Abstract
Although tumor resistance remains a significant impediment to successful radiotherapy, associated regulatory markers and detailed molecular mechanisms underlying this phenomenon are not well defined. In this study, we identified inositol polyphosphate 4-phosphatase type II (INPP4B) as a novel marker of radioresistance by systematically analyzing Unigene libraries of laryngeal cancer. INPP4B was highly expressed in radioresistant laryngeal cancer cells and was induced by treatment with either radiation or anticancer drugs in various types of cancer cells. Ectopic INPP4B overexpression increased radioresistance and anticancer drug resistance by suppressing apoptosis in HEp-2 cells. Conversely, INPP4B depletion with small interfering RNA resensitized HEp-2 as well as A549 and H1299 cells to radiation- and anticancer drug-induced apoptosis. Furthermore, radiation-induced INPP4B expression was blocked by inhibition of extracellular signal-regulated kinase (ERK). INPP4B depletion significantly attenuated radiation-induced increases in Akt phosphorylation, indicating an association of INPP4B-mediated radioresistance with Akt survival signaling. Taken together, our data suggest that ERK-dependent induction of INPP4B triggers the development of a tumor-resistance phenotype via Akt signaling and identify INPP4B as a potentially important target molecule for resolving the radioresistance of cancer cells.
Introduction
Radiotherapy in conjunction with surgery and/or chemotherapy is currently the standard adjuvant treatment for laryngeal cancer, which is the sixth most prevalent cancer worldwide.Citation1,Citation2 However, intrinsic or acquired radioresistance of tumors allows a subset of cells to survive, thus limiting the effectiveness of radiotherapy. This phenomenon contributes to poor prognosis in laryngeal cancer patients following radiotherapy.Citation3,Citation4 Several studies have investigated tumor radioresistance markers, which are useful as prognostic and therapeutic indicators for radiotherapy. Cellular components are often altered in cancer cells in a manner that serves to protect against chronic radiation damage. For example, proliferation regulatory proteins such as epidermal growth factor receptor (EGFR), phosphoinositide 3-kinase (PI3K)/Akt and Ras are constitutively activated or overexpressed in radioresistant tumors.Citation5,Citation6 The status/expression of p53 or Bcl-2/-xl, which play key roles in cell cycling and apoptosis, are involved in tumor radioresistance,Citation6,Citation7 and cellular anti-oxidant proteins such as peroxiredoxin-II and manganese superoxide dismutase are often involved in the oxidative stress tolerance of tumor cells.Citation8,Citation9 In addition, several studies have reported that fractionated radiotherapy can induce acquired radioresistance and anticancer drug resistance by regulating P-glycoprotein and multidrug resistance-associated protein.Citation10-Citation13 Although accumulating evidence has provided some understanding of radioresistance and anticancer drug resistance in cancer cells, biomarkers in specific types of cancer and detailed molecular mechanisms underlying these phenomena remain unclear.
UniGene, a National Center for Biotechnology Information (NCBI) database of the transcriptome widely used in the analysis of expressed sequence tags (ESTs) database which contains information regarding cell- and tissue-specific gene transcripts. Thus, it is an excellent resource for identifying functionally unknown or regulatory genes.Citation14-Citation16 We previously established radioresistant cells through fractionated irradiation and used these cells as a model system for studying the radioresistant phenotype.Citation17 In the present study, a laryngeal cancer EST database was systematically screened to identify gene profiles associated with tumor radioresistance. Through an in silico analysis of radioresistant HEp-2 (RR-HEp-2) cells, we identified several radioresistance-related genes. One of these genes, INPP4B, encoding inositol polyphosphate 4-phosphatase type II, had not previously been linked to tumor radioresistance and was thus examined further as a possible radioresistance regulator. INPP4B is a regulatory enzyme that selectively dephosphorylates the fourth position of phosphatidylinositol(3,4)-bisphosphate (PI(3,4)P2).Citation18-Citation20 The rapid production and/or degradation of phosphoinositides plays a critical role in cellular processes, including cell growth, differentiation, apoptosis and protein trafficking; phosphoinositide regulation is also important in the etiology of diseases, including cancer, diabetes and inflammation.Citation21-Citation23 Although the regulation of intracellular phosphoinositide profiles by other lipid phosphatases, including phosphatase and tensin homolog (PTEN) and inositol polyphosphate-5-phosphatase 1, are well characterized, little is known about the modulation of target substrates by INPP4B. The few studies that have been conducted to date have reported a physiological role for INPP4B in the regulation of erythropoietin responsiveness in proerythroblasts and a tumor-suppressive function in human basal-like breast cancer.Citation20,Citation24 Recent studies have reported that INPP4B expression pattern and genetic variants, such as single-nucleotide polymorphisms might be associated with intrinsic susceptibility to chemotherapy.Citation25,Citation26 However, the regulatory functions of INPP4B in stress responses, tumor radioresistance and anticancer drug resistance are largely unknown.
In this paper, we identified 11 radioresistance-associated genes using the previously established radioresistant cellular system and investigated the role of INPP4B in the response to radiation and anticancer drugs in various cancer cell types. Our findings show that the induction of INPP4B expression by radiation and anticancer drugs is positively correlated with tumor radioresistance and chemoresistance, respectively, via regulation of extracellular signal-regulated kinase (ERK)-Akt signaling.
Results
INPP4B expression is elevated in radioresistant HEp-2 cell lines
We screened for radioresistance-related genes by analyzing seven laryngeal cancer libraries (# 992, #6653, #6655, #6910, #6915, #7793 and #9282) and predicting function using Goblet/SMART programs (data not shown). We next identified genes that were differentially expressed between parental HEp-2 cells and RR-HEp-2 cell lines (RR-HEp-2 and RR-#-6) as potential radiation-responsive genes. As shown in (top), ERBB2 and Bcl-xl (positive control) were increased and p21 (negative control) was suppressed in RR-HEp-2 cell lines compared with parental HEp-2 cells, confirming the validity of our in vitro system. Data from conventional reverse transcription-polymerase chain reaction (RT-PCR) showed that the mRNA levels of nine genes were upregulated in parental HEp-2 cells in response to 10 Gy radiation or in RR-HEp-2 cell lines compared with untreated control or parental HEp-2 cells (, middle), whereas two genes were downregulated under the same conditions (, bottom). Information related to radioresistance- or chemoresistance-related functions of these 11 genes are summarized in .
Figure 1. Identification of INPP4B as a radioresistance-related protein in HEp-2 cells. (A) Parental HEp-2 cells and redioresistant RR-HEp-2 and RR-#6 clonal variants were left untreated or treated with 10 Gy radiation for 24 h. Gene transcripts were detected by conventional RT-PCR. ERBB2 and Bcl-xl were used as positive controls; p21 was used as a negative control; and GAPDH was used as a loading control. (B) Parental HEp-2 cells and RR-HEp-2 and RR-#6 variants were treated with the indicated doses of radiation. After 14 d, the survival fraction was determined by clonogenic survival assay; results are presented as a survival curve (top). INPP4B protein levels were determined by western blotting using β-actin as a loading control (bottom). (C) Parental (CON), radiosensitive (RS-#7, -#9 and- #18) and radioresistant (RR-#3, -#6 and -#13) HEp-2 cells were left untreated (-) or treated (+) with 4 Gy radiation. After 14 d, colony formation was quantified using an automatic colony counter; results are presented as a survival curve (top). INPP4B protein levels were determined by western blotting using β-actin was used as a loading control (bottom). The data represent typical results or mean values with standard deviations (n = 3).
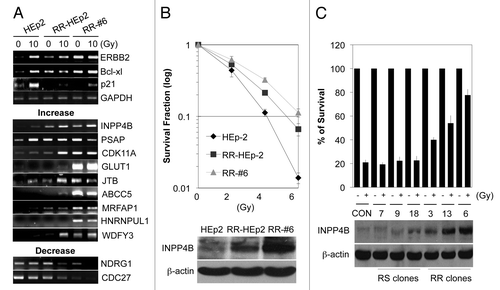
Table 1. List of genes showing altered mRNA level between parental and radioresistant HEp-2 cell lines
Because the function of INPP4B in relation to tumor radioresistance is largely unknown, we first examined the role of INPP4B, focusing on the radioresistance phenotype. RR-HEp-2 and RR-#6 cell lines showed a distinct radioresistant phenotype, as determined by clonogenic survival assays (, top) and expressed high levels of INPP4B protein compared with parental HEp-2 cells (, bottom). We confirmed the association of INPP4B with the radioresistant phenotype using radiosensitive and radioresistant HEp-2 clones. The radiation sensitivity of each clone was determined by colony formation assay after treatment with 4 Gy radiation (, top). Clones showing a radioresistant phenotype (RR-#3, -#13 and -#6) exhibited marked upregulation of INPP4B protein compared with parental HEp-2 cells, whereas INPP4B levels were only marginally altered in radiosensitive clones (RS-#7, -#9 and -#18) (, bottom). Thus, our results suggest that increased levels of INPP4B may be associated with the development of radioresistance in HEp-2 cells.
Induction of INPP4B is not limited to radiation stimulation or a specific cell type
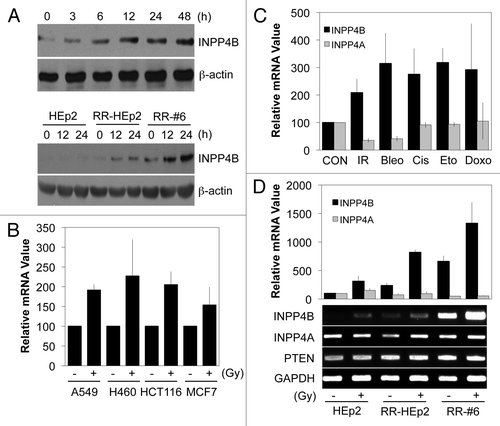
To examine INPP4B responses to radiation, we treated parental HEp-2 cells and RR-HEp-2 cell lines with 10 Gy radiation. INPP4B protein levels were increased by radiation in a time-dependent manner in parental HEp-2 cells (, top); this induction was even more striking in RR-HEp-2 cell lines (, bottom). These data indicate that INPP4B is a radiation-responsive protein and suggest that hyper-accumulation of this protein might be a mechanism for the acquisition of the radioresistant phenotype in HEp-2 cells. To examine whether radiation-mediated induction of INPP4B expression is limited to HEp-2 cells, we treated other human cancer cells, including A549, H460, HCT116 and MCF7 cells with 10 Gy radiation. Quantitative real-time PCR revealed that radiation treatment induced an increase in INPP4B transcript levels in all cancer cell lines studied (), indicating that the phenomenon is widespread in human cancer cells. Since modulation of radioresistance genes also often plays a role in the development of chemoresistance in various types of cancer,Citation10-Citation13 we investigated whether INPP4B is also regulated by anticancer drugs, including bleomycin, cisplatin, etoposide and doxorubicin, in HEp-2 cells. Interestingly, all drugs induced an increase in INPP4B mRNA levels, as determined by quantitative real-time PCR, whereas INPP4A, a paralog of INPP4B gene, was not induced by these same drugs (). We also tested whether this radiation-induced upregulation extended to other lipid phosphatases, or was largely limited to INPP4B, which regulates the 3-phosphoinositide lipid network.Citation18-Citation20 Unlike INPP4B, the lipid phosphatases INPP4A and PTEN were not induced by radiation treatment, as determined by quantitative PCR ( top) and conventional RT-PCR analyses (, bottom). These data suggest that INPP4B may be the only lipid phosphatase in mediating the development of the radioresistant phenotype in HEp-2 cells.
Figure 2. Induction of INPP4B expressionin cancer cells by treatment with radiation or anticancer drugs. (A) Parental HEp-2 cells (top) or parental HEp-2 and RR-HEp-2 and RR-#6 variants (bottom) were treated with 10 Gy radiation for the indicated times. INPP4B protein levels were determined by western blotting using β-actin was used as a loading control. (B) A549, H460, HCT116 and MCF7 cells were left untreated (-) or treated (+) with 10 Gy radiation for 24 h. INPP4B transcript levels were determined by quantitative PCR using GAPDH as an internal control. (C) HEp-2 cells were left untreated (CON) or treated with radiation (IR; 10 Gy), bleomycin (Bleo; 10 μM), cisplatin (Cis; 10 μM), etoposide (Eto; 5 μM), or doxorubicin (Doxo; 1 μM) for 24 h. INPP4A and INPP4B transcript levels were determined by quantitative PCR using GAPDH as an internal control. (D) Parental HEp-2 cells and RR-HEp-2 and RR-#6 variants were treated with 10 Gy radiation for 24 h. INPP4B, INPP4A and PTEN transcript levels were determined by quantitative PCR (top) or conventional RT-PCR (bottom). GAPDH was used as an internal control or loading control. The data represent typical results or mean values with standard deviations (n = 4).
ERK-dependent induction of INPP4B promotes the radioresistant phenotype of HEp-2 cells
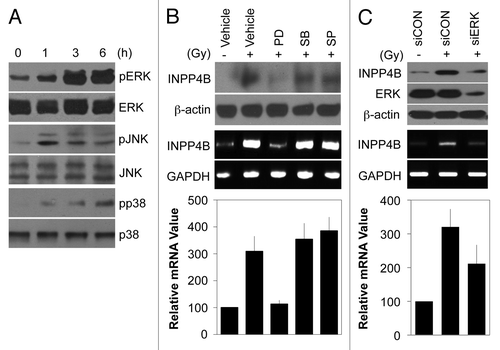
Because radiation activates mitogen-activated protein kinase (MAPK) protein,Citation27 we examined whether induction of INPP4B expression is associated with MAPK activation in HEp-2 cells. As expected, exposure to 10 Gy radiation led to an increase in the activated forms of ERK, c-Jun N-terminal kinase (JNK) and p38 kinase without altering the levels of the corresponding protein (). Next, we pretreated cells with specific inhibitors of each MAPK prior to radiation treatment. As shown in , inhibition of ERK with PD98059 blocked radiation-induced increases in the levels of INPP4B protein (top) and mRNA (middle), whereas inhibition of JNK with SP600125 or p38 kinase with SB203580 had no effect on INPP4B levels. Quantitative PCR showed that INPP4B transcript levels were increased by 3.2-fold in radiation-treated HEp-2 cells compared with untreated control cells and this induction was decreased by 1.3-fold with ERK inhibition (, bottom). To confirm that endogenous ERK regulates INPP4B expression, we used small interfering RNA (siRNA) to knock down endogenous ERK in HEp-2 cells. As shown in , siRNA treatment effectively knocked down ERK expression. Notably, depletion of endogenous ERK markedly suppressed the induction of INPP4B by irradiation, as determined by western blotting (, top) and conventional RT-PCR (, middle). Quantitative PCR analyses showed that siRNA-mediated ERK knockdown decreased radiation-induced INPP4B mRNA levels by 38% compared with cells treated with radiation alone (, bottom). These results suggest that stress-induced INPP4B expression is dependent on ERK signaling, but not on JNK or p38 kinase signaling.
Figure 3. ERK-dependent induction of INPP4B expression by radiation in HEp-2 cells. (A) HEp-2 cells were treated with 10 Gy radiation for the indicated times. Expression and phosphorylation levels of MAPK proteins were determined by western blotting. (B) HEp-2 cells were left untreated (-) or treated (+) with 10 Gy radiation in the absence (Vehicle) or presence of 10 μM PD98059 (PD), 10 μM SB203580 (SB), or SP600125 (SP) and then incubated for 24 h. INPP4B protein levels were determined by western blotting using β-actin as a loading control (top). INPP4B transcript levels were determined by conventional RT-PCR (middle) or quantitative PCR (bottom). GAPDH was used as a loading control or an internal control. (C) HEp-2 cells were transfected with control siRNA (siCON) or 100 nM ERK-1/2 siRNA (siERK) for 48 h and then left untreated (-) or treated (+) with 10 Gy radiation for an additional 24 h. INPP4B and ERK protein levels were determined by western blotting using β-actin as a loading control (top). INPP4B transcript levels were determined by conventional RT-PCR (middle) or quantitative PCR (bottom). GAPDH was used as a loading control or an internal control. The data represent typical results or mean values with standard deviations (n = 3).
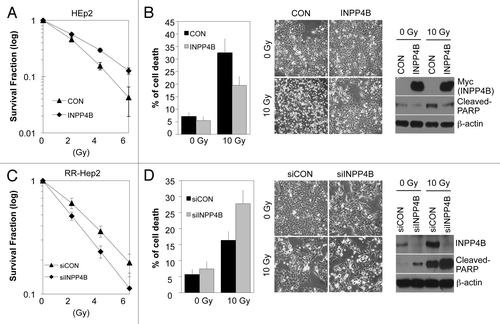
To determine whether INPP4B is essential for tumor radioresistance, we ectopically overexpressed wild-type Myc-tagged INPP4B in parental HEp-2 cells. Overexpression of INPP4B increased cell survival after exposure to radiation, as determined by clonogenic survival assays (). A flow cytometry analysis revealed that cell death induced by 10 Gy radiation was markedly lower at 48 h in INPP4B-overexpressing HEp-2 cells (~19%) compared with parental HEp-2 cells (~33%) (, left). These results were confirmed by observation of cell morphology (, middle). Consistent with these data, the level of cleaved-PARP, a marker of apoptosis, was also decreased in INPP4B-overexpressing, radiation-treated HEp-2 cells compared with radiation-treated parental HEp-2 cells (, right). Conversely, siRNA-mediated knockdown of endogenous INPP4B in RR-HEp-2 cells decreased survival following exposure to radiation (). Depletion of endogenous INPP4B synergistically increased radiation-induced RR-HEp-2 cell death from 16% (radiation alone) to 28% (radiation plus INPP4B knockdown), as determined by a flow cytometry analysis (, left), results that were confirmed by observation of cell morphology (, middle). siRNAs targeting INPP4B effectively knocked down INPP4B and substantially increased the levels of cleaved-PARP in radiation-treated-RR-HEp-2 cells (, right). Collectively, these data suggest that INPP4B has the capacity to protect HEp-2 cells against radiation-induced apoptosis, thereby increasing radioresistance.
Figure 4. INPP4B-dependent regulation of radiation sensitivity in HEp-2 cells. (A, B) Parental HEp-2 cells were transfected with an empty vector (CON) or Myc-tagged INPP4B expression vector (INPP4B) and then incubated for 24 h. The cells were treated with the indicated doses of radiation (A) or 10 Gy radiation (B). After 14 d, colony formation was quantified using an automatic colony counter; results are presented as a survival curve (A). After 48 h, cell viability was determined with a FACScan flow cytometer; data are presented as the percentage of PI-positive cells (B, left). Cell morphology was observed by light microscopy (B, middle). Levels of cleaved-PARP and INPP4B proteins were determined by western blotting using β-actin as a loading control (B, right). (C, D) RR-HEp-2 cells were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) and then cultured for 48 h. The cells were treated with the indicated doses of radiation (C) or 10 Gy radiation (D). Cell survival (C), cell viability (D, left), cell morphology (D, middle) and western blotting (D, right) experiments were performed as in A and B. The data represent typical results or mean values with standard deviations (n = 4).
INPP4B is associated with the development of chemoresistance in HEp-2 cells
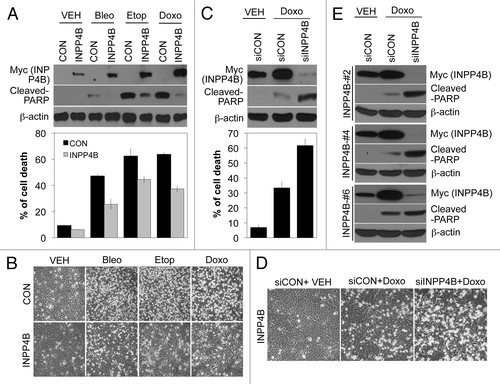
To examine whether INPP4B can modulate resistance to anticancer drugs, we established HEp-2 cells stably expressing Myc-tagged INPP4B (INPP4B-HEp-2 cells). Interestingly, INPP4B-HEp-2 cells showed markedly elevated basal INPP4B expression levels that were further induced by anticancer drugs bleomycin, etoposide and doxorubicin. These cells were resistant to cell death induced by the anticancer drugs, as detected by western blotting for cleaved-PARP (, top) and flow cytometry analysis (, bottom). Although the degree of induction of INPP4B by individual drugs differed, anti-apoptotic effects of INPP4B were clearly evident in all drug-treated cells, as demonstrated by morphological changes (). To confirm the INPP4B dependence of the anti-apoptotic phenotype in INPP4B-HEp-2 cells, we silienced INPP4B with siRNA. INPP4B siRNA effectively knocked down INPP4B expression and significantly resensitized cells to drug-induced apoptosis, as determined by western blotting (, top). INPP4B depletion synergistically promoted cell death, increasing the cell-death percentage from 33% (INPP4B-HEp-2 cells treated with doxorubicin) to 60% (doxorubicin-treated INPP4B-HEp-2 cells transfected with siRNA against INPP4B), as shown by a flow cytometry analysis (, bottom). Similar results were obtained in an examination of morphological changes (). We further confirmed this sensitization effect using other stable INPP4B-HEp-2 clones (#2, #4 and #6). These drug-resistant variants also exhibited enhanced doxorubicin-induced apoptotic cell death following knockdown of INPP4B, as determined by western blotting for cleaved-PARP (), confirming INPP4B as a positive biomarker in the development of anticancer drug resistance.
Figure 5. INPP4B-dependent regulation of anticancer drug sensitivity in stably transfected INPP4B-HEp-2 cells. (A, B) Control HEp-2 (CON) and stable INPP4B-HEp-2 cells (INPP4B) were treated with vehicle, 10 μM bleomycin, 5 μM etoposide, or 1 μM doxorubicin for 24 h. The expression levels of cleaved-PARP and Myc-tagged INPP4B were determined by western blotting using β-actin as a loading control (A, top). Cell death was determined with a FACScan flow cytometer; data are presented as the percentage of PI-positive cells (A, bottom). Cell morphology was observed by light microscopy (B). (C, D) INPP4B-HEp-2 cells were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h and then treated with vehicle or 1 μM doxorubicin for an additional 24 h. Western blotting (C, top), cell death (C, bottom) and cell morphology (D) experiments were performed as in A and B. (E) INPP4B-HEp-2 variants (INPP4B-#2, -#4 and -#6) were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h and then treated with vehicle or 1 μM doxorubicin for an additional 24 h. The expression levels of cleaved-PARP and Myc-tagged INPP4B were determined by western blotting using β-actin as a loading control. The data represent typical results or mean values with standard deviations (n = 4).
The role of INPP4B in the resistant phenotype is also applicable to lung cancer cells and may involve modulation of Akt activation
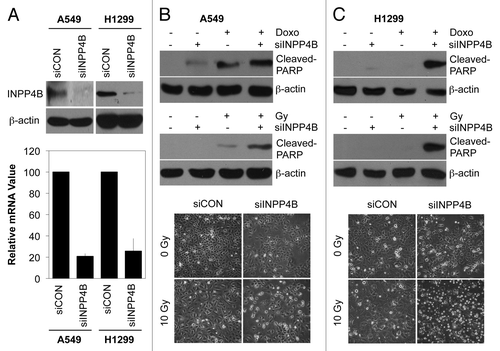
To examine whether the resistance function of INPP4B is limited to laryngeal HEp-2 cancer cells, we examined A549 and H1299 lung cancer cells, which a resistant to radiation and anticancer drugs.Citation28,Citation29 As shown in , INPP4B siRNA effectively knocked down INPP4B in A549 and H1299 cells, as determined by western blotting (top) and quantitative PCR analyses (bottom). Consistent with the results obtained in HEp-2 cells, silencing of INPP4B markedly enhanced doxorubicin- and radiation-induced cell death in A549 cells, as determined by western blotting for cleaved-PARP (, top and middle) and an examination of morphological changes (, bottom). Under the same experimental conditions, this sensitization effect was much greater in H1299 cells than in A549 cells (). Because p53 status is different between H1299 cells (p53 deficient) and A549 cells (wild-type p53),Citation29 our data imply that the role of INPP4B in resistance is likely independent of p53 signaling,
Figure 6. Induction of cell death by INPP4B depletion in lung cancer cells. (A) A549 and H1299 cells were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h. INPP4B protein levels were determined by western blotting using β-actin as a loading control (top). INPP4B mRNA levels were determined by quantitative PCR using GAPDH as an internal control (bottom). (B, C) A549 (B) and H1299 (C) cells were transfected with control siRNA (-) or 100 nM INPP4B siRNA (+) for 48 h and then left untreated (-) or treated (+) with 1 μM doxorubicin for 24 h or 10 Gy radiation for 48 h. The levels of cleaved-PARP were determined by western blotting using β-actin as a loading control (top and middle). Cell morphology was observed by light microscopy (bottom). The data represent typical results or mean values with standard deviations (n = 4).
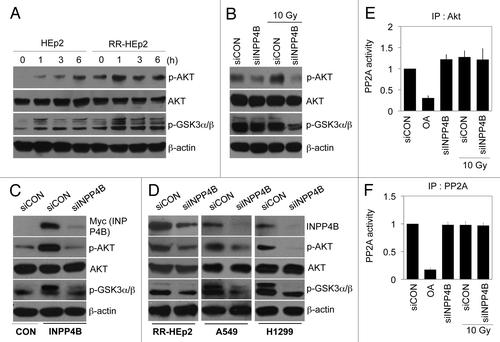
Previous reports have shown that Akt activation creates a resistant phenotype and thus is a poor prognostic factor.Citation30,Citation31 Therefore, we investigated whether radiation regulates Akt activity. Akt phosphorylation was highly induced in RR-HEp-2 cells exposed to radiation compared with parental HEp-2 cells, indicating Akt activation; consistent with this, phosphorylation of GSK3α/β, a known major target of Akt, was also increased (). To determine whether INPP4B also positively regulates Akt activation, we transfected RR-HEp-2 cells with siRNA to knock down endogenous INPP4. Although RR-HEp-2 cells contained high levels of phosphorylated Akt following radiation treatment, this activation of Akt was significantly suppressed by INPP4B depletion without altering basal Akt levels (). In addition, the levels of phosphorylated Akt were very high in INPP4B-HEp-2 cells, but were dramatically downregulated by INPP4B knockdown (), indicating a close correlation between the level of INPP4B expression and Akt activity. Consistent with the results obtained in RR-HEp-2 cells, Akt phosphorylation levels were markedly decreased by INPP4B depletion in A549 and H1299 cells with a concomitant decrease in the levels of phosphorylated GSK3α/β (). Since the phosphatase PP2A is known to antagonize Akt activity directly,Citation32,Citation33 we determined whether INPP4B regulates PP2A activity. PP2A activity was dramatically inhibited in RR-HEp-2 cells by treatment with okadaic acid, used as a positive control (). However, neither radiation treatment nor INPP4B siRNA transfection had an effect on PP2A activity in RR-HEp-2 cells, as determined by Akt and PP2A immunocomplex precipitation assays (), indicating that PP2A activity is not involved in the regulation of INPP4B-mediated Akt activation. Collectively, our data suggest that induction of INPP4B by stress (radiation, anticancer drugs) promotes a resistant phenotype in cancer cells via the Akt survival pathway.
Figure 7. Modulation of Akt activity by INPP4B expression in laryngeal and lung cancer cells. (A) Parental HEp-2 and radioresistant RR-HEp-2 cells were treated with 10 Gy radiation for the indicated times. Expression and phosphorylation levels of Akt and GSK3α/β were determined by western blotting using β-actin as a loading control. (B) RR-HEp-2 cells were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h and then left untreated or treated with 10 Gy radiation for an additional 6 h. Expression and phosphorylation levels of Akt and GSK3α/β were determined by western blotting using β-actin as a loading control. (C, D) Control HEp-2 (CON) and stably transfected INPP4B-HEp-2 (INPP4B) cells (C) or RR-HEp-2, A549 and H1299 cells (D) were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h. Expression and phophorylation levels of Akt, GSK3α/β and INPP4B were determined by western blotting using β-actin as a loading control. (E, F) RR-HEp-2 cells were transfected with control siRNA (siCON) or 100 nM INPP4B siRNA (siINPP4B) for 48 h and then left untreated or treated with 10 Gy radiation for an additional 6 h. Akt (E) or PP2A (F) was immunoprecipitated from each sample and the resulting immune complexes were used to measure PP2A phosphatase activity. The PP2A inhibitor okadaic acid was used as a positive control. The data represent typical results or mean values with standard deviations (n = 4).
Discussion
Prioritizing resistance biomarker candidates based on their biological role and molecular mechanism is an important consideration in the application of such molecular targets to tumor radiotherapy or chemotherapy. Accordingly, from an analysis of an EST database, used as the source of a laryngeal cancer-specific transcriptome, we first selected 68 candidate genes and then identified 11 radioresistance-associated genes using an in silico/in vitro analysis of established RR-HEp-2 cell lines. Although the function of these genes is not defined in laryngeal cancer cells, two identified genes, GLUT1 and ABCC5, encoding glucose transporter type 1 and ATP-binding cassette C member 5, respectively, are known as prognostic markers of radiotherapy or chemotherapy resistance in various tumors.Citation34-Citation36 Consistent with the observations, our results showed that the expression of these two genes was much higher in RR-HEp-2 cell lines than parental cells (). The products of two other identified genes, JTB (jumping translocation breakpoint) and NDRG1 (N-myc downstream regulated gene 1) are associated with UV resistance in 293FT cellsCitation37 and resistance to doxorubicin and retinoic acid in hepatocellular carcinoma cells.Citation38 However, the products of other identified genes, such as PSAP, CDK11A, MRFAP1, HNRNPUL1, WDFY3 and CDC27, have no well-defined roles in tumor radioresistance or chemoresistance. Thus, if genes identified by such screens are to serve as prognostic/diagnostic marker or provide valuable information for patient treatment, it is important that their molecular mechanisms in cancer cells be well defined (Table. 1).
In the current study, we identified INPP4B as a radioresistance and chemoresistance marker. Specifically, we found that stress (radiation, anticancer drugs)-induced increases in INPP4B expression contributed to protective role on apoptotic cell death in laryngeal HEp-2 cancer cells as well as A549 and H1299 lung cancer cells. These results are consistent with a previous report that INPP4B is a positive regulator of paclitaxel resistance in gastric cancer cells thus may be a predictive marker of chemotherapy resistance.Citation26 In addition, single-nucleotide polymorphisms signatures within the INPP4B gene have been reported to be prominent indicators of daunorubicin-induced cytotoxicity in lymphoblastoid cells.Citation25 Taken together with these previous observations, our data suggest that a specific INPP4B signature might be correlated with poor prognosis for several types of cancer patients in the context of both clinical radiotherapy and chemotherapy.
Radiation-stimulated ERK activation has been linked to cell survival via the activation of survival/DNA repair proteins or the inhibition of apoptotic proteins.Citation27 Our data also showed that radiation led to activation of MAPKs, including ERK. Moreover, the induction of INPP4B mRNA and protein was totally dependent on ERK activity, but not p38 or JNK activity, in HEp-2 cells, indicating that the INPP4B gene is a downstream target of ERK signaling. Consequently, INPP4B protein may contribute to the formation of a positive feedback loop by amplifying the INPP4B-mediated survival activity associated with the sustained radioresistance phenotype.
The INPP4A isozyme plays an important neuroprotective role by suppressing excitotoxic cell death and involuntary movements.Citation39 In addition, weeble mutant mice harboring a null mutation of the INPP4A gene exhibit massive neuronal cell death leading to neurological defects.Citation40 These facts are consistent with the suggestion that INPP4A has a pro-survival role, in contrast to the pro-apoptotic role of PTEN.Citation41 Both INPP4A and INPP4B selectively metabolize PI(3,4)P2 rather than PI(3,4,5)P3, producing PI(3)P as a product.Citation19,Citation20,Citation40 Thus, these enzymes may provide surrogate functions for each other, because they act on the same substrates and are expressed in different tissues; in particular, INPP4A is highly expressed in normal brain tissue, whereas INPP4B is highly expressed in heart and skeletal muscle.Citation19,Citation40 In this study, we demonstrated that INPP4B blocked radiation- and anticancer drug-induced apoptosis in human laryngeal and lung cancer cells, whereas INPP4B depletion potentiated cell death. Notably, the INPP4A gene was not responsive to radiation in either parental HEp-2 or RR-HEp-2 cells. These data indicate that INPP4B, alone among lipid phosphatases, is a major contributor to stress responses and/or tumor resistance. Moreover, the capacity of INPP4B to confer resistance against both radiation and anticancer drugs in human laryngeal and lung cancer cells provides evidence of a broad cytoprotective role in various types of cancer cells.
Although downstream targets and major molecular signal transduction pathways linking INPP4B with tumor radioresistance and chemoresistance are incompletely defined, we did demonstrate a strong association between the protective role of INPP4B and Akt activation, showing that levels of the phosphorylated form of Akt were positively modulated in vitro in an INPP4B expression-dependent manner (). Whether INPP4B regulates Akt activation directly or indirectly is not yet known. Akt protein acts through phosphorylation of substrates such as GSK3, mTOR and caspase-9 to play a role in cell growth and survival pathways.Citation31 Furthermore, Akt activation shows a very strong correlation with poor patient survival after radiotherapy or chemotherapy.Citation31 The increased Akt phosphorylation we observed is consistent with the positive effect of INPP4B on cell proliferation and survival in radiation-treated laryngeal and lung cancer cells. However, it should be noted that other studies have reported different finding. For example, ectopic overexpression of INPP4B in leukemic cells has been reported to reduce Akt phosphorylation without affecting cell proliferation,Citation24 and INPP4B depletion in human mammary epithelial cells using shRNA has been shown to increase insulin-stimulated Akt activation and promote cell proliferation.Citation20,Citation42 Discrepancies between our results and previous observations might reflect differences in cell types and stimuli. Indeed, it has been shown that PTEN-null and PIK3CA-mutant cancer cells exhibit different steady-state Akt pathways, resulting in differences in cell viability and growth in response to a variety of stimuli.Citation42 In this context, overexpression of INPP4A simultaneously decreases PI(3,4)P2 levels and increases PI(3,4,5)P3 levels, thereby inducing Akt phosphorylation, a phenomenon that blocks FAS-induced apoptosis.Citation43 Thus, it is conceivable that INPP4B expression may reciprocally modulate PI(3,4)P2 and PI(3,4,5)P3 levels, which could affect Akt activation during stress responses. Although Akt activation could be dependent on cancer cell types or specific stimuli, our data support the idea that INPP4B protein is a positive regulator of cell survival during radiotherapy and chemotherapy.
In summary, we identified 11 new radioresistance-related genes in a systematic screen of laryngeal cancer UniGene libraries. We also demonstrated that INPP4B expression was strongly correlated with the tumor-resistance phenotype and was induced by cytotoxic stresses such as radiation and anticancer drugs in various cancer cells. We further showed that, unlike INPP4A, stress-induced INPP4B expression exerted a cytoprotective action by suppressing apoptotic cell death via upregulation of Akt activation. Therefore, we suggest that INPP4B contributes to development of the tumor-resistance phenotype and thus, may be useful as a prognostic tool or therapeutic target of tumor radiotherapy.
Materials and Methods
Cell lines and treatment
A549, H1299 and 460 lung cancer cells, HEp-2 laryngeal cancer cells, HCT116 colon cancer cells and MCF7 breast cancer cells were purchased from American Type Culture Collection (Manassas, VA). Cells were grown in Dulbecco’s modified Eagle’s medium (HEp-2, HCT116 and MCF7 cells) or Roswell Park Memorial Institute medium 1640 (A549, H460 and H1299 cells) containing 10% fetal bovine serum, 50 μg/mL streptomycin and 50 units/mL penicillin. RR-HEp-2 and its variants were established as described previously.Citation17 Several subclones (#3, #6, #7, #9, #13 and #18) isolated from RR-HEp-2 cell population were selected as radioresistant or radiosensitive HEp-2 variants on the basis of their clonogenic survival assay after treatment of 4 Gy radiation. Parental HEp-2 cells were maintained as control cells. The cells were irradiated using a 137cesium-ray source (Atomic Energy of Canada Ltd.) at a dose rate of 3.81 Gy/min or treated with 10 μM bleomycin (Sigma), 10 μM cisplatin (Sigma), 5 μM etoposide (Sigma), or 10 μM doxorubicin (Sigma). PD98059, SB203580 and SP600125 (Calbiochem) were used to inhibit ERK, p38 and JNK, respectively.
Clonogenic assay
Cell survival after irradiation was determined by clonogenic assay as described previously.Citation17 Cells were treated with a single dose of radiation and then trypsinized, diluted and seeded into 60-mm tissue culture dishes at various cell densities (200, 400, 1500 and 3000 cells/dish for the 0, 2, 4 and 6 Gy, respectively). After 14 d, the colonies were fixed with methanol and stained with trypan blue solution. Only the colonies with 50 or more cells were counted as survivors by using a colony counter (Imaging Products).
Apoptosis analysis
Cells were seeded at a density of 2 × 105 cells per 60-mm dish and treated according to the indicated experimental conditions. Apoptotic cell death was determined by western blot analysis of the cleaved poly (ADP-ribose) polymerase (PARP) and observation of apoptotic cell morphology with light microscopy. To quantify apoptotic cell death, cells were analyzed using a FACScan flow cytometer (Becton Dickson), as described previously.Citation17
Western blot analysis
Cells were lysed in a buffer containing 50 mM TRIS-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40 and 0.1% sodium dodecyl sulfate (SDS) supplemented with protease and phosphatase inhibitors, as described previously.Citation44 The following antibodies were used to detect protein: rabbit polyclonal anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-p38, anti-p38, anti-phospho-JNK, anti-JNK and anti-cleaved-PARP (Asp214) (Cell Signaling Technology); rabbit monoclonal anti-phospho-GSK3α/β (Ser21/9) (Cell Signaling Technology); mouse monoclonal anti-ERK (BD Transduction Laboratories); mouse monoclonal anti-phospho-ERK and anti-myc (Santa Cruz Biotechnology Inc.); mouse monoclonal anti-β-actin (Sigma); goat polyclonal anti-INPP4B (Santa Cruz Biotechnology Inc.).
In silico and in vitro analyses of the UniGene library
Seven available EST libraries for human laryngeal cancer were downloaded from the UniGene transcriptome library (www.ncbi.hlm.nih.gov/UniGene) to collect the genes expressed in laryngeal cancer. The followings were used to select radioresistance-regulatory candidate genes: PubMed (www.ncbi.nlm.nih.gov/pubmed) was used to search the published function of each gene; Goblet (www.goblet.mogen.mpg.de), to predict gene ontology; SMART (www.smart.embl-heidelberg.de), to seek various protein domains. The mRNA expression of candidate genes selected by in silico analysis were subsequently screened using real-time PCR using parental and radioresistant HEp-2 cell lines. We selected genes whose expression was relatively increased or decreased in irradiated or radioresistant cells compared with control group for functional analysis.
Conventional and quantitative RT-PCR
The total RNA isolated using RNA STAT-60 (Tel-Test B Inc.) was reverse-transcribed with ImProm-IITM reverse transcriptase (Promega). Quantitative PCR was performed in triplicate by using a Chromo-4 cycler (Bio-Rad) and SYBR Premix Ex Taq™ (Takara Bio). In conventional RT-PCR experiment, the amplification signal from the target gene was normalized against the cycle threshold values of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the same reaction. The following primers and conditions were employed: Bcl-xl (151-bp product, annealing temperature 55°C, 32 cycles), sense 5′-cgggcattcagtgacctgac-3′ and antisense 5′-tcaggaaccagcggttgaag-3′; epidermal growth factor receptor 2 (ERBB2) (402-bp product, annealing temperature 55°C, 31 cycles), sense 5′-catatgtctcccgccttctg-3′ and antisense 5′-cccacacagtcacaccataac-3′; GAPDH (305-bp product, annealing temperature 55°C; 24 cycles), sense 5′-catctctgccccctctgctga-3′ and antisense 5′-ggatgaccttgcccacagcct-3′; inositol polyphosphate 4-phosphatase type I (INPP4A) (404-bp product, annealing temperature 55°C, 33 cycles), sense 5′-ggctgccagtccataatc-3′ and antisense 5′-cacactttctcccactcc-3′; INPP4B (440-bp product, annealing temperature 55°C, 31 cycles), sense 5′-aaagaatgcaggtacacag-3′ and antisense 5′-ctctgtgctgctcttagg-3′; PTEN (580-bp product, annealing temperature 55°C, 31 cycles), sense 5′-gaaagacattatgacaccg-3′ and antisense 5′-ttagcatcttgttctgtttg-3′; p21 (439-bp product, annealing temperature 55°C, 34 cycles), sense 5′-tgtccgtcagaacccatg-3′ and antisense 5′-ggagtggtagaaatctgtcatg-3′. The other primer sequences used for identification of radioresistance-regulatory genes were presented in .
Knockdown of INPP4B and ERK by siRNA
The human INPP4B siRNA 5′-cagaauguuugagucacua-3′, the human ERK-1 siRNA 5′-cucucuaaccggcccaucu-3′ and the human ERK-2 siRNA 5′-cagaucuuuacaagcucuu-3′ sequences were selected and synthesized according to the manufacturer’s notes (Bioneer, Daejeon, Korea). A scrambled siRNAs were used as a negative control, which show no significant homology to known gene sequences and did not regulate gene expression. Cells were transiently transfected with 100 nM of siRNA in a serum-free medium for 5 h using Metafectene reagent (Biontex, Munich, Germany) according to the manufacturer’s protocol. Cells were maintained for an additional 48 h and the depletion of target proteins was determined by western blotting and quantitative RT-PCR analysis.
Construction and transfection of INPP4B
A cDNA for human INPP4B was prepared by RT-PCR from pCMV-Sport6-INPP4B (Open Biosystems, Huntsville, AL) with specific primers (sense 5′-ggggtaccgagccaccatggaaattaaagaggaaggg-3′ and antisense 5′-ccgctcgagcggggtgtcagcttttccataag-3′) designed to introduce KpnI and XhoI restriction sequences at the 5′ and 3′ ends, respectively. The resulting cDNA was cloned into pcDNA3.1 (+) Myc/His vector (Invitrogen, Carlsbad, CA). HEp-2 cells were transfected with expression vector using the Metafectene reagent (Biontex), as described previously.Citation17 For establishing a stable cell line, HEp-2 cells were transfected with INPP4B cDNA vector, subcultured in standard media for 24 h and then selected transfected cells by adding 1 mg/mL G418. The INPP4B expressing stable HEp-2 cells were confirmed by western blot analysis.
Phosphatase assay
Cells were lysed in a buffer containing 50 mM TRIS-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 7.5% glycerol, 1 mM EDTA, 1 mM Na3VO4 and complete protease inhibitors. Cell lysates were precipitated with anti-Akt or anti-PP2A antibodies (Santa Cruz Biotechnology Inc.). Immune complexes were collected using protein A sepharose beads; 1/4 of the washed beads were used for western blot analysis to confirm Akt and PP2A immunoprecipitation efficiency. The remaining beads were used to analyze PP2A activity by using a RediPlate™96 EnzChek Serine/Threonine Phosphatase assay kit [Invitrogen (Molecular Probes)], according to the manufacturer’s protocol. Fluorescence was measured using a Victor2 fluorescence microplate reader (Perkin Elmer Corp.). Okadaic acid (Sigma) was used as a positive control to inhibit PP2A activity.
| Abbreviations: | ||
| ERK-1/2 | = | extracellular signal-regulated protein kinase-1/2 |
| EST | = | expressed sequence tag |
| GSK3 | = | glycogen synthase kinase 3 |
| INPP4B | = | inositol polyphosphate 4-phosphatase type II |
| PARP | = | poly ADP-ribose polymerase |
| PI(3,4)P2 | = | phosphatidylinositol(3,4)-bisphosphate |
| PI3K | = | phosphoinositide 3-kinase |
| PTEN | = | phosphatase and tensin homolog |
| RR-HEp-2 | = | radioresistant HEp-2 |
| siRNA | = | small interfering RNA |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the Nuclear Research and Development Program of the National Research Foundation grant funded by the Korean government (MEST).
References
- Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001; 94:153 - 6; http://dx.doi.org/10.1002/ijc.1440; PMID: 11668491
- Lefebvre JL. Laryngeal preservation in head and neck cancer: multidisciplinary approach. Lancet Oncol 2006; 7:747 - 55; http://dx.doi.org/10.1016/S1470-2045(06)70860-9; PMID: 16945770
- Nix PA, Greenman J, Cawkwell L, Stafford N. Radioresistant laryngeal cancer: beyond the TNM stage. Clin Otolaryngol Allied Sci 2004; 29:105 - 14; http://dx.doi.org/10.1046/j.1365-2273.2003.00796.x; PMID: 15113291
- Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 2005; 5:516 - 25; http://dx.doi.org/10.1038/nrc1650; PMID: 15965493
- Bussink J, van der Kogel AJ, Kaanders JH. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol 2008; 9:288 - 96; http://dx.doi.org/10.1016/S1470-2045(08)70073-1; PMID: 18308254
- Haffty BG, Glazer PM. Molecular markers in clinical radiation oncology. Oncogene 2003; 22:5915 - 25; http://dx.doi.org/10.1038/sj.onc.1206704; PMID: 12947398
- Kumar B, Cordell KG, D’Silva N, Prince ME, Adams ME, Fisher SG, et al. Expression of p53 and Bcl-xL as predictive markers for larynx preservation in advanced laryngeal cancer. Arch Otolaryngol Head Neck Surg 2008; 134:363 - 9; http://dx.doi.org/10.1001/archotol.134.4.363; PMID: 18427001
- Wang T, Tamae D, LeBon T, Shively JE, Yen Y, Li JJ. The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res 2005; 65:10338 - 46; http://dx.doi.org/10.1158/0008-5472.CAN-04-4614; PMID: 16288023
- Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, Ogi J, et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol 2003; 23:2362 - 78; http://dx.doi.org/10.1128/MCB.23.7.2362-2378.2003; PMID: 12640121
- Hill BT, Deuchars K, Hosking LK, Ling V, Whelan RD. Overexpression of P-glycoprotein in mammalian tumor cell lines after fractionated X irradiation in vitro. J Natl Cancer Inst 1990; 82:607 - 12; http://dx.doi.org/10.1093/jnci/82.7.607; PMID: 1968980
- Henness S, Davey MW, Harvie RM, Davey RA. Fractionated irradiation of H69 small-cell lung cancer cells causes stable radiation and drug resistance with increased MRP1, MRP2, and topoisomerase IIalpha expression. Int J Radiat Oncol Biol Phys 2002; 54:895 - 902; http://dx.doi.org/10.1016/S0360-3016(02)03037-7; PMID: 12377343
- Ryu JS, Um JH, Kang CD, Bae JH, Kim DU, Lee YJ, et al. Fractionated irradiation leads to restoration of drug sensitivity in MDR cells that correlates with down-regulation of P-gp and DNA-dependent protein kinase activity. Radiat Res 2004; 162:527 - 35; http://dx.doi.org/10.1667/RR3260; PMID: 15624307
- Bottke D, Koychev D, Busse A, Heufelder K, Wiegel T, Thiel E, et al. Fractionated irradiation can induce functionally relevant multidrug resistance gene and protein expression in human tumor cell lines. Radiat Res 2008; 170:41 - 8; http://dx.doi.org/10.1667/RR0986.1; PMID: 18582150
- Seve M, Chimienti F, Devergnas S, Favier A. In silico identification and expression of SLC30 family genes: an expressed sequence tag data mining strategy for the characterization of zinc transporters’ tissue expression. BMC Genomics 2004; 5:32; http://dx.doi.org/10.1186/1471-2164-5-32; PMID: 15154973
- Oh J, Lee J, Woo JM, Choi E, Park I, Han C, et al. Systematic identification and integrative analysis of novel genes expressed specifically or predominantly in mouse epididymis. BMC Genomics 2006; 7:314; http://dx.doi.org/10.1186/1471-2164-7-314; PMID: 17166261
- Kim JS, Ryoo ZY, Chun JS. Cytokine-like 1 (Cytl1) regulates the chondrogenesis of mesenchymal cells. J Biol Chem 2007; 282:29359 - 67; http://dx.doi.org/10.1074/jbc.M700965200; PMID: 17644814
- Kim JS, Chang JW, Yun HS, Yang KM, Hong EH, Kim DH, et al. Chloride intracellular channel 1 identified using proteomic analysis plays an important role in the radiosensitivity of HEp-2 cells via reactive oxygen species production. Proteomics 2010; 10:2589 - 604; http://dx.doi.org/10.1002/pmic.200900523; PMID: 20461716
- Norris FA, Auethavekiat V, Majerus PW. The isolation and characterization of cDNA encoding human and rat brain inositol polyphosphate 4-phosphatase. J Biol Chem 1995; 270:16128 - 33; http://dx.doi.org/10.1074/jbc.270.27.16128; PMID: 7608176
- Norris FA, Atkins RC, Majerus PW. The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J Biol Chem 1997; 272:23859 - 64; http://dx.doi.org/10.1074/jbc.272.38.23859; PMID: 9295334
- Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 2009; 16:115 - 25; http://dx.doi.org/10.1016/j.ccr.2009.06.006; PMID: 19647222
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006; 443:651 - 7; http://dx.doi.org/10.1038/nature05185; PMID: 17035995
- Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci 2009; 34:115 - 27; http://dx.doi.org/10.1016/j.tibs.2009.01.003; PMID: 19299143
- Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol 2008; 9:162 - 76; http://dx.doi.org/10.1038/nrm2335; PMID: 18216772
- Barnache S, Le Scolan E, Kosmider O, Denis N, Moreau-Gachelin F. Phosphatidylinositol 4-phosphatase type II is an erythropoietin-responsive gene. Oncogene 2006; 25:1420 - 3; http://dx.doi.org/10.1038/sj.onc.1209187; PMID: 16247441
- Duan S, Bleibel WK, Huang RS, Shukla SJ, Wu X, Badner JA, et al. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res 2007; 67:5425 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-06-4431; PMID: 17545624
- Jung JJ, Jeung HC, Chung HC, Lee JO, Kim TS, Kim YT, et al. In vitro pharmacogenomic database and chemosensitivity predictive genes in gastric cancer. Genomics 2009; 93:52 - 61; http://dx.doi.org/10.1016/j.ygeno.2008.08.002; PMID: 18804159
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene 2003; 22:5885 - 96; http://dx.doi.org/10.1038/sj.onc.1206701; PMID: 12947395
- Das AK, Sato M, Story MD, Peyton M, Graves R, Redpath S, et al. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res 2006; 66:9601 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-2627; PMID: 17018617
- Rho JK, Choi YJ, Ryoo BY, Na II, Yang SH, Kim CH, et al. p53 enhances gefitinib-induced growth inhibition and apoptosis by regulation of Fas in non-small cell lung cancer. Cancer Res 2007; 67:1163 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-2037; PMID: 17283151
- Huang WC, Hung MC. Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc 2009; 108:180 - 94; http://dx.doi.org/10.1016/S0929-6646(09)60051-6; PMID: 19293033
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489 - 501; http://dx.doi.org/10.1038/nrc839; PMID: 12094235
- Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, et al. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol 2004; 24:8778 - 89; http://dx.doi.org/10.1128/MCB.24.19.8778-8789.2004; PMID: 15367694
- Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006; 441:523 - 7; http://dx.doi.org/10.1038/nature04809; PMID: 16680151
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002; 2:48 - 58; http://dx.doi.org/10.1038/nrc706; PMID: 11902585
- Evans A, Bates V, Troy H, Hewitt S, Holbeck S, Chung YL, et al. Glut-1 as a therapeutic target: increased chemoresistance and HIF-1-independent link with cell turnover is revealed through COMPARE analysis and metabolomic studies. Cancer Chemother Pharmacol 2008; 61:377 - 93; http://dx.doi.org/10.1007/s00280-007-0480-1; PMID: 17520257
- Vordermark D, Brown JM. Endogenous markers of tumor hypoxia predictors of clinical radiation resistance?. Strahlenther Onkol 2003; 179:801 - 11; http://dx.doi.org/10.1007/s00066-003-1150-9; PMID: 14652668
- Pan JS, Cai JY, Xie CX, Zhou F, Zhang ZP, Dong J, et al. Interacting with HBsAg compromises resistance of jumping translocation breakpoint protein to ultraviolet radiation-induced apoptosis in 293FT cells. Cancer Lett 2009; 285:151 - 6; http://dx.doi.org/10.1016/j.canlet.2009.05.009; PMID: 19487072
- Jung EU, Yoon JH, Lee YJ, Lee JH, Kim BH, Yu SJ, et al. Hypoxia and retinoic acid-inducible NDRG1 expression is responsible for doxorubicin and retinoic acid resistance in hepatocellular carcinoma cells. Cancer Lett 2010; 298:9 - 15; http://dx.doi.org/10.1016/j.canlet.2010.05.020; PMID: 20573444
- Sasaki J, Kofuji S, Itoh R, Momiyama T, Takayama K, Murakami H, et al. The PtdIns(3,4)P(2) phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature 2010; 465:497 - 501; http://dx.doi.org/10.1038/nature09023; PMID: 20463662
- Nystuen A, Legare ME, Shultz LD, Frankel WN. A null mutation in inositol polyphosphate 4-phosphatase type I causes selective neuronal loss in weeble mutant mice. Neuron 2001; 32:203 - 12; http://dx.doi.org/10.1016/S0896-6273(01)00468-8; PMID: 11683991
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell 2008; 133:403 - 14; http://dx.doi.org/10.1016/j.cell.2008.04.013; PMID: 18455982
- Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget 2011; 2:321 - 8; PMID: 21487159
- Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J Biol Chem 2002; 277:6266 - 72; http://dx.doi.org/10.1074/jbc.M105969200; PMID: 11706019
- Kim JS, Park ZY, Yoo YJ, Yu SS, Chun JS. p38 kinase mediates nitric oxide-induced apoptosis of chondrocytes through the inhibition of protein kinase C zeta by blocking autophosphorylation. Cell Death Differ 2005; 12:201 - 12; http://dx.doi.org/10.1038/sj.cdd.4401511; PMID: 15665819