Abstract
We have further defined mechanism(s) by which the drug OSU-03012 (OSU) kills brain cancer cells. OSU toxicity was enhanced by the HSP90 inhibitor 17-N-Allylamino-17-demethoxygeldanamycin (17AAG) that correlated with reduced expression of ERBB1 and ERBB2. Inhibition of the extrinsic apoptosis pathway blocked the interaction between 17AAG and OSU. OSU toxicity was enhanced by the inhibitor of ERBB1/2/4, lapatinib. Knock down of ERBB1/2/4 in a cell line specific fashion promoted OSU toxicity. Combined exposure of cells to lapatinib and OSU resulted in reduced AKT and ERK1/2 activity; expression of activated forms of AKT and to a lesser extent MEK1 protected cells from the lethal effects of the drug combination. Knock down of PTEN suppressed, and expression of PTEN enhanced, the lethal interaction between OSU and lapatinib. Downstream of PTEN, inhibition of mTOR recapitulated the effects of lapatinib. Knock down of CD95, NOXA, PUMA, BIK or AIF, suppressed lapatinib and OSU toxicity. Knock down of MCL-1 enhanced, and overexpression of MCL-1 suppressed, drug combination lethality. Lapatinib and OSU interacted in vivo to suppress the growth of established tumors. Collectively our data argue that the inhibition of ERBB receptor function represents a useful way to enhance OSU lethality in brain tumor cells.
Introduction
OSU-03012, a derivative of the drug Celecoxib, lacks COX2 inhibitory activity.Citation1,Citation2 COX2 is overexpressed in several tumor types and drugs that inhibit COX2 i.e., Celecoxib have been shown to cause tumor cell specific increases in cell death, and that are also associated with a lower rate of growth.Citation3-Citation6 Prolonged treatment with COX2 inhibitors can reduce the incidence of developing cancer, which, in addition, argues that COX2 inhibitors have cancer preventative effects.Citation7,Citation8 However, it has been noted that the expression levels of COX2 do not simplistically correlate with tumor cell sensitivity to COX2 inhibitors.Citation9,Citation10 Thus, COX2 inhibitors must have at least one additional target.
Compared with Celecoxib, OSU-03012 has a similar level of bio-availability in animal models and has an order of magnitude greater efficacy at killing tumor cells.Citation11-Citation13 Based on encouraging pre-clinical data OSU-03012 is currently undergoing Phase I evaluation in patients with solid and liquid tumors. Initially, the tumoricidal effects of OSU-03012 in transformed cells were argued to be via inhibition of the enzyme PDK-1, within the PI3K pathway. And, in the micro-Molar range, it has been shown that OSU-03012 can lower AKT phosphorylation. In our previous studies, inhibition of either ERK1/2 or phosphatidyl-inositol 3 kinase signaling enhanced the toxicity of OSU-03012.Citation12-Citation14 However, our data has also strongly argued that OSU-03012 toxicity, and its radiosensitizing effects, could not easily be attributed to suppression of AKT signaling.Citation12-Citation14 Specifically, our prior studies have argued that OSU-03012 killed tumor cells through mechanisms which involved endoplasmic reticulum (ER) stress signaling, downregulation of the HSP70 family chaperone BiP/GRP78, and a caspase-independent form of cell death.Citation12-Citation14
Signaling by growth factor receptors can be regulated by the actions of paracrine ligands, plasma membrane receptor density and mutational activation of the receptor.Citation15 This is particularly true for ERBB family receptors. In glioblastoma cells expression of a truncated activated form of ERBB1 (ERBB1 vIII) is often observed as are activating point mutations in the ERBB1 catalytic domain. Because of these changes in cell biology, the kinase domains of ERBB receptors have been targeted by drug companies with the intention of developing kinase specific inhibitors.Citation16 Inhibition of such receptors can lower the steady-state activities of downstream pathways such as ERK1/2 and PI3K/AKT, whose inhibition would a priori be predicted to enhance OSU-03012 lethality. In particular, the drug lapatinib (Tykerb), a dual ERBB1 and ERBB2 inhibitor has shown particular promise and is an approved therapeutic in combination with capecitabine for recurrent breast cancer patients.Citation17 Whether multi-kinase inhibitory agents such as lapatinib and OSU-03012 interact is presently unknown.
Tumors of the brain are notoriously difficult to therapeutically control. Untreated adult glioblastoma (GBM) patients have a mean survival of several months that is only prolonged up to 12–18 mo by aggressive therapeutic intervention.Citation18 In pediatric medulloblastoma patients up to an 80% 5 y survival rate has been achieved using traditional cytotoxic radio- and chemotherapies however this effect in parallel results in multiple debilitating negative sequelae for the child.Citation19 Thus for both GBM and medulloblastoma patients new therapeutic approaches that could translate to the clinic are urgently required.
In the present studies using GBM and medulloblastoma cells, we initially defined whether inhibition of HSP90 using 17-N-Allylamino-17-demethoxygeldanamycin (17AAG) can enhance OSU-03012 lethality and second whether inhibition of ERBB family growth factor receptors can enhance OSU-03012 lethality in primary human GBM cell isolates. Our findings demonstrate that inhibition of multiple ERBB receptors by either 17AAG or by lapatinib can play a role in enhancing OSU-03012 toxicity through activation of the extrinsic apoptosis pathway.
Results
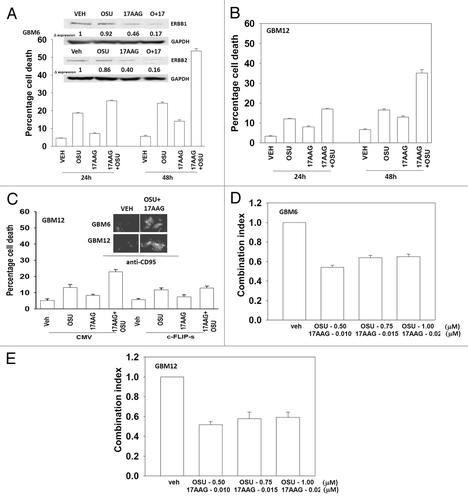
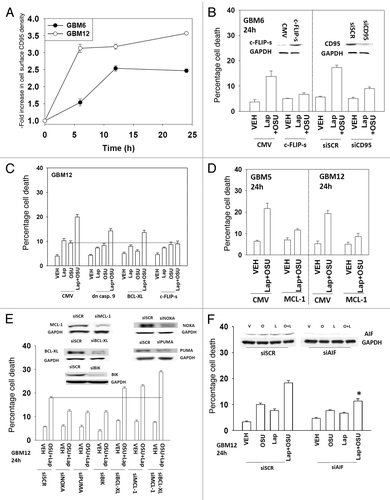
The present studies were designed to develop novel drug combinations for the agent OSU-03012. Initial studies examined the interaction between OSU-03012 and the HSP90 inhibitor 17AAG. Treatment of GBM cells with OSU-03012 and 17AAG caused a time dependent increase in drug combination toxicity (). Treatment of cells with 17AAG reduced expression of ERBB1 and ERBB2, an effect that was magnified in cells treated with the drug combination (, upper blots). In contrast to our findings with ERBB family receptors, the drug combination increased plasma membrane / cell surface levels of the death receptor CD95 on un-permeabilized cells (, upper IHC). Expression of the caspase 8 inhibitor c-FLIP-s reduced drug combination lethality but did not alter the toxicity of OSU-03012 as a single agent (, lower graph). In colony formation assays OSU-03012 and 17AAG synergized to kill GBM cells ().
Figure 1. OSU-03012 toxicity in transformed cells is enhanced by 17AAG. (A) GBM6 cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or 17AAG (50 nM) as indicated. Cells were isolated 24h and 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). Upper blots: The expression of ERBB1 and ERBB2 was determined 24h after drug exposure. (B) GBM12 cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or 17AAG (50 nM) as indicated. Cells were isolated 24h and 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (C) GBM12 cells were infected with empty vector virus (CMV) or a virus to express c-FLIP-s. Twenty four h later cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or 17AAG (50 nM) as indicated. Cells were isolated 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). Upper IHC: Cells were treated with drugs and 24h later fixed. Immunohistochemistry was performed on fixed un-permeabilized cells to determine the level of plasma membrane localization of CD95. (D and E) GBM6 and GBM12 cells were plated as single cells in sextuplicate. Twelve h after plating cells were treated with 17AAG (0–20 nM) and OSU (0–1.0 μM) at a fixed concentration ratio. After 48h media was removed, the cells washed and drug free media added. Colonies formed over ~14 d. A colony was defined as a group of > 50 cells. A combination index of < 1.00 indicates a synergy of drug interaction (n = 3 ± SEM).
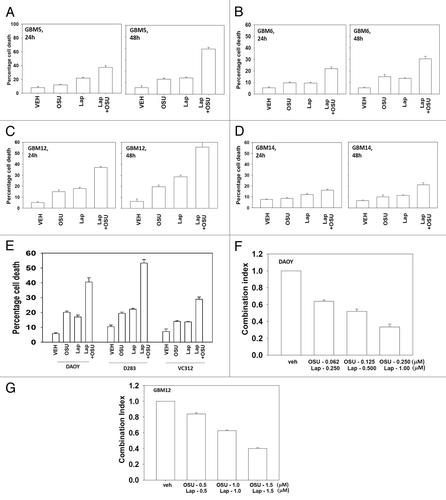
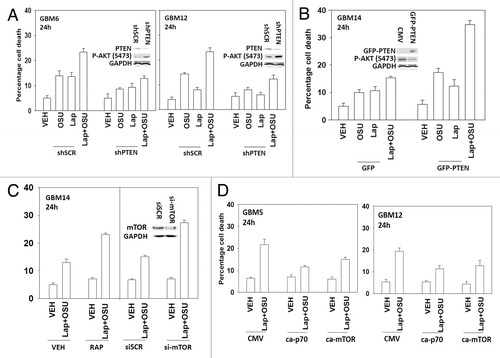
Based on the data in with respect to ERBB1 and ERBB2 expression, we next determined whether an inhibitor of ERBB1 and ERBB2, lapatinib, interacted with OSU-03012 to kill GBM cells. Lapatinib interacted with OSU-03012 in multiple GBM isolates to enhance tumor cell killing (−D). In GBM cells lacking PTEN function, i.e., GBM14, the relative increase in killing following drug treatment was apparently lower than in cells expressing a wild type PTEN protein. Similar data to that in GBM cells were obtained in pediatric medulloblastoma cell lines (). In colony formation assays OSU-03012 and lapatinib synergized to kill GBM and medulloblastoma cells ().
Figure 2. OSU-03012 toxicity in transformed cells is enhanced by Lapatinib. (A) GBM5; (B) GBM6; (C) GBM12; (D) GBM14 cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or Lapatinib (1.0 μM) as indicated. Cells were isolated 24h and 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (E) DAOY, D283 and VC312 cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or Lapatinib (1.0 μM) as indicated. Cells were isolated 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (F and G) GBM6 and GBM12 cells were plated as single cells in sextuplicate. Twelve h after plating cells were treated with OSU (0–250 nM) and Lapatinib (0–1.0 μM) at a fixed concentration ratio. After 48h media was removed, the cells washed and drug free media added. Colonies formed over ~14 d. A colony was defined as a group of > 50 cells. A combination index of < 1.00 indicates a synergy of drug interaction (n = 3 ±SEM).
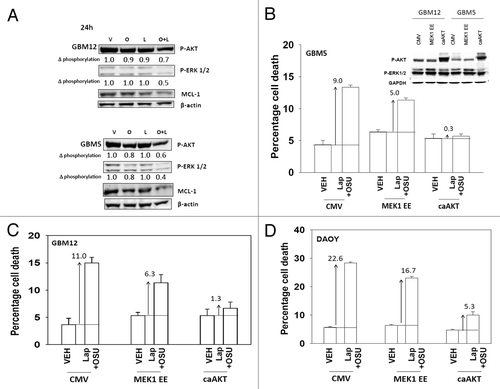
Based on our data with lapatinib we next defined, using an siRNA knock down approach, the relative survival / regulatory roles of each ERBB receptor in our systems. In GBM12 cells knock down of ERBB1 and ERBB2 enhanced OSU-03012 toxicity to a greater extent than knock down of the individual receptors (). In DAOY cells knock down of ERBB1 and ERBB4 but not the individual receptors enhanced OSU-03012 toxicity (). Downstream of the ERBB receptors are signal transduction pathways; e.g., “protective” pathways such as ERK1/2 and AKT. Drug combination treatment reduced both ERK1/2 and AKT activity (). This reduction correlated with lower MCL-1 expression. Expression of activated AKT, to a significantly greater extent than expression of activated MEK1 EE, protected cells from drug combination lethality (−D).
Figure 3. Molecular inhibition of ERBB receptors recapitulates the effects of lapatinib. (A) GBM12 cells were transfected with scrambled control siRNA (siSCR, 20 nM) or with siRNA molecules to knock down expression of ERBB1-3. Twenty four h after transfection cells were treated with vehicle (DMSO) or with OSU-03012 (1 μM). Cells were isolated 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (B) DAOY cells were transfected with scrambled control siRNA (siSCR, 20 nM) or with siRNA molecules to knock down expression of ERBB1–4. Twenty four h after transfection cells were treated with vehicle (DMSO) or with OSU-03012 (1 μM). Cells were isolated 48h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM).
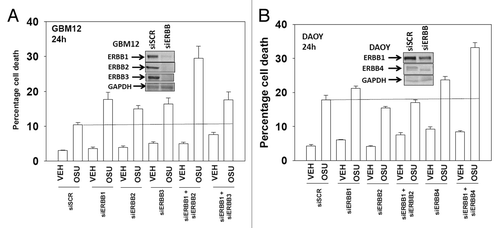
Figure 4. OSU-03012 and lapatinib treatment lowers ERK1/2 and AKT activity that correlates with lower MCL-1 expression. (A) GBM12 and GBM5 cells were treated with vehicle (DMSO); OSU-03012 (O, 1 μM); lapatinib (L, 1 μM) or the drugs combined. Cells were isolated 24h later and the phosphorylation of ERK1/2 and AKT (S473) determined, and the expression of MCL-1 determined. (B–D) GBM5, GBM12 and DAOY cells were infected with empty vector control (CMV) or with viruses to express active MEK1 (MEK1EE) or active AKT (caAKT). Twenty four h after infection cells were treated with vehicle (DMSO) or with lapatinib and OSU-03012. Cells were isolated after 48h and viability determined by trypan blue exclusion (n = 3, ± SEM). Arrows indicate the true percentage difference between vehicle and lap+OSU treatment.
Based on our observation that PTEN null GBM cells were apparently less sensitive to being killed by the drug combination than were cells expressing the wild type protein, and our viability data in cells expressing activated AKT, we determined whether altered PTEN status impacted on drug combination lethality. Knock down of PTEN resulted in cells that were relatively resistant to OSU-03012 and lapatinib treatment (). Expression of PTEN in GBM14 cells, that are PTEN null, enhanced drug combination lethality (). In agreement with the PI3K pathway playing a key role in survival, treatment of GBM14 cells with rapamycin or knock down of mTOR facilitated drug combination toxicity (). Expression of activated p70 S6K or of activated mTOR also protected cells that expressed wild type PTEN from the drug combination ().
Figure 5. OSU-03012 lethality is regulated by PTEN expression. (A) GBM6 and GBM12 cells were transfected with a scrambled control plasmid (shSCR) or a plasmid to knock down PTEN expression (shPTEN). Twenty four h after transfection cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or Lapatinib (1.0 μM) as indicated. Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (B) GBM14 cells were transfected to express GFP or GFP-PTEN Twenty four h after transfection cells were treated with vehicle (DMSO); OSU-03012 (OSU) (1 μM) and/or Lapatinib (1.0 μM) as indicated. Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 +/− SEM). (C) GBM14 cells were transfected with scrambled control siRNA (siSCR, 20 nM) or with an siRNA molecule to knock down expression of mTOR. Twenty four h after transfection cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). In parallel untransfected cells were treated with vehicle (DMSO) or with rapamycin (10 nM); then cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (D) GBM5 and GBM12 cells were transfected with empty vector plasmid (CMV) or plasmids to express activated forms of p70 S6K or of mTOR. Twenty four h after transfection cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM).
Lapatinib and OSU-03012 treatment increased cell surface levels of CD95 (). Lapatinib and OSU-03012 toxicity, as was previously observed with 17AAG and OSU-03012, was inhibited by knock down of the death receptor CD95, by overexpression of the caspase 8 inhibitor c-FLIP-s or by overexpression of the mitochondrial protective proteins BCL-XL or MCL-1 (−D). To further refine our data we knocked down expression of pro- and anti-apoptotic effector proteins. Knock down of NOXA, PUMA or BIK protected cells whereas knock down of MCL-1 and BCL-XL facilitated drug lethality (). Knock down of AIF also significantly protected cells from the drug combination ().
Figure 6. Lapatinib and OSU-03012 interact to activate the extrinsic apoptosis pathway. (A) GBM6 and GBM12 cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were fixed at the indicated time points (5–24h) and the plasma membrane levels of CD95 determined by immunohistochemistry. Data are presented as the –Fold increase in CD95 density (n = 3, ± SEM). (B) GBM6 cells were either transfected with siRNA to knock down CD95 expression or were infected with a virus to express c-FLIP-s. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (C) Cells were infected with empty vector virus (CMV) or with viruses to express dominant negative caspase 9, BCL-XL or c-FLIP-s. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and/or OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (D) GBM5 and GBM12 cells were transfected with empty vector plasmid (CMV) or with a plasmid to express MCL-1. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and/or OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). (E) GBM12 cells were transfected with scrambled siRNA (siSCR) or siRNA molecules to knock down the expression of NOXA, PUMA, BIK, BCL-XL, MCL-1 and BCL-XL + MCL-1. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ±SEM). (F) GBM12 cells were transfected with scrambled siRNA (siSCR) or an siRNA molecule to knock down the expression of AIF. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and/or OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM). *p < 0.05 less than corresponding value in siSCR cells.
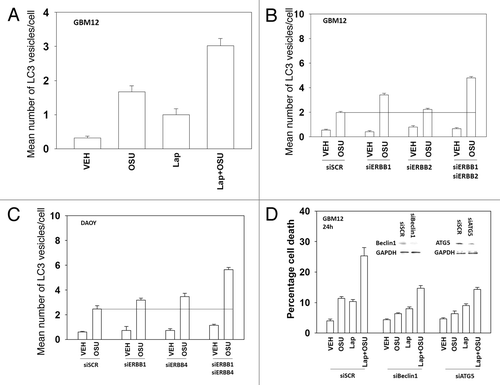
Prior studies from our laboratory have linked OSU-03012 toxicity to increased levels of a toxic form of autophagy. In contrast, other findings have argued that autophagy downstream of CD95 is a protective event. Lapatinib enhanced the mean number of autophagic vesicles induced by OSU-03012 (); knock down of either Beclin1 or ATG5 abolished the increase in vesicle numbers (data not shown). Knock down of ERBB1/2 in GBM12 cells and ERBB1/4 in DAOY cells also enhanced the generation of autophagic vesicles after OSU treatment (). Knock down of Beclin1 or ATG5 reduced OSU-03012 toxicity by > 70% (). In contrast, knock down of Beclin1 or ATG5 only reduced OSU-03012 and lapatinib toxicity by ~50%. Treatment of cells with a pan-caspase inhibitor (zVAD) did not alter OSU-03012-induced killing but also reduced drug combination lethality by ~50% (data not shown). These findings argue that inhibition of ERBB receptors recruits apoptotic pathways that interact with drug-induced autophagy to cause tumor cell killing.
Figure 7. Lapatinib and OSU-03012 interact to increase a toxic form of autophagy. (A) GBM12 cells were transfected with a plasmid to express GFP-LC3. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and/or OSU-03012 (1 μM). Cells were examined 24h later under a fluorescent microscope to determine the number of GFP punctae per cell from 40 cells (n = 3, ± SEM). (B) GBM12 cells were transfected with a plasmid to express GFP-LC3 and with siRNA molecules to knock down ERBB1 and/or ERBB2. Cells were examined 24h later under a fluorescent microscope to determine the number of GFP punctae per cell from 40 cells (n = 3, ± SEM). (C) DAOY cells were transfected with a plasmid to express GFP-LC3 and with siRNA molecules to knock down ERBB1 and/or ERBB4. Cells were examined 24h later under a fluorescent microscope to determine the number of GFP punctae per cell from 40 cells (n = 3, ± SEM). (D) GBM12 cells were transfected with siRNA molecules to knock down Beclin1 or ATG5. Twenty four h later cells were treated with vehicle (DMSO) or with lapatinib (1 μM) and/or OSU-03012 (1 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion assay (n = 3 ± SEM).
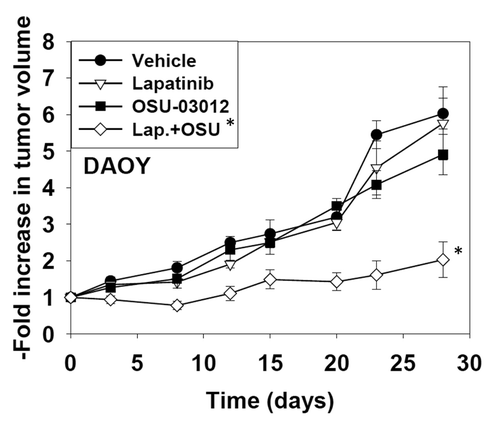
Finally we determined whether lapatinib and OSU-03012 interacted in vivo to blunt the growth of DAOY tumors. Exposure of the tumor to low doses of either lapatinib or OSU-03012 had no effect on tumor growth. Combined exposure to both drugs resulted in a significant reduction in the growth of tumors (). Collectively our findings demonstrate that OSU-3012 toxicity in vitro and in vivo can be enhanced by ERBB1/2 inhibition.
Figure 8. OSU-03012 and lapatinib interact in vivo to suppress tumor growth. DAOY cells (1 × 107) were injected into the rear flanks of athymic mice. Tumors formed over the following month (~200 mm3, per treatment group, day 0). Animals were doses with drugs: vehicle (DMSO and Cremophore); Lapatinib (100 mg/kg, BID); OSU-03012 (25 mg/kg QD) or both drugs combined. Tumor volumes were assessed by caliper on the indicated days (n = 2 experiments with 3 animals per group in each experiment: total 6 animals per group ± SEM). * p < 0.05 less than other treatment values.
Discussion
The present studies were initiated to further understand the molecular mechanisms by which OSU-03012 in combination with other signaling modulatory agents kills brain cancer cells. OSU-03012 combined with the geldanamycin derivative 17AAG to reduce expression of ERBB family receptors and to increase activity of the death receptor CD95.Citation20 OSU-03012 combined with the ERBB1/ERBB2 inhibitor lapatinib, or with molecular knock down of these receptors, to inhibit ERBB family receptor function and to increase activity of the death receptor CD95. Treatment with OSU-03012 combined with inhibition of ERBB receptors lead to decreases in both ERK1/2 and AKT activity, with loss of AKT/mTOR/p70 S6K representing the pathway central to maintaining viability.Citation21 Drug treatment reduced expression of protective MCL-1 and BCL-XL proteins and expression/overexpression of c-FLIP-s, BCL-XL or MCL-1 protected cells from drug combination lethality.Citation22 Collectively our findings argue that OSU-03012 interacts with agents that facilitate activation of the death receptor CD95 and inactivation of the PI3K-AKT pathway to cause tumor cell death.
Growth factor receptor signaling is known to be activated in many GBM cells, with expression of truncated active ERBB1 vIII the best characterized.Citation23 Inhibition of ERBB receptor function by use of 17AAG or lapatinib predisposed tumor cells to the toxic effects of OSU-03012. Inhibition of growth factor receptor signaling has been shown to promote cell killing through apoptotic pathways and also, through loss of mTOR activity, autophagy.Citation24 In our cell system the combination of OSU-03012 and lapatinib enhanced activation of CD95 that was a pro-apoptotic stimulus. The drug combination promoted autophagy that also played a prominent role in inducing cell death. The crossroads between apoptosis and autophagy has not been fully defined with some investigators arguing that autophagy antagonizes apoptosis whereas others arguing the opposite.Citation25 Prior studies by this group had shown that autophagy downstream of CD95 was protective however in our present studies with OSU-03012, autophagy was a toxic event.Citation26,Citation27
The loss of PTEN function is a known therapy resistance factor in GBM.Citation28 Knock down of PTEN reduced the toxic effect of OSU-03012 and lapatinib treatment whereas expression of PTEN in a PTEN null GBM cell facilitated drug combination lethality. Furthermore, knock down of mTOR facilitated drug combination toxicity in a PTEN null cell. OSU-03012 was originally described as a PDK-1 inhibitor although prior studies from this laboratory argued that inhibition of the PI3K/PDK-1 pathway was a secondary pathway in the induction of autophagic tumor cell death.Citation12-Citation14 In general agreement with these findings, however, expression of activated AKT, activated p70 S6K or activated mTOR, to a greater extent than that of active MEK1, protected cells from lapatinib and OSU-03012 lethality. Collectively these findings argue that should this drug combination be translated to the clinic, the PTEN status of a patients’ tumor should be assessed.
Prior studies have demonstrated that autophagy played a key role in OSU-03012 lethality; knock down of ATG5 or Beclin1 suppressed OSU-03012 and OSU-03012 and lapatinib toxicity.Citation12-Citation14 Inhibition of caspase 9 function did not alter the toxicity of OSU-03012 as a single agent but blunted drug combination lethality. In contrast expression of BCL-XL or of MCL-1 suppressed both OSU-03012 and OSU-03012 and lapatinib toxicity. Knock down of BCL-XL and MCL-1 enhanced OSU-03012 and lapatinib toxicity. Knock down of CD95, NOXA, PUMA or BIK suppressed the toxicity of the drug combination. Inhibition of the extrinsic apoptosis pathway by expression of c-FLIP-s did not alter OSU-03012 lethality but abolished the potentiation of OSU-03012 lethality by lapatinib. Collectively these findings argue that two pro-death responses are induced by OSU-03012 and lapatinib treatment: (1) a toxic form of autophagy; (2) the extrinsic apoptosis pathway.
Based on our in vitro data we attempted to determine whether OSU-03012 and lapatinib interacted in vivo to suppress tumor growth. We have previously published that OSU-03012 and ionizing radiation exposure interact in vivo to suppress GBM growth and to prolong survival. Initial studies attempted to grow medulloblastoma tumors in the brain however these approaches proved unsuccessful. Studies instead grew tumors on the flanks of athymic mice. Low doses of OSU-03012 did not significantly alter tumor growth. Treatment with lapatinib at 100 mg/kg BID also did not alter tumor growth. However, the drug combination profoundly suppressed the growth of medulloblastoma tumors. These data, together with our prior analyses argues that OSU-03012 has potential to be a therapeutic agent against CNS tumors.
Materials and Methods
Materials
Phospho-/total- antibodies, were purchased from Cell Signaling Technologies. All the secondary antibodies were purchased from Santa Cruz Biotechnology. TUNEL kits were purchased from NEN Life Science Products Boehringer Mannheim. Commercially available validated short hairpin RNA molecules to knock down RNA / protein levels were from Qiagen. Primary human GBM cells and information on the genetic background of such cells were very kindly supplied for our use by Dr. C. David James, University of California San Francisco, CA. Other reagents and techniques were as described in refs. Citation12–Citation14.
Culture and in vitro exposure of cells to drugs
All established cell lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. Primary human glioma cells were cultured in 2% (v/v) fetal calf serum to prevent growth of contaminating rodent fibroblasts during in vitro analyses. For short-term cell killing assays and immunoblotting studies, cells were plated at a density of 3 × 103 per cm2 (~2 × 105 cells per well of a 12 well plate) and 48h after plating treated with various drugs, as indicated. In vitro OSU-03012 treatment was from a 10 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
In vitro cell treatments, microscopy, SDS-PAGE and western blot analysis
For in vitro analyses of short-term cell death effects, cells were treated with Vehicle or OSU-03012 for the indicated times in the Figure legends. For apoptosis assays where indicated, cells were isolated at the indicated times, and either subjected to trypan blue cell viability assay by counting in a light microscope or fixed to slides, and stained using a commercially available Diff Quick (Geimsa) assay kit.Citation12-Citation14 OSU-03012 lethality, as judged in trypan blue exclusion assays or by Geimsa assays, was first evident ~12h after drug exposure (not shown).
For SDS PAGE and immunoblotting, cells were plated and treated with drugs at the indicated concentrations and after the indicated time of treatment, lysed in whole-cell lysis buffer (0.5 M TRIS-HCl, pH 6.8, 2%SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue) and the samples were boiled for 30 min. The boiled samples were loaded onto 10–14% SDS-PAGE and electrophoresis was run overnight. Proteins were electrophoretically transferred onto 0.22 μm nitrocellulose, and immunoblotted with various primary antibodies against different proteins. All immunoblots were visualized using fluorescent secondary antibodies and an Odyssey Infrared imaging machine.
Infection of cells with recombinant adenoviruses
Cells were plated at 3 × 103 per cm2 in each well of a 12 well, 6 well or 60 mm plate. After plating (24h), cells were infected (at a multiplicity of infection of 50) with a control empty vector virus (CMV) or an adenovirus to express c-FLIP-s (Vector Biolabs). Twenty four hours after infection cells were treated with the indicated concentrations of OSU-03012 and/or drugs, and cell survival or changes in expression/protein phosphorylation determined 0–48h after drug treatment by trypan blue assay and immunoblotting, respectively.
Transfection of cells with siRNA or with plasmids
For plasmids
Cells were plated as described above and 24h after plating, transfected. Cells we transfected with (0.5 μg) plasmid(s) as indicated expressing a specific mRNA (or siRNA) or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium (1 portion for each sample). Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well/dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl, and the cells were incubated for 4 h at 37°C. An equal volume of 2× medium was then added to each well. Cells were incubated for 48h then treated with OSU-03012. For analysis of cells transfected with GFP-LC3 constructs, the GFP-LC3-positive vesicle containing cells were examined under the 40× objective of a Zeiss Axiovert fluorescent microscope. Forty LC3-GFP-positive cells were analyzed per condition. Each vesicle was counted and the average number of vesicles per cell for each, including cells that did not exhibit vesicularization, was calculated.
Transfection with siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA targeting ATG5, Beclin 1 or BiP/GRP78, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used (predominantly Qiagen; occasional alternate siRNA molecules were purchased from Ambion, Inc.). Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added dropwise to each dish. The medium in each dish was swirled gently to mix then incubated at 37°C for 2h. One ml of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37°C for 48h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with OSU-03012 (0–48h). Trypan blue exclusion assays and SDS PAGE/immunoblotting analyses were then performed at the indicated time points.
Flank inoculation of DAOY cells
Athymic female NCr-nu/nu mice (NCI-Fredrick) weighing ~20 g, were used for this study. Mice were maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US. Department of Agriculture, Washington, DC, the US. Department of Health and Human Services, Washington, DC and the National Institutes of Health, Bethesda, MD. DAOY cells (1 × 107) were injected into the rear flanks of athymic mice. Tumors formed over the following month (~200 mm3, per treatment group, day 0). Two-four weeks after tumor cell implantation animals were segregated into treatment groups of approximately the same mean tumor volume ± SEM. For animal administration, Lapatinib and OSU-03012 were first dissolved in DMSO and an equal volume of 50:50 Cremophor/Ethanol (Sigma) was added. After mixing, a 1:10 dilution was made with sterile PBS. Animals were treated with vehicle (PBS/Cremophor/Ethanol/DMSO), Lapatinib, OSU-03012 or combination of Lapatinib and OSU-03012 using oral gavage to a final concentration of 25 mg/kg body mass QD for OSU-03012 and 100 mg/kg BID for lapatinib.
Data analysis
Comparison of the effects of various treatments was performed following ANOVA using the Student’s t test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points (± SEM).
| Abbreviations: | ||
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| EGF | = | epidermal growth factor |
| OSU | = | OSU-03012 |
| PARP | = | poly ADP ribosyl polymerase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| −/− | = | null/gene deleted |
| MAPK | = | mitogen activated protein kinase, PTEN, phosphatase and tensin homolog |
| R | = | receptor |
| JNK | = | c-Jun NH2-terminal kinase |
| dn | = | dominant negative |
| COX | = | cyclooxygenase |
| P | = | phospho- |
| ca | = | constitutively active |
| WT | = | wild type |
| PERK | = | PKR like endoplasmic reticulum kinase |
| HSP | = | heat shock protein |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded; to P.D. from PHS grants (R01-DK52825, P01-CA104177, R01-CA141188), Department of Defense Awards (DAMD17–03–1-0262), the Lind-Lawrence Foundation; to S.G. from PHS grants (R01-CA63753; R01-CA77141) and a Leukemia Society of America grant 6405–97. P.D. is the holder of the Universal Inc. Professorship in Signal Transduction Research.
References
- Hawkey CJ, Fortun PJ. Cyclooxygenase-2 inhibitors. Curr Opin Gastroenterol 2005; 21:660 - 4; PMID: 16220041
- Kiefer W, Dannhardt G. Novel insights and therapeutical applications in the field of inhibitors of COX-2. Curr Med Chem 2004; 11:3147 - 61; PMID: 15579005
- Kang SG, Kim JS, Park K, Kim JS, Groves MD, Nam DH. Combination celecoxib and temozolomide in C6 rat glioma orthotopic model. Oncol Rep 2006; 15:7 - 13; PMID: 16328028
- Klenke FM, Gebhard MM, Ewerbeck V, Abdollahi A, Huber PE, Sckell A. The selective Cox-2 inhibitor Celecoxib suppresses angiogenesis and growth of secondary bone tumors: an intravital microscopy study in mice. BMC Cancer 2006; 6:9; http://dx.doi.org/10.1186/1471-2407-6-9; PMID: 16409625
- Koehne CH, Dubois RN. COX-2 inhibition and colorectal cancer. Semin Oncol 2004; 31:Suppl 7 12 - 21; http://dx.doi.org/10.1053/j.seminoncol.2004.03.041; PMID: 15252926
- Cui W, Yu CH, Hu KQ. In vitro and in vivo effects and mechanisms of celecoxib-induced growth inhibition of human hepatocellular carcinoma cells. Clin Cancer Res 2005; 11:8213 - 21; http://dx.doi.org/10.1158/1078-0432.CCR-05-1044; PMID: 16299255
- Kashfi K, Rigas B. Is COX-2 a ‘collateral’ target in cancer prevention?. Biochem Soc Trans 2005; 33:724 - 7; http://dx.doi.org/10.1042/BST0330724; PMID: 16042585
- Narayanan BA, Narayanan NK, Pttman B, Reddy BS. Adenocarcina of the mouse prostate growth inhibition by celecoxib: downregulation of transcription factors involved in COX-2 inhibition. Prostate 2006; 66:257 - 65; http://dx.doi.org/10.1002/pros.20331; PMID: 16175586
- Patel MI, Subbaramaiah K, Du B, Chang M, Yang P, Newman RA, et al. Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res 2005; 11:1999 - 2007; http://dx.doi.org/10.1158/1078-0432.CCR-04-1877; PMID: 15756026
- Kulp SK, Yang YT, Hung CC, Chen KF, Lai JP, Tseng PH, et al. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res 2004; 64:1444 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-03-2396; PMID: 14973075
- Zhu J, Huang JW, Tseng PH, Yang YT, Fowble JW, Shiau CW, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res 2004; 64:4309 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-03-4063; PMID: 15205346
- Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, et al. OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol 2008; 73:1168 - 84; http://dx.doi.org/10.1124/mol.107.042697; PMID: 18182481
- Yacoub A, Park MA, Hanna D, Hong Y, Mitchell C, Pandya AP, et al. OSU-03012 promotes caspase-independent but PERK-, cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Mol Pharmacol 2006; 70:589 - 603; http://dx.doi.org/10.1124/mol.106.025007; PMID: 16622074
- Booth L, Cazanave SC, Hamed HA, Yacoub A, Ogretmen B, Chen CS, et al. OSU-03012 suppresses GRP78/BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biol Ther 2012; 13:224 - 36; http://dx.doi.org/10.4161/cbt.13.4.18877; PMID: 22354011
- Valerie K, Yacoub A, Hagan MP, Curiel DT, Fisher PB, Grant S, et al. Radiation-induced cell signaling: inside-out and outside-in. Mol Cancer Ther 2007; 6:789 - 801; http://dx.doi.org/10.1158/1535-7163.MCT-06-0596; PMID: 17363476
- Berezowska S, Schlegel J. Targeting ErbB receptors in high-grade glioma. Curr Pharm Des 2011; 17:2468 - 87; http://dx.doi.org/10.2174/138161211797249233; PMID: 21827413
- Murphy CG, Morris PG. Recent advances in novel targeted therapies for HER2-positive breast cancer. Anticancer Drugs 2012; 23:765 - 76; http://dx.doi.org/10.1097/CAD.0b013e328352d292; PMID: 22824822
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 2012; 26:756 - 84; http://dx.doi.org/10.1101/gad.187922.112; PMID: 22508724
- Massimino M, Giangaspero F, Garrè ML, Gandola L, Poggi G, Biassoni V, et al. Childhood medulloblastoma. Crit Rev Oncol Hematol 2011; 79:65 - 83; http://dx.doi.org/10.1016/j.critrevonc.2010.07.010; PMID: 21129995
- Walker T, Mitchell C, Park MA, Yacoub A, Rahmani M, Häussinger D, et al. 17-allylamino-17-demethoxygeldanamycin and MEK1/2 inhibitors kill GI tumor cells via Ca2+-dependent suppression of GRP78/BiP and induction of ceramide and reactive oxygen species. Mol Cancer Ther 2010; 9:1378 - 95; http://dx.doi.org/10.1158/1535-7163.MCT-09-1131; PMID: 20442308
- Fasolo A, Sessa C. Targeting mTOR pathways in human malignancies. Curr Pharm Des 2012; 18:2766 - 77; http://dx.doi.org/10.2174/138161212800626210; PMID: 22475451
- Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs 2011; 20:1397 - 411; http://dx.doi.org/10.1517/13543784.2011.609167; PMID: 21851287
- Camara-Quintana JQ, Nitta RT, Li G. Pathology: commonly monitored glioblastoma markers: EFGR, EGFRvIII, PTEN, and MGMT. Neurosurg Clin N Am 2012; 23:237 - 46, viii; http://dx.doi.org/10.1016/j.nec.2012.01.011; PMID: 22440867
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2 - 11; http://dx.doi.org/10.1128/MCB.06159-11; PMID: 22025673
- Shen S, Kepp O, Kroemer G. The end of autophagic cell death?. Autophagy 2012; 8:1 - 3; http://dx.doi.org/10.4161/auto.8.1.16618; PMID: 22082964
- Zhang G, Park MA, Mitchell C, Walker T, Hamed H, Studer E, et al. Multiple cyclin kinase inhibitors promote bile acid-induced apoptosis and autophagy in primary hepatocytes via p53-CD95-dependent signaling. J Biol Chem 2008; 283:24343 - 58; http://dx.doi.org/10.1074/jbc.M803444200; PMID: 18614532
- Park MA, Zhang G, Martin AP, Hamed H, Mitchell C, Hylemon PB, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther 2008; 7:1648 - 62; http://dx.doi.org/10.4161/cbt.7.10.6623; PMID: 18787411
- Höland K, Salm F, Arcaro A. The phosphoinositide 3-kinase signaling pathway as a therapeutic target in grade IV brain tumors. Curr Cancer Drug Targets 2011; 11:894 - 918; http://dx.doi.org/10.2174/156800911797264743; PMID: 21861842