino JR, Schimke RT. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem 1978; 253:1357 - 70; PMID: 627542
- Göker E, Kheradpour A, Waltham M, Banerjee D, Tong WP, Elisseyeff Y, et al. Acute monocytic leukemia: a myeloid leukemia subset that may be sensitive to methotrexate. Leukemia 1995; 9:274 - 6; PMID: 7532768
- Banerjee D, Mayer-Kuckuk P, Capiaux G, Budak-Alpdogan T, Gorlick R, Bertino JR. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim Biophys Acta 2002; 1587:164 - 73; http://dx.doi.org/10.1016/S0925-4439(02)00079-0; PMID: 12084458
- Matherly LH, Goldman DI. Membrane transport of folates. Vitam Horm 2003; 66:403 - 56; http://dx.doi.org/10.1016/S0083-6729(03)01012-4; PMID: 12852262
- Zhao R, Diop-Bove N, Visentin M, Goldman ID. Mechanisms of membrane transport of folates into cells and across epithelia. Annu Rev Nutr 2011; 31:177 - 201; http://dx.doi.org/10.1146/annurev-nutr-072610-145133; PMID: 21568705
- Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev 2007; 26:153 - 81; http://dx.doi.org/10.1007/s10555-007-9049-z; PMID: 17333344
- Gorlick R, Goker E, Trippett T, Waltham M, Banerjee D, Bertino JR. Intrinsic and acquired resistance to methotrexate in acute leukemia. N Engl J Med 1996; 335:1041 - 8; http://dx.doi.org/10.1056/NEJM199610033351408; PMID: 8793930
- Mishra PJ, Humeniuk R, Mishra PJ, Longo-Sorbello GS, Banerjee D, Bertino JR. A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc Natl Acad Sci U S A 2007; 104:13513 - 8; http://dx.doi.org/10.1073/pnas.0706217104; PMID: 17686970
- Pankiewicz KW, Watanabe KA, Lesiak-Watanabe K, Goldstein BM, Jayaram HN. The chemistry of nicotinamide adenine dinucleotide (NAD) analogues containing C-nucleosides related to nicotinamide riboside. Curr Med Chem 2002; 9:733 - 41; http://dx.doi.org/10.2174/0929867024606920; PMID: 11966436
- Sandhu SK, Yap TA, de Bono JS. The emerging role of poly(ADP-Ribose) polymerase inhibitors in cancer treatment. Curr Drug Targets 2011.; 12:2034 - 44; http://dx.doi.org/10.2174/138945011798829438; PMID: 21777194
- Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Soto M, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors in cancer chemotherapy. Recent Pat Anticancer Drug Discov 2006; 1:39 - 53; http://dx.doi.org/10.2174/157489206775246430; PMID: 18221025
- Krohn K, Dörner H, Zukowski M. Chemical synthesis of benzamide riboside. Curr Med Chem 2002; 9:727 - 31; http://dx.doi.org/10.2174/0929867024606876; PMID: 11966435
- Jäger W, Salamon A, Szekeres T. Metabolism of the novel IMP dehydrogenase inhibitor benzamide riboside. Curr Med Chem 2002; 9:781 - 6; http://dx.doi.org/10.2174/0929867024606830; PMID: 11966442
- Saunders PP, Arimilli S, Krohn K, Muhs MA, Alvarez E, Surve-lyer R. Metabolism and action of benzamide riboside in Chinese hamster ovary cells. Anticancer Drugs 1996; 7:93 - 9; http://dx.doi.org/10.1097/00001813-199601000-00011; PMID: 8742104
- Gharehbaghi K, Grünberger W, Jayaram HN. Studies on the mechanism of action of benzamide riboside: a novel inhibitor of IMP dehydrogenase. Curr Med Chem 2002; 9:743 - 8; http://dx.doi.org/10.2174/0929867024606902; PMID: 11966437
- Hsieh Y-C, Lawal RAL, Scotto K, Banerjee D, Ercikan Abali E, Bertino JR. Targeting the NADPH binding site of dihydrofolate reductase for cancer therapy AACR Meeting Abstracts 2010; 2010.
- Perkins JP, Bertino JR. Dihydrofolate reductase from the L1210R murine lymphoma. Fluorometric measurements of the interaction of the enzyme with coenzymes, substrates, and inhibitors. Biochemistry 1966; 5:1005 - 12; http://dx.doi.org/10.1021/bi00867a028; PMID: 4380350
- Hakala MT, Suolinna EM. Specific protection of folate reductase against chemical and proteolytic inactivation. Mol Pharmacol 1966; 2:465 - 80; PMID: 6008166
- Bonnac L, Chen L, Pathak R, Gao G, Ming Q, Bennett E, et al. Probing binding requirements of NAD kinase with modified substrate (NAD) analogues. Bioorg Med Chem Lett 2007; 17:1512 - 5; http://dx.doi.org/10.1016/j.bmcl.2007.01.012; PMID: 17258457
- Peñuelas S, Noé V, Ciudad CJ. Modulation of IMPDH2, survivin, topoisomerase I and vimentin increases sensitivity to methotrexate in HT29 human colon cancer cells. FEBS J 2005; 272:696 - 710; http://dx.doi.org/10.1111/j.1742-4658.2004.04504.x; PMID: 15670151
- Yalowitz JA, Jayaram HN. Modulation of cytotoxicity of benzamide riboside by expression of NMN adenylyltransferase. Curr Med Chem 2002; 9:749 - 58; http://dx.doi.org/10.2174/0929867024606867; PMID: 11966438
- Jayaram HN, Yalowitz JA, Arguello F, Greene JF Jr.. Toxicity and efficacy of benzamide riboside in cancer chemotherapy models. Curr Med Chem 2002; 9:787 - 92; http://dx.doi.org/10.2174/0929867024606858; PMID: 11966443
- Mini E, Moroson BA, Franco CT, Bertino JR. Cytotoxic effects of folate antagonists against methotrexate-resistant human leukemic lymphoblast CCRF-CEM cell lines. Cancer Res 1985; 45:325 - 30; PMID: 3855284
- Skacel N, Menon LG, Mishra PJ, Peters R, Banerjee D, Bertino JR, et al. Identification of amino acids required for the functional up-regulation of human dihydrofolate reductase protein in response to antifolate Treatment. J Biol Chem 2005; 280:22721 - 31; http://dx.doi.org/10.1074/jbc.M500277200; PMID: 15817466
- Felczak K, Pankiewicz KW. Synthesis of methylenebis(phosphonate) analogues of 2-, 4-, and 6-pyridones of nicotinamide adenine dinucleotide. Nucleosides Nucleotides Nucleic Acids 2011; 30:512 - 23; http://dx.doi.org/10.1080/15257770.2011.575909; PMID: 21888543