Abstract
Human oncogene DEK has been shown to be upregulated in a number of neoplasms. The purpose of this study was to investigate DEK expression level in chronic lymphocytic leukemia (CLL), analyze the correlation between DEK expression and CLL prognostic markers, and characterize the role of DEK in the response to either chemotherapeutic drugs or nongenotoxic activators of the p53 pathway. DEK mRNA was evaluated by real-time quantitative reverse transcriptase-polymerase chain reaction (qPCR), and primary CLL samples were treated in vitro with either fludarabine or Nutlin-3 to explore the interaction of p53 status and DEK mRNA expression. The median expression levels of DEK mRNA were 6.792 × 10−2 (1.438 × 10−2−3.201 × 10−1) in 65 patients with CLL. A marked increase of DEK mRNA expression was observed in the CLL patients with unmutated immunoglobulin heavy chain variable (IGHV) gene (p = 0.025), CD38-positive (p = 0.047), del(17p13) (p = 0.006). Both fludarabine and Nutlin-3 significantly downregulated DEK in the primary CLL cells which were with normal function of p53, or without deletion or mutation of p53 (p = 0.042, p = 0.038; p = 0.021, p = 0.017; p = 0.037, p = 0.017). However, the downregulation of DEK was not observed in the primary CLL cells which were with dysfunction of p53, or with deletion or mutation of p53 (p = 0.834, p = 0.477; p = 0.111, p = 0.378; p = 0.263, p = 0.378). These data show that DEK might be applied for the assessment of prognosis in patients with CLL, and fludarabine and Nutlin-3 regulate DEK expression depended on p53 status.
Introduction
Human oncogene DEK located on chromosome 6p22-23.Citation1,Citation2 It is a 375 amino acid (43 kDa) abundant nuclear protein with important functions in the architectural regulation of chromatin assembly.Citation3,Citation4 DEK was initially identified as a fusion protein with CAN nucleoporin in a subtype of acute myeloid leukemia (AML) involving the t(6;9) translocation.Citation5 Subsequent studies have repeatedly identified DEK as a frequently overexpressed gene independent of the t(6;9) translocation in a number of neoplasms including melanoma, hepatocellular carcinoma, glioblastoma, retinoblastoma, uterine cervical cancers, ovarian cancers, and bladder cancer.Citation1,Citation6-Citation12 Furthermore, autoantibodies to DEK have been detected in juvenile rheumatoid arthritis, systemic lupus erythematosus and sarcoidosis.Citation13,Citation14 Though precise cellular function of DEK remains unclear, several studies have implicated DEK in a variety of cellular processes, such as DNA replication, splice site recognition, and gene transcription, as well as in the control of cell viability, differentiation, and cell-to-cell signaling.Citation15-Citation23 The mechanisms through DEK mediates its oncogenic effects are only partially understood. However, it has been proposed that the oncogenic role of DEK is mediated by its ability to destabilize p53 protein and to inhibit p53 activity and p53-mediated apoptosis.Citation10,Citation24-Citation26 DEK can cooperate with the oncogenes E6 and E7 to overcome senescence,Citation27 and promote epithelial transformation in vitro and in vivo when overexpressed.Citation28,Citation29
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the Western world, but less frequent in Eastern countries. The clinical course of CLL is highly variable. One third of CLL patients require therapy as soon as they are diagnosed, one third survive for many years without therapy, and one third have disease progression over the years and require treatment at some point.Citation30 Previous study reported upregulation of DEK in a subset of CLL with del(11q23) and advanced clinical stage.Citation31
In this study, we detected DEK expression by real-time quantitative reverse transcriptase-polymerase chain reaction (qPCR) in 65 Chinese patients with CLL to investigate the DEK expression level in CLL, and analyze the correlation between DEK expression and CLL prognostic markers such as clinical stage, immunoglobulin heavy-chain variable region (IGHV) mutational status, ZAP-70, CD38, and chromosomal abnormalities. Furthermore, primary CLL samples were treated in vitro with either fludarabine, which represented the treatment of choice for CLL, or Nutlin-3, which showed promising cytotoxic activity against CLL,Citation32-Citation36 to explore the role of DEK in the response to either chemotherapeutic drugs or nongenotoxic activators of the p53 pathway.
Results
Clinical characteristics of CLL patients
The characteristics of 65 CLL patients are summarized in . Forty-three patients were male and 22 were female (male: female, 2.0), and the median age was 60 y (rang: 44–84). According to the Binet staging system,Citation37 29 (44.6%) patients were in stage A, 10 (15.4%) in stage B, and 26 (40.0%) in stage C.
Table 1. Clinical and biological characteristics of 65 patients with chronic lymphocytic leukemia
DEK mRNA expression in 65 CLL patients and the correlations between DEK expression and prognostic factors
The median expression levels of DEK mRNA were 6.792 × 10−2 (1.438 × 10−2−3.201 × 10−1) in 65 patients with CLL. The correlations between DEK expression and prognostic factors were shown in . A marked increase of DEK mRNA expression was observed in CLL patients with unmutated IGHV (p = 0.025), CD38-positive (p = 0.047), del(17p13) (p = 0.006).
Table 2. The differences of DEK mRNA expression level between various groups of patients
The clinical characteristics of the CLL patients with cell culture
For preparation of primary cell cultures, CLL cells were from 22 untreated CLL patients. The clinical and biological characteristics of these patients are detailed in . Four cases showed p53 mutations paired to del(17p13), one cases showed del(17p13) in the absence of p53 mutations. The function of p53/p21 was detected by flow cytometry both at the beginning of isolation and after 24 h fludarabine treatment. The level of p53 protein was not increased after treatment with fludarabine in five patients. The level of p21 protein of eight patients showed no increase after fludarabine-treatment, implying p53 dysfunction. In the rest cases, the p21 protein was increased after fludarabine-treated, suggesting p53 normal function.Citation38
Table 3. Clinical and biological characteristics in 22 CLL patients with cell culture
Fludarabine and Nutlin-3 regulate DEK expression depended on p53 status
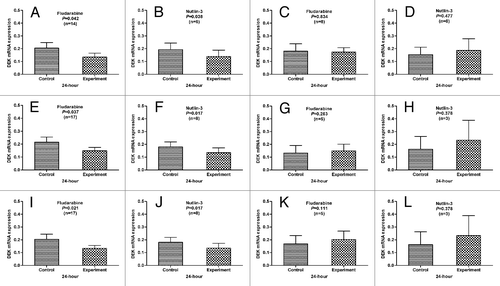
To assay the interaction of p53 status and DEK mRNA expression, we detected the mRNA expression in fludarabine-treated (n = 22) or Nutlin-3-treated (n = 11) CLL cells by qPCR. The primary CLL cells with normal p53 function, without deletion or mutation of p53, the level of DEK expression was significantly decreased after 24 h treatment with fludarabine and Nutlin-3 compared with the cells in medium only (p = 0.042, p = 0.038; p = 0.021, p = 0.017; p = 0.037, p = 0.017) (). However, the DEK was not downregulated in the primary CLL cells with dysfunction of p53, with deletion or mutation of p53 (p = 0.834, p = 0.477; p = 0.111, p = 0.378; p = 0.263, p = 0.378) (). These results indicated that fludarabine and Nutlin-3 regulated DEK expression depended on p53 status, but certainly not consistently.
Figure 1. The correlation between the p53 status and the level of DEK expression. In the primary CLL cells with normal p53 function (A and B), or without deletion (E and F) or mutation (I and J) of p53, DEK expression was significantly decreased after 24 h treatment with fludarabine or Nutin-3 compared with the cells in medium only. However, this downregulation of DEK expression was not observed in the primary CLL cells with p53 dysfunction (C and D), or deletion (G and H) or mutation (K and L) of p53.
Discussion
The chromatin architectural factor DEK is an oncogene located on the region of 6p22–23.Citation1,Citation2 It was discovered by the identification of the t(6;9) in a subset of patients with AML, and was named based on the initials of the patient DEK.Citation5 The observation that this chromosomal change was associated with an accelerated tumor onset and poor prognosis prompted a series of studies that ultimately support a causative role of DEK in tumor development.Citation3,Citation4 DEK has been shown to be upregulated in AML,Citation5,Citation39,Citation40 retinoblastoma,Citation2,Citation41,Citation42 glioblastoma,Citation6 hepatocellular carcinoma,Citation7 melanoma,Citation9 and in an increasing list of other tumor types.Citation1,Citation8,Citation10 However, the mechanisms leading to this preferential accumulation of DEK in cancer cells are not completely understood.
With the increasing list of tumor types, high expression of DEK raises the exciting possibility of using DEK as a tumor marker. The finding that DEK expression levels can distinguish benign nevi from malignant melanomas is a prime example of a clinically relevant setting in which DEK may prove to be highly useful.Citation10 Moreover, as DEK may be present at higher levels in immature cells than in their differentiated counterparts,Citation43 it could also aid in gauging the differentiation potential of tumor cells. In fact, t(6;9) translocation has been suggested to be considered in AML prognostic stratification.Citation44 In this study, the correlations between DEK expression and prognostic factors of CLL were analyzed. A marked increase of DEK mRNA expression was observed in CLL patients with unmutated IGHV, CD38-positive, and del(17p13). This is the first study to correlate DEK expression levels in CLL with prognostic factors to understand the role of DEK upregulation in CLL progression. Our results suggest that the expression level of DEK might be applied for the assessment of prognosis in patients with CLL.
The crosstalk between DEK, p53 and the apoptotic machinery deserves attention, as this information may guide the development of improved therapies. In response to a variety of stimuli, such as cellular stress induced by chemotherapeutic drugs, the p53-MDM2 interaction is disrupted and p53 rapidly accumulates within the cells.Citation32 Alternatively, p53 can accumulate in response to selective small-molecule inhibitors of the p53-MDM2 interaction, which binds MDM2 in the p53 binding pocket with high selectivity and can release p53 from negative control leading to effective stabilization of p53 and activation of the p53 pathway.Citation36 In this study, we have shown that both chemotherapeutic drugs (fludarabine) and nongenotoxic activators of the p53 pathway (Nutlin-3) significantly downregulated DEK in the primary CLL cells with normal p53 function, or without deletion or mutation of p53. However, the DEK was not downregulated in the primary CLL cells with p53 dysfunction, or with deletion or mutation of p53. Although these data clearly indicate that a p53 status is necessary to observe the DEK downregulation in response to either fludarabine or Nutlin-3 in CLL cells, the exact role of p53 in DEK regulation remains to be determined.
In conclusion, we provide evidence that increased expression of DEK correlates with IGHV mutational status, CD38-positive and del(17p13), and DEK can therefore be considered as potential prognostic factor. As the result that fludarabine and Nutlin-3 regulate DEK expression depended on p53 status, therapeutic strategies able to downregulate DEK expression should be further explored to improve the antileukemic activity of both conventional and novel antileukemic drugs.
Materials and Methods
Patients
Our study population consisted of 65 consecutive patients with newly diagnosed and untreated CLL between December 2004 and January 2011. All patients provided their informed consent and the research project was approved by the University and Institutional Review Boards. The diagnosis was based on the revised NCI criteria.Citation45 The staging of CLL was performed according to the Binet stage system.Citation37 Data collected at diagnosis included: age, gender, Binet stages, β2-microglobulin (β2-MG) and lactate dehydrogenase (LDH). A range of other prognostic markers was also analyzed for the majority of patients: IGHV and p53 mutational status, CD38 and ZAP-70 expression and cytogenetics by florescence in situ hybridization (FISH).
Detection of CD38 and ZAP-70 by flow cytometry
Flow cytometric analysis of CD38 and ZAP-70 was performed as previously described.Citation46 Cut-off points of 30% and 20% were used to define positivity for CD38 and ZAP-70, respectively.
IGHV mutational status analysis
IGHV mutational status was detected by IGHV gene primer and IGH Somatic Hypermutation Assay for Gel Detection kit (InVivoScribe Company). The multiplex PCR products of IGHV were detected by direct sequencing as previously described.Citation47 A germline homology of 98% was used as the cut-off between IGHV mutated and unmutated cases.
p53 mutational status analysis
p53 mutational status was studied by PCR and direct sequencing. We used the same primers as those mentioned in the previous study.Citation48 PCR products of p53 were purified by standard methods (Invitrogen) and directly sequenced using the ABI3730XL 96-capillary DNA Analyzer (Applied Biosystems).
Detection of molecular cytogenetic aberrations by FISH
FISH analysis was performed on the sample for conventional cytogenetic studies. In order to detect prognostically relevant anomalies of chromosomal regions 6q, 11q, 13q, 14q, 17p and chromosome 12, the following fluorescent-labeled probes were used in interphase cytogenetic analyses: LSI MYB (6q23), LSI ATM (11q22), LSI D13S319 (13q14), LSI IGHC/IGHV (14q32), LSI p53 (17p13) and CEP12 (centromere 12) (all probes were purchased from Vysis, Downers Grove, IL, USA). FISH was performed as described.Citation49 The cut-off levels for positive values (mean of normal control ± 3 SD), determined from samples of 8 cytogenetically normal persons, was 7.5%, 7.7%, 10.3%, 8.9%, 5.2% and 3.0% for del(6q23), del(11q22), del(13q14), 14q32 translocation, del(17p13) and trisomy 12, respectively.
The protocol of primary cell culture
For preparation of primary cell cultures, CLL cells from 26 untreated patients were isolated from heparinized venous blood by density gradient centrifugation. The isolated cells were predominantly CLL B cells (> 90% CD5+CD19+), as assessed by flow cytometry (FACScan, Becton Dickinson). Freshly isolated CLL cells were seeded in 6-well plates (5−10 × 106 cells/well), treated by 3.5 μmol/L fludarabine (Sigma) or 10 μmol/L Nutlin-3 (Sigma) or not, and cultured in RPMI-1640 medium supplemented with 10% fetal calf serum in a humidified atmosphere containing 5% CO2 at 37°C for 24 h.
Detection of the p53/p21 function by flow cytometry
The function of p53/p21 gene was detected by flow cytometry. Cells were harvested after in vitro culture 24 h with 3.5 μmol/L fludarabine (Sigma) treatment. 5 × 106 cells were fixed in 2% paraformaldehyde, −4°C 30 min, washed with PBS, and overnight in 80% ethanol at −20°C. Fixed cells were washed with PBS and cell membrane ruptured with cell permebilization kit (BD Biosciences FIX&PERM) at room temperature for 30 min. CLL cells were labeled with CD19-allophycocyanin away from light for 15 min, and then washed with PBS. Fixed cells were stained with p53-phycoerythrin antibody (BD Biosciences) and p21-fluorescein isothiocyanate (Calbiochem) or the corresponding isotype controls. After incubation at ambient temperature for 15 min away from light, cells were detected on the FACSCalibur and data were analyzed using the CellQuest Pro software.Citation38
qPCR analysis for DEK
DEK mRNA expression was investigated by qPCR. Total RNA was isolated from peripheral blood mononuclear cells or culture cells, which had > 90% CD5+CD19+ cells measured by flow cytometry. RNA (1μg) was reverse transcribed using random hexamers, and then amplification was performed with fluorescent dye SYBR Green I, PCR Master Mix and primers (). The β-actin was used as internal reference. Cycle conditions for DEK and β-actin were 1 cycle for 5 min at 95°C, 35 cycles for 10 sec at 95°C, 30 sec at 62°C, 30 sec at 72°C, and finally, 1 cycle for 10 min at 72°C. The threshold cycle (Ct) was defined as the fractional cycle number at which the fluorescence passes the fixed threshold, and each sample was normalized based on its endogenous β-actin RNA content. Sequences of amplified products were verified by DNA sequencing. Each sample was replicated for two times.
Table 4. The sequences of qRT-PCR primers of DEK and β-actin
Statistical analysis
All statistical analyses were performed using the SPSS program for Windows (version 17.0). ΔCt was calculated by subtracting the Ct of β-actin from the Ct of DEK. The relative quantitative value of DEK mRNA was calculated by the equation 2−ΔCt. The difference of DEK mRNA expression between groups with different prognostic factors was described using the Mann-Whitney U test. Differences of gene expression levels between primary CLL cells treated with or without fludarabine and Nutlin-3 were analyzed by matched-pairs t test. For all tests, a p value of 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by National Natural Science Foundation of China (30971296, 81170485, 81170488, 81200360), Natural Science Foundation of Jiangsu Province (BK2010584, BK2012484), Key Projects of Health Department of Jiangsu Province (K201108), Jiangsu Province’s Medical Elite Program (RC2011169), Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institute (JX10231801), National Public Health Grand Research Foundation (201202017), the Program for Development of Innovative Research Team in the First Affiliated Hospital of NJMU, Jiangsu Province Higher Education Institute Foundation of Science and Technology Innovation Team Program, the Project for State Key Clinical Department construction.
References
- Evans AJ, Gallie BL, Jewett MA, Pond GR, Vandezande K, Underwood J, et al. Defining a 0.5-mb region of genomic gain on chromosome 6p22 in bladder cancer by quantitative-multiplex polymerase chain reaction. Am J Pathol 2004; 164:285 - 93; http://dx.doi.org/10.1016/S0002-9440(10)63118-5; PMID: 14695341
- Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer 2006; 45:72 - 82; http://dx.doi.org/10.1002/gcc.20263; PMID: 16180235
- Kappes F, Burger K, Baack M, Fackelmayer FO, Gruss C. Subcellular localization of the human proto-oncogene protein DEK. J Biol Chem 2001; 276:26317 - 23; http://dx.doi.org/10.1074/jbc.M100162200; PMID: 11333257
- Waldmann T, Baack M, Richter N, Gruss C. Structure-specific binding of the proto-oncogene protein DEK to DNA. Nucleic Acids Res 2003; 31:7003 - 10; http://dx.doi.org/10.1093/nar/gkg864; PMID: 14627833
- von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol 1992; 12:1687 - 97; PMID: 1549122
- Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res 1999; 59:4990 - 6; PMID: 10519413
- Kroes RA, Jastrow A, McLone MG, Yamamoto H, Colley P, Kersey DS, et al. The identification of novel therapeutic targets for the treatment of malignant brain tumors. Cancer Lett 2000; 156:191 - 8; http://dx.doi.org/10.1016/S0304-3835(00)00462-6; PMID: 10880769
- Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle 2006; 5:1202 - 7; http://dx.doi.org/10.4161/cc.5.11.2801; PMID: 16721057
- Wu Q, Li Z, Lin H, Han L, Liu S, Lin Z. DEK overexpression in uterine cervical cancers. Pathol Int 2008; 58:378 - 82; http://dx.doi.org/10.1111/j.1440-1827.2008.02239.x; PMID: 18477217
- Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res 2009; 69:6405 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-09-1063; PMID: 19679545
- Han S, Xuan Y, Liu S, Zhang M, Jin D, Jin R, et al. Clinicopathological significance of DEK overexpression in serous ovarian tumors. Pathol Int 2009; 59:443 - 7; http://dx.doi.org/10.1111/j.1440-1827.2009.02392.x; PMID: 19563407
- Shibata T, Kokubu A, Miyamoto M, Hosoda F, Gotoh M, Tsuta K, et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene 2010; 29:4671 - 81; http://dx.doi.org/10.1038/onc.2010.217; PMID: 20543864
- Szer IS, Sierakowska H, Szer W. A novel autoantibody to the putative oncoprotein DEK in pauciarticular onset juvenile rheumatoid arthritis. J Rheumatol 1994; 21:2136 - 42; PMID: 7869324
- Dong X, Michelis MA, Wang J, Bose R, DeLange T, Reeves WH. Autoantibodies to DEK oncoprotein in a patient with systemic lupus erythematosus and sarcoidosis. Arthritis Rheum 1998; 41:1505 - 10; http://dx.doi.org/10.1002/1529-0131(199808)41:8<1505::AID-ART23>3.0.CO;2-N; PMID: 9704652
- Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev 2000; 14:1308 - 12; PMID: 10837023
- Campillos M, García MA, Valdivieso F, Vázquez J. Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res 2003; 31:1571 - 5; http://dx.doi.org/10.1093/nar/gkg247; PMID: 12595566
- Sitwala KV, Mor-Vaknin N, Markovitz DM. Minireview: DEK and gene regulation, oncogenesis and AIDS. Anticancer Res 2003; 23:3A 2155 - 8; PMID: 12894590
- Kappes F, Scholten I, Richter N, Gruss C, Waldmann T. Functional domains of the ubiquitous chromatin protein DEK. Mol Cell Biol 2004; 24:6000 - 10; http://dx.doi.org/10.1128/MCB.24.13.6000-6010.2004; PMID: 15199153
- Soares LM, Zanier K, Mackereth C, Sattler M, Valcárcel J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science 2006; 312:1961 - 5; http://dx.doi.org/10.1126/science.1128659; PMID: 16809543
- Mor-Vaknin N, Punturieri A, Sitwala K, Faulkner N, Legendre M, Khodadoust MS, et al. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol 2006; 26:9484 - 96; http://dx.doi.org/10.1128/MCB.01030-06; PMID: 17030615
- Gamble MJ, Fisher RP. SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol 2007; 14:548 - 55; http://dx.doi.org/10.1038/nsmb1248; PMID: 17529993
- Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N, et al. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol 2008; 28:3245 - 57; http://dx.doi.org/10.1128/MCB.01921-07; PMID: 18332104
- Kim DW, Chae JI, Kim JY, Pak JH, Koo DB, Bahk YY, et al. Proteomic analysis of apoptosis related proteins regulated by proto-oncogene protein DEK. J Cell Biochem 2009; 106:1048 - 59; http://dx.doi.org/10.1002/jcb.22083; PMID: 19229864
- Sammons M, Wan SS, Vogel NL, Mientjes EJ, Grosveld G, Ashburner BP. Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem 2006; 281:26802 - 12; http://dx.doi.org/10.1074/jbc.M600915200; PMID: 16829531
- Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol 2006; 26:7506 - 19; http://dx.doi.org/10.1128/MCB.00430-06; PMID: 16894028
- Secchiero P, Voltan R, di Iasio MG, Melloni E, Tiribelli M, Zauli G. The oncogene DEK promotes leukemic cell survival and is downregulated by both Nutlin-3 and chlorambucil in B-chronic lymphocytic leukemic cells. Clin Cancer Res 2010; 16:1824 - 33; http://dx.doi.org/10.1158/1078-0432.CCR-09-3031; PMID: 20215548
- Wise-Draper TM, Allen HV, Thobe MN, Jones EE, Habash KB, Münger K, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol 2005; 79:14309 - 17; http://dx.doi.org/10.1128/JVI.79.22.14309-14317.2005; PMID: 16254365
- Wise-Draper TM, Morreale RJ, Morris TA, Mintz-Cole RA, Hoskins EE, Balsitis SJ, et al. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am J Pathol 2009; 174:71 - 81; http://dx.doi.org/10.2353/ajpath.2009.080330; PMID: 19036808
- Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA, et al. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res 2009; 69:1792 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-2304; PMID: 19223548
- Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol 1999; 17:399 - 408; PMID: 10458259
- Aalto Y, El-Rifa W, Vilpo L, Ollila J, Nagy B, Vihinen M, et al. Distinct gene expression profiling in chronic lymphocytic leukemia with 11q23 deletion. Leukemia 2001; 15:1721 - 8; http://dx.doi.org/10.1038/sj.leu.2402282; PMID: 11681413
- Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A, et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood 2006; 107:4122 - 9; http://dx.doi.org/10.1182/blood-2005-11-4465; PMID: 16439677
- Coll-Mulet L, Iglesias-Serret D, Santidrián AF, Cosialls AM, de Frias M, Castaño E, et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006; 107:4109 - 14; http://dx.doi.org/10.1182/blood-2005-08-3273; PMID: 16439685
- Kojima K, Konopleva M, McQueen T, O’Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006; 108:993 - 1000; http://dx.doi.org/10.1182/blood-2005-12-5148; PMID: 16543464
- Secchiero P, Corallini F, Gonelli A, Dell’Eva R, Vitale M, Capitani S, et al. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res 2007; 100:61 - 9; http://dx.doi.org/10.1161/01.RES.0000253975.76198.ff; PMID: 17138942
- Saddler C, Ouillette P, Kujawski L, Shangary S, Talpaz M, Kaminski M, et al. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood 2008; 111:1584 - 93; http://dx.doi.org/10.1182/blood-2007-09-112698; PMID: 17971485
- Binet JL, Auquier A, Dighiero G, Chastang C, Piguet H, Goasguen J, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981; 48:198 - 206; http://dx.doi.org/10.1002/1097-0142(19810701)48:1<198::AID-CNCR2820480131>3.0.CO;2-V; PMID: 7237385
- Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114:2589 - 97; http://dx.doi.org/10.1182/blood-2009-05-224071; PMID: 19643983
- Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J, Vihinen M, et al. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica 2002; 87:569 - 77; PMID: 12031912
- Casas S, Nagy B, Elonen E, Aventín A, Larramendy ML, Sierra J, et al. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma 2003; 44:1935 - 41; http://dx.doi.org/10.1080/1042819031000119299; PMID: 14738146
- Grasemann C, Gratias S, Stephan H, Schüler A, Schramm A, Klein-Hitpass L, et al. Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene 2005; 24:6441 - 9; PMID: 16007192
- Paderova J, Orlic-Milacic M, Yoshimoto M, da Cunha Santos G, Gallie B, Squire JA. Novel 6p rearrangements and recurrent translocation breakpoints in retinoblastoma cell lines identified by spectral karyotyping and mBAND analyses. Cancer Genet Cytogenet 2007; 179:102 - 11; http://dx.doi.org/10.1016/j.cancergencyto.2007.08.014; PMID: 18036396
- Ageberg M, Gullberg U, Lindmark A. The involvement of cellular proliferation status in the expression of the human proto-oncogene DEK. Haematologica 2006; 91:268 - 9; PMID: 16461319
- Garçon L, Libura M, Delabesse E, Valensi F, Asnafi V, Berger C, et al. DEK-CAN molecular monitoring of myeloid malignancies could aid therapeutic stratification. Leukemia 2005; 19:1338 - 44; http://dx.doi.org/10.1038/sj.leu.2403835; PMID: 15973457
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al, International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446 - 56; http://dx.doi.org/10.1182/blood-2007-06-093906; PMID: 18216293
- Xu W, Li JY, Wu YJ, Yu H, Shen QD, Tian T, et al. CD38 as a prognostic factor in Chinese patients with chronic lymphocytic leukaemia. Leuk Res 2009; 33:237 - 43; http://dx.doi.org/10.1016/j.leukres.2008.06.026; PMID: 18674817
- Chen L, Zhang Y, Zheng W, Wu Y, Qiao C, Fan L, et al. Distinctive IgVH gene segments usage and mutation status in Chinese patients with chronic lymphocytic leukemia. Leuk Res 2008; 32:1491 - 8; http://dx.doi.org/10.1016/j.leukres.2008.02.005; PMID: 18359082
- Dong HJ, Zhou LT, Zhu DX, Wang DM, Fang C, Zhu HY, et al. The prognostic significance of TP53 mutations in Chinese patients with chronic lymphocytic leukemia is independent of del(17p13). Ann Hematol 2011; 90:709 - 17; http://dx.doi.org/10.1007/s00277-010-1125-8; PMID: 21113594
- Qiu HX, Xu W, Cao XS, Zhou M, Shen YF, Xu YL, et al. Cytogenetic characterisation in Chinese patients with chronic lymphocytic leukemia: a prospective, multicenter study on 143 cases analysed with interphase fluorescence in situ hybridisation. Leuk Lymphoma 2008; 49:1887 - 92; http://dx.doi.org/10.1080/10428190802308710; PMID: 18949612