Abstract
Cancer cells convert glucose preferentially to lactate even in the presence of oxygen (aerobic glycolysis–Warburg effect). New concepts in cancer treatment aim at inhibition of aerobic glycolysis. Pyruvate dehydrogenase converts pyruvate to acetylCoA thus preventing lactate formation. Therefore, the aim of this study was to evaluate compounds that could activate pyruvate dehydrogenase in cancer cells. We investigated the effects of (R)-(+)-α-lipoic acid (LPA) and dichloroacetate (DCA), possible activators of pyruvate dehydrogenase, on suppression of aerobic glycolysis and induction of cell death.
The neuroblastoma cell lines Kelly, SK-N-SH, Neuro-2a and the breast cancer cell line SkBr3 were incubated with different concentrations (0.1–30 mM) of LPA and DCA. The effects of both compounds on cell viability/proliferation (WST-1 assay), [18F]-FDG uptake, lactate production and induction of apoptosis (flow cytometric detection of caspase-3) were evaluated. Furthermore, NMRI nu/nu mice that had been inoculated s.c. with SkBr3 cells were treated daily for four weeks with LPA (i.p, 18.5 mg/kg) starting at day 7 p.i.. Tumor development was measured with a sliding calliper and monitored via [18F]-FDG-PET. Residual tumors after therapy were examined histopathologically.
These data suggests that LPA can reduce (1) cell viability/proliferation, (2) uptake of [18F]-FDG and (3) lactate production and increase apoptosis in all investigated cell lines. In contrast, DCA was almost ineffective. In the mouse xenograft model with s.c. SkBr3 cells, daily treatment with LPA retarded tumor progression. Therefore, LPA seems to be a promising compound for cancer treatment.
Introduction
Many cancer cells preferentially use aerobic glycolysis, i.e., conversion of glucose to lactate, for generation of ATP even in the presence of oxygen. This phenomenon, known as the Warburg effect, has been discovered already in the 1920s.Citation1,Citation2 Since then aerobic glycolysis has been repeatedly verified in cancer cells of different origin.Citation3 Nevertheless, aerobic glycolysis is inefficient, producing only 2 molecules of ATP per molecule of glucose. In contrast, complete oxidation of glucose to CO2 and H2O produces > 32 molecules of ATP. Accordingly, cancer cells have to metabolise large amounts of glucose to meet their high-energy demand. This increased glucose consumption is exploited in visualization of tumor nodules via [18F]-FDG PET.Citation4 Though it does not seem evident at first glance, cancer cells using aerobic glycolysis have a significant proliferative advantage compared with normal cells using complete oxidation of glucose. One advantage might be due to adaption to intermittent hypoxia.Citation3 This key feature of glucose metabolism of cancer cells opens up promising ways for cancer therapy. Inhibition of aerobic glycolysis by activation of enzymes that cause a shift in metabolism toward complete oxidation of glucose should be effective in eradication of tumor cells. In this regard (R)-(+)-α-lipoic acid and dichloroacetate that both support pyruvate dehydrogenase reaction have turned out as promising compounds.
The dithiol compound α-lipoic acid (LPA) is synthesized from octanoic acid in the mitochondria and also absorbed from dietary sources. The disulfide group of LPA can be readily reduced to form DHLPA. LPA (oxidized form) and DHLPA (reduced form) constitute an effective redox pair. Accordingly, both LPA and DHLPA have been reported to scavenge a variety of oxygen species.Citation5-Citation7 LPA is found as lipoamide covalently bound to the pyruvate dehydrogenase complex.Citation8 Moreover, beneficial effects of α-lipoic acid on atherosclerosis and diabetic peripheral neuropathy have been described.Citation9-Citation12 The pharmacological impact of the LPA/DHLPA redox couple is due to its metal-chelating properties and the already mentioned scavenging of reactive oxygen species (ROS).Citation13 Additionally LPA can inhibit pyruvate dehydrogenase kinase, thus increasing the activity of the pyruvate dehydrogenase complex.Citation14,Citation15 Similarly, efficacy of dichloroacetate (DCA) in cancer therapy most probably also results from inhibition of mitochondrial pyruvate dehydrogenase kinase hence shifting cell metabolism toward oxidative turnover.Citation16,Citation17 DCA has been reported to induce apoptosis, probably caused by enhanced production of ROS during respiration, to inhibit proliferation of tumor cells and to sensitize tumor cells to radiation.Citation17-Citation19 The aim of our study was to investigate the effects of LPA and DCA on glucose metabolism of cancer cells, particularly with regard to induction of cell death, both in vitro and in vivo. For this purpose different parameters indicative of changes in cellular metabolism were investigated. LPA and DCA induced impact on aerobic glycolysis was demonstrated by determination of cellular lactate concentrations and [18F]-FDG uptake. Analysis of changes in cell viability after LPA and DCA treatment was done using the WST-1 proliferation assay and by quantification of caspase-3, an indicator of apoptosis. To verify the promising results obtained in vitro, therapeutic efficacy of LPA was additionally assayed in an animal tumor model. Nude mice that had been s.c. inoculated with SkBr3 breast carcinoma cells received daily i.p. injections of LPA for four weeks starting one week after tumor cell inoculation. Tumor development was measured with a calliper and monitored via [18F]-FDG PET. Additionally, residual tumors after LPA therapy were subjected to immunohistochemical analysis with regard to proliferation (Ki-67 staining), cell death (detection of necrosis or apoptosis), hypoxia (HIF-1α staining) and glucose metabolism (transketolase like-1 staining). The results obtained within this study are in favor of pursuing LPA therapy for future clinical application.
Results
Effects of LPA and DCA on cell viability/proliferation
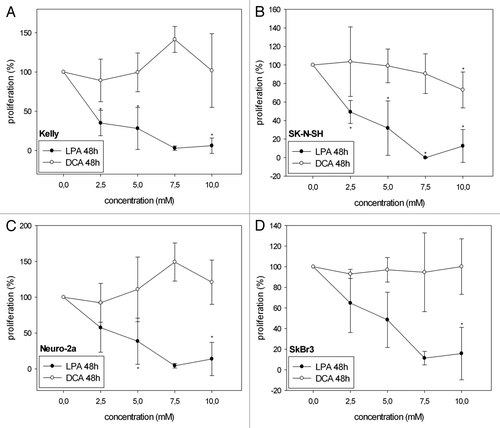
LPA significantly decreased cell viability/proliferation in all four cell lines analyzed both at 24 h () and at 48h () as investigated with the WST-1 proliferation assay. In Kelly and SK-N-SH cells, treatment with 2.5 mM, 5 mM and 7.5 mM LPA reduced cell viability/proliferation more effectively after 48 h than after 24 h. In contrast, DCA did not cause a significant decrease of cell viability/proliferation at the same concentrations. Only in SK-N-SH cells proliferation slightly decreased with increasing concentrations of DCA at 48h (). In Kelly and Neuro-2a cells even increased cell viability/proliferation was observed at 48h with 7.5 mM DCA () (Kelly: 141.4% ± 16.5%; Neuro-2a: 149.1% ± 26.5%). Statistical analysis of the effects of LPA observed at 24 h and 48 h revealed a significant decrease in cell viability/proliferation at 48 h compared with 24 h with 2.5 mM, 5 mM and 7.5 mM for Kelly and with 2.5 mM for SK-N-SH (). No significant differences in cell viability/proliferation at 24 h and 48 h were observed in Neuro-2a and SkBr3 after treatment with LPA and in all cell lines analyzed after treatment with DCA.
Figure 1A–D. Cell viability/proliferation of Kelly (A, E), SK-N-SH (B, F), Neuro-2a (C, G) and SkBr3 (D, H) cells 24 h and 48 h after treatment with different concentrations of LPA or DCA (WST-1 cell proliferation assay). Controls were set to 100%. Values below 100% indicate decreased and values above 100% increased cell viability/proliferation. Treatment with LPA resulted in decreased cell viability/proliferation both at 24h and 48h. In contrast, DCA did not significantly decrease cell viability/proliferation. Moreover, DCA treatment with and 7.5 mM and 10 mM resulted in increased cell viability/proliferation in Neuro-2a cells at 48h (G). Similarly, treatment of Kelly cells with 7.5 mM DCA resulted in increased cell viability/proliferation at both 24h and 48h (A, E). * indicates p < 0.0125 compared with the untreated controls (after Bonferroni adjustment). Means ± standard deviations (n = 3).
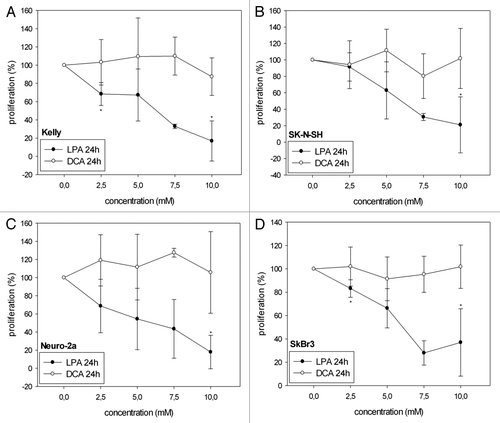
Figure 1E–H. See previous page for figure legend.
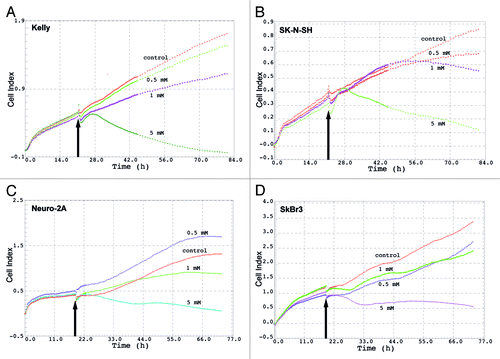
LPA induced decrease of cell proliferation as measured by changes of electrical impedance
Continuous real time measurement over 80 h of electrical impedance of cells revealed that LPA significantly reduces the cell viability/proliferation in all four cell lines analyzed both at 1 mM and 5 mM. The highest reduction of cell proliferation was observed at 5 mM LPA. The effect of LPA on cell proliferation could be observed already 4–6 h after addition of LPA to the culture medium. ().
Figure 2. Cell Index (electrical impedance) of different cell lines after treatment with LPA. Kelly (A), SK-N-SH (B), Neuro-2a (C) and SkBr3 (D) cells were treated with different concentrations of LPA (0.5 mM, 1 mM, 5 mM). Electrical impedance of cells expressed as Cell Index, indicative of cell viability, was measured continuously over 80 h using the xCELLigence system. Arrows indicate time-points of LPA addition.
Effects of LPA and DCA on [18F]-FDG uptake
In Kelly, Neuro-2a and SK-N-SH cells treatment with increasing LPA concentrations resulted in decreasing uptake of [18F]-FDG (). For SkBr3 cells [18F]-FDG uptake was observed to increase at 1 mM and 2.5 mM LPA but to decrease at concentrations above 5 mM LPA (). In contrast, treatment with DCA did not significantly reduce [18F]-FDG uptake at concentrations of up to 10 mM in all cell lines analyzed (). However, at DCA concentrations of 25 mM and 30 mM [18F]- FDG uptake was significantly reduced in Kelly and Neuro-2a cells, respectively. Treatment of SkBr3 cells with 30 mM of DCA caused a relevant decrease of cell proliferation but did not reach the level of statistical significance. In SK-N-SH and SkBr3 cells reduction of [18F]-FDG uptake observed at 25 mM and 30 mM DCA, respectively, was not statistically significant (data not shown).
Figure 3. Effects of different concentrations of LPA and DCA on [18F]-FDG uptake (CPM/μg protein) of Kelly (A), SK-N-SH (B), SkBr3 (C) and Neuro-2a (D) cells. Controls were set to 100%. Treatment with LPA significantly reduced [18F]-FDG uptake in a concentration dependent manner. In contrast, DCA did not affect glucose uptake at concentrations up to 10 mM. * indicates p < 0.01 compared with the untreated controls (after Bonferroni adjustment). Means ± standard deviations (n = 3).
![Figure 3. Effects of different concentrations of LPA and DCA on [18F]-FDG uptake (CPM/μg protein) of Kelly (A), SK-N-SH (B), SkBr3 (C) and Neuro-2a (D) cells. Controls were set to 100%. Treatment with LPA significantly reduced [18F]-FDG uptake in a concentration dependent manner. In contrast, DCA did not affect glucose uptake at concentrations up to 10 mM. * indicates p < 0.01 compared with the untreated controls (after Bonferroni adjustment). Means ± standard deviations (n = 3).](/cms/asset/973bc4d3-bd9d-4de7-a748-b7417395c90e/kcbt_a_10922003_f0004.gif)
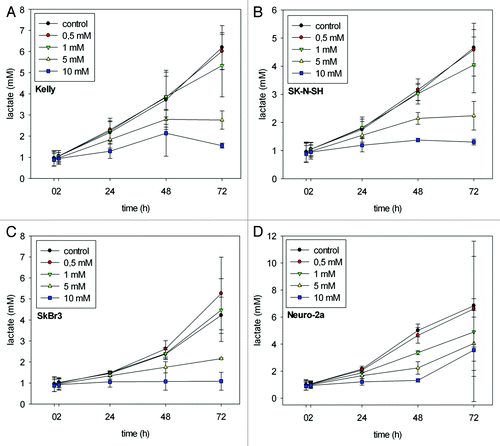
LPA induced changes in lactate production
As demonstrated in treatment with 0.5 mM and 1 mM LPA did not impair the increase in lactate production as observed in the untreated controls after 72 h. In contrast, treatment with 5 mM and 10 mM LPA did prevent the increase in lactate production. Lactate production remained at the level observed at time point 0 h throughout the period of the experiment (up to 72 h). Moreover, in SkBr3 cells 0.5 mM and 1 mM LPA even slightly enhanced lactate concentration compared with controls (). In Neuro-2a cells differences in lactate concentration between control and treatment with 10 mM LPA reached a maximum already at 48 h and slightly decreased at 72 h ().
Figure 4. L-lactate production (mM) in Kelly (A), SK-N-SH (B), SkBr3 (C) and Neuro-2a (D) cells at 0, 2, 24, 48 and 72 h after initiation of treatment with different concentrations of LPA. Time-dependent increase in lactate concentrations as observed in untreated controls was reduced subject to the applied LPA concentrations. Means ± standard deviations (n = 3).
Detection of LPA induced apoptosis via caspase-3
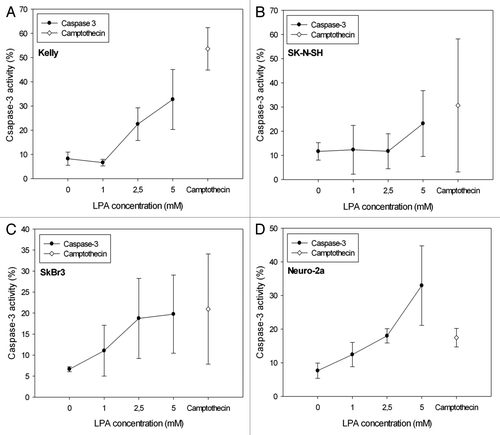
LPA treatment of cells increased caspase-3 activity subject to the applied LPA concentrations, however differently in the various cell lines analyzed (). Neuro-2a cells showed an almost linear LPA concentration dependent increase in caspase-3 activity (). However, incubation of Neuro-2a cells with camptothecin, known to induce apoptosis, resulted in a comparatively low induction of caspase-3 (17%), considerably lower than in Kelly cells (55%) (). Moreover, camptothecin (1 μg/ml) induced caspase-3 activity achieved higher values compared with incubation with 5 mM LPA in all cell lines except for Neuro-2a cells ().
Figure 5. Detection of apoptosis subject to the LPA concentration applied by quantification of active caspase-3 in Kelly (A), SK-N-SH (B), SkBr3 (C) and Neuro-2a (D) cells 24 h after initiation of treatment. The graphs show an increase of active caspase-3 with increasing LPA concentrations in all cell lines analyzed. Activity of caspase-3 after treatment of cells with camptothecin (1 μg/ml) was used as a positive control, because camptothecin is known to induce apoptosis. Means ± standard deviations (n = 3).
Detection of pyruvate dehydrogenase (PDH) activity
As investigated exemplarily for Kelly cells DCA treatment (1mM and 5mM) did not cause statistically significant alterations of PDH activity both after 24h and 48 h compared with untreated controls (data not shown). Treatment with 1 mM LPA did not alter PDH activity compared with untreated controls at 24 h. However, 24 h after treatment with 2.5 mM and 5 mM LPA almost no PDH activity could be detected. 48 h after treatment with 1 mM LPA, PDH activity slightly increased compared with the control sample. After treatment with 2.5 mM and 5 mM LPA PDH activity remained at a very low level also after 48 h (data not shown).
Monitoring of tumor development in animals treated with LPA
Daily therapy with LPA and mock-treatment with PBS were initiated 7 d after s.c. inoculation of SkBr3 tumor cells. Tumor volumes were measured regularly between days 1 and 29 after start of treatment. As shown in in the mock-treated controls (PBS) tumors continuously grew until they reached a maximum of approximately 400 mm3 29 d after start of mock-treatment (i.e., 36 d after s.c. inoculation of tumor cells). In contrast, treatment with LPA significantly retarded tumor growth (p = 0.006 at day 29; ). These data could be confirmed by PET imaging with [18F]-FDG of representative mice. As shown in mock-treated and LPA treated mice revealed almost equal tumor sizes at day 1 after start of treatment. At days 14 and 28, retardation of tumor growth in the LPA treated animals was clearly visible compared with the mock-treated animals (). Accordingly, T/M ratios determined from [18F]-FDG PET images of mock-treated and LPA treated animals significantly differed both at days 14 and 28 after initiation of treatment (data not shown).
Figure 6. Monitoring of tumor growth at different time points after initiation of treatment with LPA. Seven days after s.c. inoculation of SkBr3 tumor cells animals were mock treated with PBS or injected daily with LPA (18.5 mg/kg) (n = 7 per group). Tumor volumes were measured biweekly with a sliding calliper beginning at day 1 after initiation of treatment (A) (means ± standard deviations). Additionally, tumor sizes and locations were monitored with [18F]FDG-PET at days 7, 14 and 28 both in mock-treated (B) and LPA treated mice (C). Tumors are marked by arrows. Treatment of animals with LPA significantly retarded tumor growth compared with mock-treated animals.
![Figure 6. Monitoring of tumor growth at different time points after initiation of treatment with LPA. Seven days after s.c. inoculation of SkBr3 tumor cells animals were mock treated with PBS or injected daily with LPA (18.5 mg/kg) (n = 7 per group). Tumor volumes were measured biweekly with a sliding calliper beginning at day 1 after initiation of treatment (A) (means ± standard deviations). Additionally, tumor sizes and locations were monitored with [18F]FDG-PET at days 7, 14 and 28 both in mock-treated (B) and LPA treated mice (C). Tumors are marked by arrows. Treatment of animals with LPA significantly retarded tumor growth compared with mock-treated animals.](/cms/asset/1d0997ed-4ce5-44e7-9936-a4ad75e5badf/kcbt_a_10922003_f0007.gif)
Histological analysis of residual tumors after LPA therapy
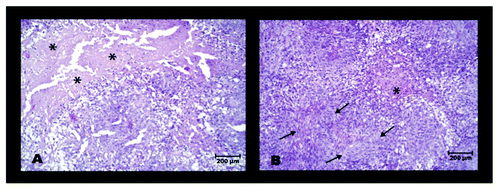
Slices of SkBr3 tumors from animals that had been mock treated with PBS or received daily injections of LPA were subjected to various histological stainings. H&E staining revealed, that necrotic areas were significantly reduced in LPA treated tumors compared with mock-treated tumors (). In contrast, statistically significant differences between LPA treated and mock-treated tumors could not be detected in terms of proliferation (Ki-67 stain), the hypoxia inducible factor HIF-1α and induction of apoptosis (caspase-3 detection) (data not shown). Moreover, expression of transketolase TKTL1, a marker for the activation of the pentose phosphate pathway, could be detected neither in LPA treated nor in mock-treated tumors (data not shown).
Figure 7. H&E stained slices of residual SkBr3 tumors 29 d after initiation of mock- treatment (A) or LPA treatment (B). * indicates areas of necrosis, typical for mock-treated tumors; arrows mark areas with fibrotic strings, typical for LPA treated tumors.
Discussion
Use of LPA in vitro and in vivo revealed its potential to interact with tumor cell metabolism and to negatively influence cancer growth. In vitro investigations of proliferation in these four cell lines depicted a dose dependent decrease of proliferation after 24 h and 48 h. It is likely that even lower concentrations than those used might negatively influence cell proliferation when continuously administered over a longer period of time. Inhibition of proliferation is in line with results from Na, et al. and Pack, et al. who reported dose dependent anti-proliferative effects of LPA in MDA-MB-231 human breast cells as well as Jurkat and CCRF-CEM cells.Citation20,Citation21 It seems very likely that LPA has the potential to interact with a variety of different cell lines.
The hypothesis that LPA could possibly change the glucose uptake in Kelly, Neuro-2a, SK-N-SH and SkBr3 cells was investigated by [18F]-FDG uptake experiments. The data support this idea because in our in vitro experiments all cells showed a dose dependent decrease of uptake. The impaired uptake might be in part due to decreased cell proliferation and/or cell death caused by LPA. However, it is also possible that a shift to oxidative turnover caused cells to reduce uptake since less glucose is needed to meet the demands for energy by switching over to oxidative respiration.Citation3,Citation22 In SK-N-SH cells uptake was decreased by up to 78% of the uptake of untreated cells (at 10 mM LPA). In vivo cell proliferation, as assessed by measuring of the tumor volumes, was reduced in mice treated with LPA. Furthermore, there was also a difference in the amount of [18F]-FDG uptake as observed in PET scans. T/M ratios dropped by day 14 and were approx. twice as low compared with animals that did not receive any LPA treatment. In a report of three cases, Berkson, et al. depicted that a LPA/low dose naltrexone protocol in people with metastatic and nonmetastatic pancreatic cancer resulted in [18F]-FDG PET scans without evidence of disease approximately 4 mo after treatment.Citation23 Therefore, LPA holds the promise to have anti-proliferative properties when applied continuously. The incubation of cancer cells with 5 mM and 10 mM LPA resulted in decreased lactate levels compared with untreated controls. This decrease could be due to the shift from non-oxidative (pyruvate - lactate) to oxidative respiration induced by LPA. Usually, the decreased rate of oxidative respiration in cancer cells results in lower ROS production. Therefore, a sudden shift toward oxidative metabolism could increase the amount of ROS and therefore selectively hit cancer cells by induction of apoptosis.Citation24 As proposed by Deubzer, et al., cellular defense systems against oxidative stress could be diminished in cells of the Warburg type so that cells become more sensitive to oxidative stress.Citation25 As we could show in this study, treatment of cells with LPA induced a dose dependent increase of the apoptosis marker Caspase-3. This result is in agreement with similar findings by other groups concerning induction of apoptosis.Citation17,Citation24,Citation26-Citation31
In addition to the in vitro experiments, studies with LPA were performed in a mouse model after inoculation of SkBr3 tumor cells. LPA was clearly able to reduce the tumor volume and T/M ratios compared with untreated controls. However, the exact mechanism of how LPA influences tumor growth has not yet been clarified. Histopathological analysis of the tumor sample neither showed an increased activity of caspase 3 nor a reduced cell proliferation as indicated by ki-67 activity. Furthermore, staining of TKTL1, an indicator of the pentosephosphate pathway which is activated in a number of human cancers and which has been described to be associated with a poor patients’ survival did not reveal differences in LPA treated tumors and untreated tumors.Citation32-Citation34 The only remarkable histological difference between LPA treated and untreated animals was a reduced rate of necrosis in the LPA group. In contrast to LPA, DCA did not significantly impair proliferation in Kelly, SK-N-SH, SkBr3 and Neuro-2a cells. Nor did it affect [18F]-FDG uptake at concentrations of up to 10 mM. Only very high concentrations of DCA resulted in a statistically significant decrease of [18F]-FDG uptake. A low susceptibility of cells toward DCA might be due to variations in the subtypes of the pyruvate dehydrogenase complex or different properties of the mitochondrial membranes that could limit the anti-proliferative capacity of DCA.Citation35,Citation36
In order to directly quantify the impact of LPA (or DCA) on the amount of pyruvate that is metabolized via pyruvate dehydrogenase, a novel technique, using pyruvate as a tracer for hyperpolarized carbon-13 magnetic resonance imaging, could open up new insights.Citation37 Using this technique could sustain our findings from the ELISA-based PDH assay that LPA incerases the PDH activity after 48h of LPA treatment (data not shown). Alternatively, the amount of oxygen that is produced during treatment of cells with LPA could be quantified. If there is a LPA triggered shift toward oxidative metabolism the amount of oxygen produced should decrease. The question whether the effects of LPA are caused by an increase of pyruvate dehydrogenase activity or by other mechanisms associated with the redox-properties of this compound could be clarified in future experiments by using racemic or (S)-α lipoic acid.
Materials and Methods
Cell lines
The neuroblastoma cell lines Neuro-2a (ATCC®: CCL-131TM), Kelly (DSMZ: ACC 355) and SK-N-SH (ATCC®: HTB-11TM) as well as the breast carcinoma cell line SkBr3 (ATCC®: HTB-30TM) were used for the in vitro experiments. Cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2 in RPMI 1640 (Biochrom AG) plus 10% FCS (Kelly and SK-N-SH), in Dulbeccos MEM with 1 g/l glucose, GlutaMAX-ITM and pyruvate (Gibco Invitrogen) plus 10% FCS (Neuro-2a) or in Dulbeccos MEM (Biochrom) plus 10% FCS (SkBr3).
WST-1 cell viability assay
(R)-(+)-α-lipoic acid (LPA) was obtained from Ratiopharm and dichloroacetate (DCA) from Sigma-Aldrich. For quantification of anti- proliferative effects of LPA and DCA treatment, 5·104 cells (Kelly, SK-N-SH), 2.5·104 cells (Neuro-2a) and 5·103 cells (SkBr3) were seeded in triplicate into 96 well plates 24 h before treatment. Cells were left untreated (controls) or incubated with LPA and DCA at concentrations of 2.5 mM, 5 mM, 7.5 mM and 10 mM for either 24h or 48h. Metabolic activity of cells, indicative of cell proliferation, was measured after addition of 10μl (per 100μl medium) of a liquid reagent containing tetrazolium salt (WST-1 reagent, Roche Diagnostics). Changes in the formazan dye development – compared with untreated controls – indicative of changes in cellular metabolic activity were quantified 60 min after addition of the WST-1 reagent in an ELISA Reader (Bio- Tek). Results of controls were set to 100%. Therefore, results above 100% are indicative of increased proliferation whereas those below 100% represent decreased proliferation.
Assessment of proliferation/viability by measurement of electrical impedance (xCELLigence assay)
Neuro-2a (6000/well), SkBr3 (2000 cells/well), Kelly (6000 cells/well) and SK-N-SH cells (6000 cells/well) were seeded in E-plates 16 (Roche, Mannheim, Germany), placed on the real time cell analyzer (RTCA) of the xCELLigence system (Roche, Mannheim, Germany) and allowed to adhere overnight in a humidified atmosphere (37°C) containing 5% CO2. Subsequently, cells were treated with 0.5 mM, 1 mM and 5 mM LPA. The xCELLigence system measures electrical impedance across micro-electrodes integrated on the bottom of the tissue culture E-Plates. The impedance measurement (expressed as Cell Index) provides quantitative information about the biological status of the cells, including cell number, viability and morphology. Measurement of cell impedance was performed continuously over 80 h.
Determination of lactate production after LPA treatment
For investigation of lactate production of cells after treatment with different concentrations of LPA, 2·105 Kelly and SK-N-SH cells, 5·104 Neuro-2a cells and 2.5·104 SkBr3 cells were seeded in 6 well plates (Greiner Bio One) and cultivated for 24h. During incubation with LPA the different cell lines analyzed were grown in the same culture medium (DMEM plus 10% FCS, 1g/l glucose, GlutaMAX- ITM, pyruvate; Gibco Invitrogen) to assure optimal conditions for comparison of results based on colorimetric determination. Cells were incubated without LPA (control) or with 0.1 mM, 0.5 mM, 5 mM and 10 mM LPA for different time points: at 0 h, 2 h, 24 h, 48 h and 72 h after start of incubation samples (cells plus culture media) were taken and frozen at -20°C. Before analysis, samples were heated at 80°C for 15 min and centrifuged. Lactate concentrations in the supernatants were measured at 340 nm using a lactate kit (R-Biopharm AG) according to the manufacturer’s instructions.
Determination of [18F]-FDG uptake subject to LPA and DCA treatment
For evaluation of possible effects of LPA and DCA treatment on glucose metabolism, [18F]-FDG uptake was determined. Because both compounds are expected to cause a shift toward oxidative phosphorylation, the demand for glucose is thought to decrease. For that purpose Kelly and SK-N-SH cells (1·105), Neuro-2a cells (5·104) and SkBr3 cells (4·104) per well were seeded in 24 well plates (Greiner Bio One) and cultivated for 24 h. Cells were then incubated as triplicates without LPA and DCA (controls), and 1 mM, 2.5 mM, 5 mM, 7.5 mM, 10 mM, 25 mM or 30 mM LPA or DCA for 24h (LPA) and 48h (DCA). Subsequently culture medium was removed and uptake of [18F]-FDG (3.7·102 kBq) was assayed in 500 μl of DMEM supplemented with 1g/l glucose, GlutaMAX-ITM, pyruvate and 10% FCS for 30 min. Cells were then washed with PBS and detached with 1 M NaOH. [18F]-FDG uptake was measured in a gamma counter (1480 Wizard; Wallac). Protein concentrations were determined using the Bradford assay (BioRad).Citation38 Uptake of controls (CPM/μg protein) was set to 100%.
Detection of apoptosis after LPA treatment via caspase-3 activity
To investigate the potential of LPA to induce apoptosis, 3·105 cells of each cell line were seeded in 6 well plates and were allowed to adhere for 24 h. LPA was then added at concentrations of 1 mM, 2.5 mM and 5 mM and cells were incubated for 24 h. Since camptothecin has been reported to induce apoptosis, treatment of cells with camptothecin (1 μg/ml) served as a positive control. After detachment with 1 mM EDTA in PBS cells were repeatedly washed in PBS and fixed with cytofix/cytoperm (BD PharmingenTM) for 20 min on ice. Subsequently, cells were washed twice with perm/wash (BD PharmingenTM) and incubated with anti- caspase-3 antibody labeled with FITC (4 μl of antibody in 20 μl perm/wash buffer; BD PharmingenTM) for 30min in the dark at room temperature. Cells were again washed with perm/wash and finally resuspended in 0.5 ml perm/wash. Flow cytometric analysis was performed with a FACS Calibur (Beckton Dickinson) according to the manufacturers’ instructions (BD PharmingenTM).Citation39
Measurement of Pyruvate Dehydrogenase (PDH) Activity
Three ·107 Kelly cells (per 175 cm2 flask) were seeded at 37°C and 5% CO2 in a humified atmosphere and allowed to adhere overnight. Cells were treated with 1 mM and 5 mM DCA or 1 mM, 2.5 mM and 5 mM LPA for up to 48 h. PDH activity was measured at 0 h, 24 h and 48 h after addition of LPA. For that purpose cells were lysed with 300 µl of a lysis buffer (M-PER containig protease inhibitors) and processed according to the manufactors’ instructions (PDH Protein Quantity Microplate Assay Kit, Abcam, Cambridge, UK). PDH activity was measured using an ELISA reader at 450 nm (Bio-Tek 800).
Animals and tumor model
Female NMRI nu/nu mice (Charles River Laboratories), 6–8 weeks old, were used for studies investigating therapeutic efficacy of LPA following s.c. inoculation of SkBr3 tumor cells. Mice (five per cage) were housed in ventilated cages under standard conditions at 26°C and 50–60% humidity. Mice had free access to food and water. Approximately 6 h before [18F]-FDG PET imaging access to food was abandoned. Mice were inoculated s.c. 5·106 SkBr3 tumor cells each, suspended in 50μl of PBS close to the right shoulders. One week after tumor cell inoculation the treatment group (n = 7) received daily i.p. injections of LPA over a period of four weeks (18.5 mg/kg in 50 μl PBS). The animals of the control group (n = 7) were repeatedly injected i.p. with 50 μl of sterile PBS. The tumor volume was determined biweekly by measuring height, width and depth with a sliding caliper. All animal experiments were conducted in accordance with the German federal law and the guidelines for the protection of animals according to the permission of the government of Upper Bavaria.
Tumor staging during LPA treatment with PET and CT
To monitor tumor development during daily treatment of animals with LPA, four mice per group were imaged using PET at days 7, 14 and 28 after tumor cell inoculation (i.e., at days 0, 7 and 21 after start of daily LPA therapy) (microPET Focus 120 scanner, Siemens). Two animals of each group additionally received a CT scan (CT Scanner Inveon, Siemens). For PET imaging the mice were anesthetized with isoflurane for the length of the imaging procedure. After i.v. injection of 11.1 MBq [18F]-FDG mice were imaged in the prone position for 15 min. To exclude discrepancies derived from differences in injected activities and body weights, tumor/muscle (T/M) ratios were calculated, as defined by the ratios of ROItumor and ROImuscle (measured at the psoas muscle).
Histological analysis
Residual tumors after LPA treatment of mice were subjected to histological analysis in terms of proliferation, hypoxia, cell death and glucose metabolism. Because LPA may induce a shift toward oxidative phosphorylation, changes induced by LPA in tumors should preferentially affect proliferation and glucose metabolism. Moreover, LPA could possibly modify activities of enzymes involved in the Warburg effect. Therefore tumors were also investigated for transketolase TKTL1 activity.Citation34 Tumors were fixed in 4% neutrally buffered formalin and embedded in paraffin. Tumor slices were subjected to H&E staining for analysis of morphological changes in tumor cells. Ki-67 staining (anti-Ki-67, Dakocytomation) was performed as a marker for detection of proliferation.Citation40 Hypoxia was revealed by HIF-1α staining (anti-HIF-1α, Novus Biologicals). Caspase-3 staining (anti-caspase-3 cleaved, Cell Signaling Technology) was applied to detect apoptotic cells. Immunohistochemical stainings were processed using a Roche Ventana Discovery device (Roche) according to the manufacturer’s instructions. All slices were scanned and digitalized by a slide scanner (Olympus). For each of the resulting digital slides, regions of interests (ROIs) were defined according to areas with and without necrosis, expression of Ki-67, HIF-1α, Caspase-3 and Transketolase TKTL1. Digital slides were analyzed with Definiens Enterprise Imaging Intelligence Suite Software (Definiens).
Statistical methods
Statistical analysis was performed using SPSS (version 18.0, IBM Corporation) and SigmaPlot 11.0 (Systat Software Inc.). Means and standard deviations were calculated from at least 3 independent experiments. A t-test was used to compare means of two independent samples and tests were Bonferroni adjusted. The level of statistical significance was set at p < 0.05.
Acknowledgments
We would like to thank Mathias Gehrmann at the Department of Radiotherapy, Technische Universitaet Muenchen, for supporting the FACS analysis and Axel Walch at the Department of Pathology, Helmholtz Zentrum Muenchen, for supervising the immunohistochemical analyses.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Z 1924; 152:309 - 44
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol 1927; 8:519 - 30; http://dx.doi.org/10.1085/jgp.8.6.519; PMID: 19872213
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 9; http://dx.doi.org/10.1038/nrc1478; PMID: 15516961
- Czernin J, Phelps ME. Positron emission tomography scanning: current and future applications. Annu Rev Med 2002; 53:89 - 112; http://dx.doi.org/10.1146/annurev.med.53.082901.104028; PMID: 11818465
- Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta 2009; 1790:1149 - 60; http://dx.doi.org/10.1016/j.bbagen.2009.07.026; PMID: 19664690
- Handelman GJ, Han D, Tritschler H, Packer L. Alpha-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem Pharmacol 1994; 47:1725 - 30; http://dx.doi.org/10.1016/0006-2952(94)90298-4; PMID: 8204089
- Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med 1995; 19:227 - 50; http://dx.doi.org/10.1016/0891-5849(95)00017-R; PMID: 7649494
- Packer L, Roy S, Sen CK. Alpha-lipoic acid: a metabolic antioxidant and potential redox modulator of transcription. Adv Pharmacol 1997; 38:79 - 101; http://dx.doi.org/10.1016/S1054-3589(08)60980-1; PMID: 8895805
- Ziegler D, Hanefeld M, Ruhnau KJ, Meissner HP, Lobisch M, Schütte K, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38:1425 - 33; http://dx.doi.org/10.1007/BF00400603; PMID: 8786016
- Mijnhout GS, Alkhalaf A, Kleefstra N, Bilo HJ. Alpha lipoic acid: a new treatment for neuropathic pain in patients with diabetes?. Neth J Med 2010; 68:158 - 62; PMID: 20421656
- Zhang WJ, Bird KE, McMillen TS, LeBoeuf RC, Hagen TM, Frei B. Dietary alpha-lipoic acid supplementation inhibits atherosclerotic lesion development in apolipoprotein E-deficient and apolipoprotein E/low-density lipoprotein receptor-deficient mice. Circulation 2008; 117:421 - 8; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.725275; PMID: 18158360
- de Oliveira AM, Rondó PH, Luzia LA, D’Abronzo FH, Illison VK. The effects of lipoic acid and α-tocopherol supplementation on the lipid profile and insulin sensitivity of patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled trial. Diabetes Res Clin Pract 2011; 92:253 - 60; http://dx.doi.org/10.1016/j.diabres.2011.02.010; PMID: 21371770
- Biewenga GP, Haenen GR, Bast A. The pharmacology of the antioxidant lipoic acid. Gen Pharmacol 1997; 29:315 - 31; http://dx.doi.org/10.1016/S0306-3623(96)00474-0; PMID: 9378235
- Novotny L, Rauko P, Cojocel C. alpha-Lipoic acid: the potential for use in cancer therapy. Neoplasma 2008; 55:81 - 6; PMID: 18237244
- Korotchkina LG, Sidhu S, Patel MS. R-lipoic acid inhibits mammalian pyruvate dehydrogenase kinase. Free Radic Res 2004; 38:1083 - 92; http://dx.doi.org/10.1080/10715760400004168; PMID: 15512796
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99:989 - 94; http://dx.doi.org/10.1038/sj.bjc.6604554; PMID: 18766181
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11:37 - 51; http://dx.doi.org/10.1016/j.ccr.2006.10.020; PMID: 17222789
- Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008; 109:394 - 402; http://dx.doi.org/10.1016/j.ygyno.2008.01.038; PMID: 18423823
- Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008; 68:1223 - 31; http://dx.doi.org/10.1002/pros.20788; PMID: 18465755
- Na MH, Seo EY, Kim WK. Effects of alpha-lipoic acid on cell proliferation and apoptosis in MDA-MB-231 human breast cells. Nutr Res Pract 2009; 3:265 - 71; http://dx.doi.org/10.4162/nrp.2009.3.4.265; PMID: 20098578
- Pack RA, Hardy K, Madigan MC, Hunt NH. Differential effects of the antioxidant alpha-lipoic acid on the proliferation of mitogen-stimulated peripheral blood lymphocytes and leukaemic T cells. Mol Immunol 2002; 38:733 - 45; http://dx.doi.org/10.1016/S0161-5890(01)00110-9; PMID: 11841833
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 33; http://dx.doi.org/10.1126/science.1160809; PMID: 19460998
- Berkson BM, Rubin DM, Berkson AJ. Revisiting the ALA/N (alpha-lipoic acid/low-dose naltrexone) protocol for people with metastatic and nonmetastatic pancreatic cancer: a report of 3 new cases. Integr Cancer Ther 2009; 8:416 - 22; http://dx.doi.org/10.1177/1534735409352082; PMID: 20042414
- Wenzel U, Nickel A, Daniel H. alpha-Lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2-*-generation. Apoptosis 2005; 10:359 - 68; http://dx.doi.org/10.1007/s10495-005-0810-x; PMID: 15843897
- Deubzer B, Mayer F, Kuçi Z, Niewisch M, Merkel G, Handgretinger R, et al. H(2)O(2)-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells: potential role of lactate and ferritin. Cell Physiol Biochem 2010; 25:767 - 74; http://dx.doi.org/10.1159/000315098; PMID: 20511723
- van de Mark K, Chen JS, Steliou K, Perrine SP, Faller DV. Alpha-lipoic acid induces p27Kip-dependent cell cycle arrest in non-transformed cell lines and apoptosis in tumor cell lines. J Cell Physiol 2003; 194:325 - 40; http://dx.doi.org/10.1002/jcp.10205; PMID: 12548552
- Selvakumar E, Hsieh TC. Regulation of cell cycle transition and induction of apoptosis in HL-60 leukemia cells by lipoic acid: role in cancer prevention and therapy. J Hematol Oncol 2008; 1:4; http://dx.doi.org/10.1186/1756-8722-1-4; PMID: 18577252
- Sen CK, Sashwati R, Packer L. Fas mediated apoptosis of human Jurkat T-cells: intracellular events and potentiation by redox-active alpha-lipoic acid. Cell Death Differ 1999; 6:481 - 91; http://dx.doi.org/10.1038/sj.cdd.4400514; PMID: 10381641
- Choi SY, Yu JH, Kim H. Mechanism of alpha-lipoic acid-induced apoptosis of lung cancer cells. Ann N Y Acad Sci 2009; 1171:149 - 55; http://dx.doi.org/10.1111/j.1749-6632.2009.04708.x; PMID: 19723049
- Kim JI, Cho SR, Lee CM, Park ES, Kim KN, Kim HC, et al. Induction of ER Stress-Mediated Apoptosis by α-Lipoic Acid in A549 Cell Lines. Korean J Thorac Cardiovasc Surg 2012; 45:1 - 10; http://dx.doi.org/10.5090/kjtcs.2012.45.1.1; PMID: 22363901
- Dozio E, Ruscica M, Passafaro L, Dogliotti G, Steffani L, Marthyn P, et al. The natural antioxidant alpha-lipoic acid induces p27(Kip1)-dependent cell cycle arrest and apoptosis in MCF-7 human breast cancer cells. Eur J Pharmacol 2010; 641:29 - 34; http://dx.doi.org/10.1016/j.ejphar.2010.05.009; PMID: 20580704
- Xu X, Zur Hausen A, Coy JF, Löchelt M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int J Cancer 2009; 124:1330 - 7; http://dx.doi.org/10.1002/ijc.24078; PMID: 19065656
- Ramos-Montoya A, Lee WN, Bassilian S, Lim S, Trebukhina RV, Kazhyna MV, et al. Pentose phosphate cycle oxidative and nonoxidative balance: A new vulnerable target for overcoming drug resistance in cancer. Int J Cancer 2006; 119:2733 - 41; http://dx.doi.org/10.1002/ijc.22227; PMID: 17019714
- Langbein S, Zerilli M, Zur Hausen A, Staiger W, Rensch-Boschert K, Lukan N, et al. Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br J Cancer 2006; 94:578 - 85; http://dx.doi.org/10.1038/sj.bjc.6602962; PMID: 16465194
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab 2003; 284:E855 - 62; PMID: 12676647
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 1988; 4:155 - 81; http://dx.doi.org/10.1146/annurev.cb.04.110188.001103; PMID: 3058159
- Schroeder MA, Cochlin LE, Heather LC, Clarke K, Radda GK, Tyler DJ. In vivo assessment of pyruvate dehydrogenase flux in the heart using hyperpolarized carbon-13 magnetic resonance. Proc Natl Acad Sci U S A 2008; 105:12051 - 6; http://dx.doi.org/10.1073/pnas.0805953105; PMID: 18689683
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72:248 - 54; http://dx.doi.org/10.1016/0003-2697(76)90527-3; PMID: 942051
- Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407:770 - 6; http://dx.doi.org/10.1038/35037710; PMID: 11048727
- Bullwinkel J, Baron-Lühr B, Lüdemann A, Wohlenberg C, Gerdes J, Scholzen T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J Cell Physiol 2006; 206:624 - 35; http://dx.doi.org/10.1002/jcp.20494; PMID: 16206250