Abstract
The tumor suppressor p53 transcriptionally regulates a number of genes that are involved in cell-cycle inhibition, apoptosis and the maintenance of genetic stability. Recent studies suggest that p53 also contributes to the regulation of cell migration and invasion. Here, we show that human chloride channel accessory-2 (CLCA2) is a target gene of the p53 family (p53, p73 and p63). CLCA2 is induced by DNA damage in a p53-dependent manner. The p53 family proteins activate the CLCA2 promoter by binding directly to the conserved consensus p53-binding site present in the CLCA2 promoter. In terms of function, ectopic expression of CLCA2 inhibited cancer cell migration. In contrast, silencing CLCA2 with siRNA stimulated cancer cell migration and invasion. We also found that inactivation of CLCA2 enhanced the expression of focal adhesion kinase (FAK), as well as its promoter activation. A small-molecule FAK inhibitor reduced the effect of CLCA2 siRNA on cell migration and invasion, suggesting that CLCA2 inhibits cancer cell migration and invasion through suppression of the FAK signaling pathway. Furthermore, there was an inverse correlation between CLCA2 and FAK expression in 251 human breast cancer tissues. These results strongly suggest that CLCA2 is involved in the p53 tumor suppressor network and has a significant effect on cell migration and invasion.
Keywords: :
Introduction
Alterations to the p53 gene are among the most common and important events in human carcinogenesis. In response to inappropriate growth signals or various cellular stresses, p53 is activated and inhibits cell growth through activation of both cell cycle arrest and apoptosis, maintaining genome stability and preventing cancer development.Citation1,Citation2 p53 achieves these functions mainly through the transactivation of its downstream target genes. Therefore, the identification of new p53 downstream genes has shed light on how p53 exerts its tumor-suppressing effects under different cellular stresses. The p53 master regulatory network is composed of a vast number of genes that are direct targets for p53-mediated transactivation.
p73 and p63, two p53 family proteins, share a high degree of structural homology with p53, especially in their DNA-binding domains.Citation3-Citation5 p73 and p63 are mainly involved in normal development and differentiation.Citation6,Citation7 Unlike p53, p73 and p63 are expressed as multiple isoforms that either contain an N-terminal p53-homologous transactivation domain (TA) or that lack this domain (ΔN). TAp73 and TAp63 can transactivate a subset of p53 target genes and induce apoptosis of cancer cells. In contrast, the ∆N isoforms are thought to play a role in blocking the transactivation of target genes of p53 and their respective TA isoforms. The dominant negative ∆N isoforms of p63 and p73 are overexpressed in several types of cancers. At least 30% of head and neck squamous cell carcinomas harbor mutations in genes that regulate squamous differentiation, including p63.Citation8 In general, the TA isoforms might be expected to have a role in tumor suppression, whereas an increased expression of the ∆N isoforms might be oncogenic, suggesting that p73 and p63 have the potential to become interesting cancer therapeutic targets.Citation9 Recently, researchers have tested small-molecule strategies that stimulate TAp73 or TAp63 to trigger p53-like transcriptional responses even in p53-deficient tumor cells.Citation10-Citation12
In an attempt to identify novel p53-inducible genes involved in tumor suppression, we profiled the gene expression of Saos-2 human osteosarcoma cells. We identified CLCA2 as a direct target of the p53 family and also found that inactivation of CLCA2 by siRNA-stimulated cancer cell migration and invasion though the transcriptional activation of FAK is a key driving force for cancer progression. A small-molecule FAK inhibitor suppresses CLCA2 siRNA-enhanced cell migration and invasion. Taken together, our data provide strong evidence that CLCA2 is one of the downstream mediators of p53 that regulate cancer cell migration and invasion.
Results
The p53 family upregulates the expression of the CLCA2 mRNA and protein
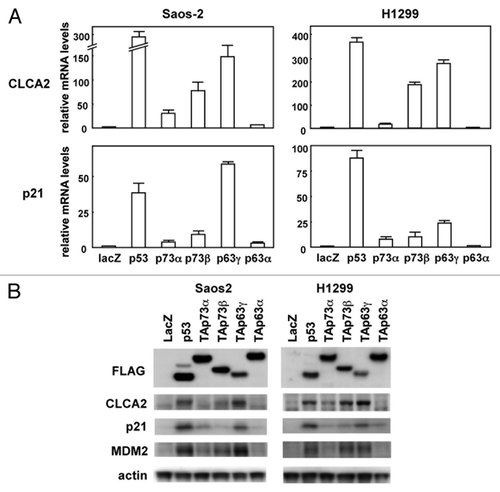
In an effort to identify specific targets regulated by the p53 family, Saos-2 human osteosarcoma cells were infected at a multiplicity of infection (MOI) of 25 with recombinant adenovirus vectors carrying an expression cassette for human p53 (Ad-p53), human TAp63γ (Ad-p63γ), or bacterial lacZ gene (Ad-lacZ). RNAs were isolated from the infected cells 24 h post-infection and subjected to microarray analysis.Citation13 Microarray data are available in the GEO database (GSE35512). Using this approach, we found that the CLCA2 gene was highly induced in p53- or p63-transfected Saos-2 cells compared with lacZ-transfected cells. The results from the microarray were confirmed by real-time RT-PCR with human cancer cell lines, Saos-2 and H1299. As shown in , CLCA2 mRNA was markedly increased by p53, p73α, p73β and p63γ in both cells. CLCA2 mRNA induction by p63α was less pronounced in H1299 cells. As a positive control, we also performed real-time RT-PCR analysis of the known p53-target gene p21 (, lower panel). The level of CLCA2 protein, as examined by immunoblot, shows that total cellular CLCA2 protein was increased in p53-, p73- and p63-transfected cells, consistent with real-time RT-PCR analysis (). A recent report showed that CLCA2 can be induced in a p53-dependent manner in breast cancer cells.Citation14 These results indicate that the p53 family of transcription factors regulates CLCA2 in a number of different cell types.
Figure 1. Upregulation of the CLCA2 mRNA and protein by the p53 family. (A) Upregulation of CLCA2 mRNA by p53 family members in human cancer cell lines. Saos-2 and H1299 cells were infected with replication-deficient recombinant adenoviruses encoding human p53 family proteins with an N-terminal FLAG epitope or the bacterial lacZ at an MOI of 25 for 24 h. CLCA2 and p21 mRNA levels were assayed using real-time PCR. Values shown are the mean ± standard error of three independent experiments normalized to their respective controls as 1. (B) Upregulation of CLCA2 protein by p53 family members in human cancer cell lines. Cells were infected with adenoviruses as described above, and an immunoblot analysis of FLAG, CLCA2, p21, MDM2 and β-actin was performed.
CLCA2 is upregulated after DNA damage in a p53-dependent manner
To confirm that CLCA2 is a true target of p53, we examined whether activation of endogenous p53 was able to induce CLCA2. To test this, HCT116 cells expressing wild-type p53 [HCT116-p53(+/+)] and isogenic mutant cells lacking p53 [HCT116-p53(−/−)] were exposed to adriamycin (ADR) or Mdm2 inhibitor Nutlin-3. Treatment of HCT116-p53(+/+) cells with ADR (0.2 µg/ml) or Nutlin-3 (10 µM) resulted in the upregulation of p53 and its target p21, with an increase in CLCA2 (Fig. S1). However, the increase in CLCA2 expression was not observed in HCT116-p53(−/−) cells following the same treatment. If the CLCA2 gene has an important role downstream of p53, we might expect that CLCA2 induction by p53 would be conserved among species. We compared the effects of UV-induced DNA damage on the expression of the mouse ortholog of CLCA2, mClca5, in isogenic p53(+/+) MEFs and p53(−/−) MEFs. As expected, UV treatment caused p53 activation with the induction of p21 in p53(+/+) MEFs but not in p53(−/−) MEFs (Fig. S2A). Similar to p21 induction, mClca5 expression was significantly induced in p53(+/+) MEFs. Moreover, rat CLCA2 (rClca2) expression was induced in rat fibroblasts in a wild-type p53-dependent manner (Fig. S2B). These results indicate that CLCA2 is upregulated by activating endogenous p53 in cancer and normal cells.
A conserved response element in the CLCA2 gene in the p53 family
To determine whether the transcription of hCLCA2 is directly regulated by the p53 family, we searched for candidate p53-binding sequence(s)Citation15 in the genomic region of the CLCA2 gene. We found two possible candidates, designated CLCA2-RE1 and CLCA2-RE2 (hereafter referred to as RE1 and RE2, respectively), which are located approximately 73 and 2394 bp upstream of the first exon, respectively. RE1 (GGACTTGTagAGGCAgGTCC) and RE2 (AAGCcTGagTGAGCTTGTTC) sequences both contain three-mismatch nucleotides (lowercase letters) in noncritical positions within the 20-bp consensus p53-binding sequences. Thus, we analyzed the interaction of the p53 protein with these candidate p53-binding sequences using the chromatin immunoprecipitation (ChIP) assayCitation16 with Saos-2 cells infected with adenoviruses expressing FLAG-tagged p53 family genes (, left; Ad-p53, Ad-p73α, Ad-p73β, Ad-p63β and Ad-p63α) and a control adenovirus Ad-LacZ. The cross-linked chromatin complex from these cells was immunoprecipitated with an anti-FLAG monoclonal antibody. We then measured the abundance of candidate sequences within the immunoprecipitated complexes using PCR amplification. The ChIP assay revealed that a DNA fragment containing RE1 was reproducibly present in the immunoprecipitated complex containing p53, TAp73α, TAp73β, or TAp63γ protein (, top row, lanes 9, 11, 13 and 15). In contrast, the p63α protein binding to this site was insignificant (, right, upper panel, lane 9). We designated this binding sequence for the p53 family RE-CLCA2 (for response element in CLCA2). Another DNA fragment containing RE2 was amplified in the input chromatin-positive control for PCR but not in the immunoprecipitates with the anti-FLAG antibody (data not shown). As Walia, et al. also reported that endogenous and exogenous p53 can bind to the same site,Citation14 we considered RE-CLCA2 to be a true responsive site for the p53 family. As a positive control for the ChIP assay, we analyzed the interaction of p53 family proteins with the MDM2 promoter that contains a p53 response element. As expected, p53, p73 and p63 proteins resided at the MDM2 promoter, which contained a p53-response element and (, bottom row, lanes 9, 11, 13, 15 and 17).
Figure 2. Regulation of CLCA2 transcription by the p53 family. (A) The position and nucleotide sequence of a response element for the p53 family in the CLCA2 gene RE-CLCA2. Open boxes represent exons. RE-CLCA2 is located 73-bp upstream of the first exon and consists of two copies of the consensus 10-bp motif of the p53-binding sequence. The consensus sequences are indicated by uppercase letters. Lowercase letters identify mismatches with the consensus sequence. The alignment of the conserved binding sites in rat, mouse, dog, horse and platypus sequences from the CLCA2 gene is shown at the top. A mutated sequence corresponding to potentially critical nucleotides of RE-CLCA2 used in the luciferase assay is also indicated (RE-CLCA2-mut). R represents purine; Y, pyrimidine; W, adenine or thymine. (B) p53 family proteins bind to the RE-CLCA2 site in vivo. Expression of FLAG-tagged p53, p73 and p63 is shown in adenovirus-infected Saos-2 cells. Cells were infected with adenovirus at an MOI of 25 and were harvested 24 h after infection. Immunoblot analysis was performed on lysates (10 µg) of cells infected with Ad-lacZ, Ad-p53, Ad-p73α, Ad-p73β, Ad-p63γ and Ad-p63α. In vivo recruitment of p53 family proteins to the RE-CLCA2 site (right). Cross-linked chromatin was extracted from Saos-2 cells after infection with Ad-LacZ, Ad-p53, Ad-p73α, Ad-p73β, Ad-p63γ and Ad-p63α and immunoprecipitated with an anti-FLAG antibody. The immunoprecipitated material was amplified using primers specific for RE-CLCA2 (1st panel) and the MDM2 promoter (2nd panel). “Input chromatin,” which represents a portion of the sonicated chromatin prior to immunoprecipitation, was used as a positive PCR control (lanes 1 to 6). Immunoprecipitates in the absence of antibody (Ab (-), lanes 8, 10, 12, 14, 16 and 18) served as negative controls. (C) The RE-CLCA2 sequence is responsive to p53 family proteins. Saos-2, SW480 or H1299 cells were transiently transfected with the pGL3-promoter vector containing the RE-CLCA2 (pGL3-RE-CLCA2) or its mutant (pGL3-RE-CLCA2-mut) along with a transfection-control plasmid expressing Renilla luciferase, pRL-TK. Cells were cotransfected with a control pCMV-Tag2 vector or a vector that expresses p53 family members 24 h prior to performing the luciferase assay. Luciferase activity was measured using the dual-luciferase reporter assay system with the Renilla luciferase activity as an internal control. All experiments were performed in triplicate, and the means and standard deviations are indicated by bars and brackets, respectively. (D) The regulation of CLCA2 transcription by endogenous p53. HCT116-p53(+/+) and HCT116-p53(−/−) cells were co-transfected with pGL3-RE-CLCA2 or pGL3-RE-CLCA2-mut together with pRL-TK. At 4 h after transfection, cells were treated with 0, 0.5 or 1.0 µg/ml ADR for 24 h and subjected to a dual luciferase assay. Firefly luciferase activity relative to the Renilla luciferase control is given. Experiments were conducted in triplicate with standard deviations indicated. Activity in the control HCT116-p53(+/+) cells was set to 100%.
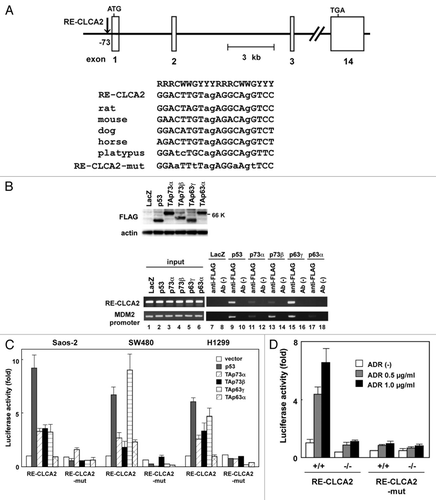
If the CLCA2 gene has an important role as a downstream mediator of p53 family members, the response element within the human CLCA2 gene would be conserved among species. We thus performed a sequence comparison for the response element RE-CLCA2 among orthologous mammalian CLCA2 genes. A nucleotide sequence comparison revealed that the RE-CLCA2 sequence is well conserved among rat, mouse, dog, horse and platypus at nearly identical positions within their genomes (see ), suggesting that CLCA2 is an evolutionarily conserved transcriptional target of the p53 family members.
To further confirm whether the RE-CLCA2 sequence confers transcriptional activity in a manner that depends on p53 family members, we performed a heterologous promoter-reporter assay using a luciferase vector, pGL3-RE-CLCA2, prepared by cloning the oligonucleotide corresponding to RE-CLCA2 upstream of a minimal promoter (see Materials and Methods). Saos-2, SW480 and H1299 cells were transiently co-transfected with pGL3-RE-CLCA2 together with a p53-, p63α-, p73β-, p63γ- or p63α-expressing plasmid. shows an induction of luciferase activity from pGL3-RE-CLCA2 by the p53 family compared with an empty vector. A mutant reporter plasmid, pGL3-RE-CLCA2-mut, was constructed by altering potentially critical nucleotides of RE-CLCA2 as a negative control. In contrast, the mismatches in RE-CLCA2 (pGL3-RE-CLCA2-mut) significantly abolished transactivation by the p53 family (). We next examined whether the RE-CLCA2 sequence is responsive to transcriptional activation by endogenous p53. HCT116 cells expressing wild-type p53 [HCT116-p53(+/+)] and isogenic mutant cells lacking p53 [HCT116-p53(−/−)] were transiently transfected with pGL3-RE-CLCA2 or pGL3-RE-CLCA2-mut and incubated with 0, 0.5 or 1.0 µg/ml ADR. The basal luciferase activity was higher in HCT116-p53(+/+) cells than in HCT116-p53(−/−) cells (, compare lane 4 with lane 1). When pGL3-RE-CLCA2 was transiently transfected into HCT116-p53(+/+) cells, treatment with ADR significantly induced luciferase expression from this construct (). In contrast, ADR treatment did not substantially induce luciferase activity in HCT116-p53(−/−) cells (, lane 6). These data suggest that the RE-CLCA2 sequence responds to DNA-damaging agents in a p53-dependent manner.
CLCA2 negatively regulates cancer cell migration and invasion
Previous studies have shown that CLCA2 is downregulated in human colorectal and breast cancers.Citation17,Citation18 Initially, we analyzed the endogenous mRNA expression of CLCA2 in various human cancer cell lines using semiquantitative RT-PCR (Fig. S3), and a downregulation of CLCA2 was observed in certain cancer cell lines. To examine the effects of CLCA2 in cancer cell growth, we transfected the CLCA2 expression vector into Saos-2 osteosarcoma and DLD1 colorectal cancer cells; in both cells, endogenous CLCA2 expressions were at negligible levels. After 24 h, cell viability was measured using the MTT assay. There was no major difference in proliferation between CLCA2-transfected cells and the control (Fig. S4), suggesting that CLCA2 has an insufficient effect on the proliferation of both cell lines tested. Recent studies have reported that plasma membrane ion channels and transporters are important for cell functions including cell migration, proliferation and apoptosis.Citation19,Citation20 Chloride channels also contribute to the cell-shape and volume changes required for glioma cell migration.Citation21 Thus, we examined the effects of CLCA2 in cancer cell motility. We established a stable transfectant expressing CLCA2 using Saos-2 cells selected by growth in G418-containing medium (Fig. S5). To assess the effect of CLCA2 overexpression on cancer cell migration, we conducted wound-healing assays using established Saos-2 cells expressing CLCA2. We found that CLCA2 overexpression markedly inhibited cancer cell migration. The overexpression of CLCA2 in Saos2 and DLD1 cells also increased cell adhesiveness (Fig. S6), suggesting that CLCA2 inhibits signaling from focal adhesions.
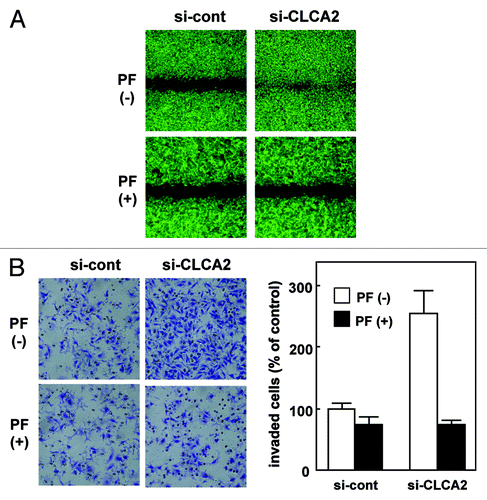
To further our understanding of the function of CLCA2, we investigated the consequences of CLCA2 deficiency in human tumor cells. We chose to study CHC-Y1 cells, in which CLCA2 is expressed at a relatively high level. We introduced an siRNA vector targeting CLCA2 (iLenti-si-CLCA2) into CHC-Y1 cells. After selection in G418-containing medium, three independent transfectants were established (si-CLCA2–1, 2 and 3). CLCA2 protein expression was totally silenced in these three clones as opposed to parental CHC-Y1 cells and mock transfectants [transfection (-) and si-cont, respectively, ]. To assess the effect of CLCA2 knockdown on cell migration, we conducted wound-healing assays. As shown in , CLCA2 knockdown in CHC-Y1 cells stimulated cell migration. Moreover, CLCA2-silenced CHC-Y1 cells had higher invasive activity than control cells as assessed by Matrigel invasion assays (). These results indicate that p53-inducible CLCA2 negatively regulates cancer cell migration and invasion.
Figure 3. CLCA2 siRNA promotes cancer cell migration and invasion. (A) western blot analysis after stable transfection of GFP-tagged CLCA2 siRNA plasmid (iLenti-si-CLCA2) or empty plasmid (si-cont) in CHC-Y1 colorectal cells. The results from untransfected CHC-Y1 controls, a pooled control, and three independent transfectants [si-CLCA2 (clone-1, 2 and 3)] are shown. (B) Wound-healing assay of CLCA2 siRNA plasmid-transfected human cancer cells. Phase contrast images were taken 0, 18 and 28 h after wounding. Representative images showing increased cell migration to the wounded area in CLCA2 siRNA-transfected CHC-Y1 cells. (C) Cell invasion was measured in a Matrigel invasion assay following stable transduction. The experiments were repeated three times with similar results. Representative images showing increased cell invasion in CLCA2 siRNA-transfected CHC-Y1 cells. Quantification of invasion as a percentage of the control is also shown (right).
![Figure 3. CLCA2 siRNA promotes cancer cell migration and invasion. (A) western blot analysis after stable transfection of GFP-tagged CLCA2 siRNA plasmid (iLenti-si-CLCA2) or empty plasmid (si-cont) in CHC-Y1 colorectal cells. The results from untransfected CHC-Y1 controls, a pooled control, and three independent transfectants [si-CLCA2 (clone-1, 2 and 3)] are shown. (B) Wound-healing assay of CLCA2 siRNA plasmid-transfected human cancer cells. Phase contrast images were taken 0, 18 and 28 h after wounding. Representative images showing increased cell migration to the wounded area in CLCA2 siRNA-transfected CHC-Y1 cells. (C) Cell invasion was measured in a Matrigel invasion assay following stable transduction. The experiments were repeated three times with similar results. Representative images showing increased cell invasion in CLCA2 siRNA-transfected CHC-Y1 cells. Quantification of invasion as a percentage of the control is also shown (right).](/cms/asset/45e66d8e-af5f-484a-8d45-a747d4a1d9f3/kcbt_a_10922280_f0003.gif)
The molecular mechanism through which CLCA2 suppresses cell migration and invasion
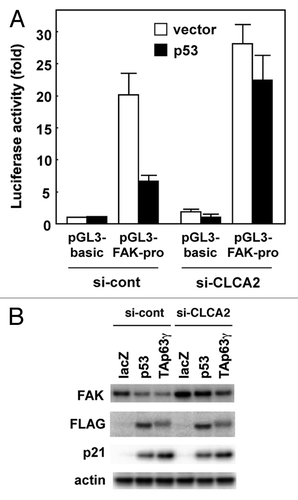
Abdel-Ghany, et al. reported that focal adhesion kinase (FAK) signaling in cancer cells is regulated by β4 integrin ligation to the mouse endothelial Ca(2+)-activated chloride channel protein mCLCA1.Citation22 Activation of FAK and the small GTP-binding protein Rac is necessary for cell migration.Citation23 FAK is activated via autophosphorylation at Tyr397, which is initiated by integrin engagement with its ligand.Citation24,Citation25 Here, we show that FAK activation was enhanced by silencing CLCA2 in CHC-Y1 cells, compared with parental cells and mock transfectants (). Surprisingly, total levels of FAK protein were increased by CLCA2 siRNA (). Furthermore, we found that FAK mRNA was higher in CLCA2 knockdown CHC-Y1 cells than in control cells (), suggesting that endogenous FAK expression is enhanced at the transcriptional level. To validate whether CLCA2 silencing resulted in transcriptional activation of the FAK gene, we performed dual luciferase assays with FAK promoter-reporter constructs. Consistent with the results from real-time PCR, FAK promoter activities were increased at least 2-fold in HEK293 cells after CLCA2 siRNA (). Conversely, CLCA2 expression downregulated FAK promoter activity in HCT116 and Saos-2 cells (), which both expressed endogenous CLCA2 at negligible levels. These results clearly show that CLCA2 transcriptionally activates the FAK gene. In addition to FAK activation, extracellular signal-regulated kinase (ERK), downstream of FAK, was activated in CLCA2 knockdown cells ().
Figure 4. Regulation of FAK transcription by CLCA2. (A) western blot analysis after stable transfection of CLCA2 siRNA plasmid or empty control plasmid (si-cont) in CHC-Y1 cells. The results from untransfected CHC-Y1 cells, a pooled control, two control clones [si-cont (clone-1 and -2)] and four independent CLCA2 siRNA transfectants [si-CLCA2 (clone-1, to -4)] are shown. Inhibition of CLCA2 expression by siRNA results in the induction of FAK protein levels and activated ERK. (B) FAK mRNA levels were assayed using real-time PCR. (C) CLCA2 siRNA upregulated FAK promoter activity. HEK293 cells were co-transfected with the FAK promoter-reporter construct P1020 together with CLCA2 siRNA plasmid (si-CLCA2) or empty plasmid (si-cont). After 24 h, cells were lysed in a plate and subjected to the dual luciferase assay. The pGL3-basic vector without the inserts was used as a control. Histograms show firefly luciferase activity relative to Renilla luciferase control. Experiments were conducted in triplicate with SD indicated. (D) The repression of FAK promoter activity by CLCA2. HCT116 and Saos-2 cells were co-transfected with the P1020 FAK-promoter construct together with a CLCA2 expression vector or empty vector plasmid (vector). At 24 h after transfection, cells were lysed in a plate and subjected to the dual luciferase assay.
![Figure 4. Regulation of FAK transcription by CLCA2. (A) western blot analysis after stable transfection of CLCA2 siRNA plasmid or empty control plasmid (si-cont) in CHC-Y1 cells. The results from untransfected CHC-Y1 cells, a pooled control, two control clones [si-cont (clone-1 and -2)] and four independent CLCA2 siRNA transfectants [si-CLCA2 (clone-1, to -4)] are shown. Inhibition of CLCA2 expression by siRNA results in the induction of FAK protein levels and activated ERK. (B) FAK mRNA levels were assayed using real-time PCR. (C) CLCA2 siRNA upregulated FAK promoter activity. HEK293 cells were co-transfected with the FAK promoter-reporter construct P1020 together with CLCA2 siRNA plasmid (si-CLCA2) or empty plasmid (si-cont). After 24 h, cells were lysed in a plate and subjected to the dual luciferase assay. The pGL3-basic vector without the inserts was used as a control. Histograms show firefly luciferase activity relative to Renilla luciferase control. Experiments were conducted in triplicate with SD indicated. (D) The repression of FAK promoter activity by CLCA2. HCT116 and Saos-2 cells were co-transfected with the P1020 FAK-promoter construct together with a CLCA2 expression vector or empty vector plasmid (vector). At 24 h after transfection, cells were lysed in a plate and subjected to the dual luciferase assay.](/cms/asset/c940600d-1dd5-480b-a3ac-8ce60ccac7a5/kcbt_a_10922280_f0004.gif)
Next, we addressed whether FAK activity is required for CLCA2 siRNA-mediated cancer-cell migration and invasion. We tested whether PF-573228, a small-molecule FAK inhibitor,Citation26 could inhibit cancer cell migration. In a wound-healing assay, CLCA2-silencing CHC-Y1 cells were allowed to migrate in normal medium without PF-573228, and wound closure resulted after 24 h [, upper row: PF(-)]; this migration was inhibited by the addition of PF-573228. PF-573228 completely suppressed the migration of CLCA2-silencing cells in scratch-wound cultures after 24 h (: compare the first to the second row). In contrast, PF-573228 had less of an effect on control cell migration (, bottom row). Similar results were observed in cell invasion assays (). These results suggest that CLCA2 regulates cancer cell migration and invasion partially through the FAK signal pathway.
Figure 5. The migration- and invasion-enhancing effects of CLCA2 siRNA were inhibited by the addition of FAK inhibitor. (A) The migration-enhancing effect of CLCA2 siRNA was inhibited by the addition of FAK inhibitor. Control and CLCA2-silenced CHC-Y1 cell monolayers (clone-1) on collagen-coated coverslips were scratched manually with a plastic pipette tip. Cells were washed with fresh medium to remove floating cells and cultured with or without 10 µM PF-573228. Wound healing was monitored at 24 h after scratching. (B) The invasion-enhancing effect of CLCA2 siRNA was inhibited by the addition of FAK inhibitor. Cell invasion was measured using a Matrigel invasion assay. Cells were suspended in medium containing 0 or 10 µM PF-573228 (0.5 mL/chamber) and loaded at a density of 1 × 105 cells/mL to an insert of a transwell chamber as indicated. Cells that had invaded to the underside of the inserts after 24 h of incubation were stained and counted by light microscopy. The experiments were repeated three times with similar results. Quantification of invasion as a percentage of the control is also shown (right).
p53 inhibits FAK expression in human cancer cells.Citation27 Thus, to determine whether p53 downregulates the FAK gene in a CLCA2-dependent manner, control and CLCA2-silencing CHC-Y1 cells were transiently co-transfected with FAK promoter-reporter constructs together with a p53-expressing plasmid. As shown in , p53 downregulated FAK promoted activity in control CHC-Y1 cells. In contrast, a decrease in FAK promoter activity was not observed in CLCA2-silencing cells. Furthermore, FAK expression was markedly decreased in p53 and TAp63γ in control CHC-Y1 cells (, si-cont). In contrast, this effect was not substantial in CLCA2-silencing cells (, si-CLCA2), suggesting that CLCA2 is a critical mediator downstream of p53 that regulates FAK expression.
Figure 6. p53 downregulates FAK expression, which is inhibited by CLCA2 knockdown. (A) Knockdown of CLCA2 prevents p53-mediated suppression of FAK promoter activity. Control and CLCA2-silenced CHC-Y1 cells were co-transfected with the P1020 FAK-promoter construct together with a p53 expression vector or empty vector plasmid (vector). After 24 h, the cells were lysed in a plate and subjected to the dual luciferase assay. (B) The p53-mediated downregulation of FAK was partially abrogated by CLCA2 siRNA. Control and CLCA2-silenced CHC-Y1 cells were treated with Ad-lacZ, Ad-p53 or Ad-p63γ. After 24 h, cell lysates were prepared, and FAK protein expression was detected by western blotting.
Expression of CLCA2 in primary breast cancer tissue
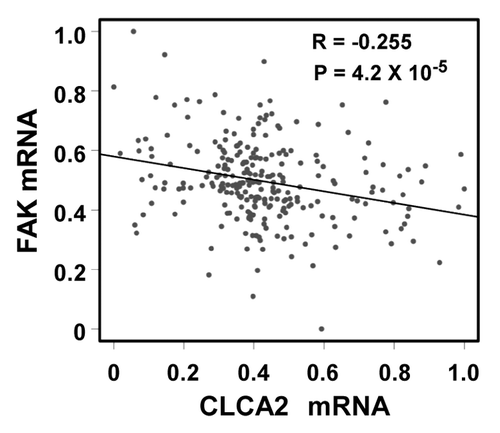
We were intrigued to discover that p53-inducible CLCA2 is involved in cancer cell migration and invasion. Because cancer-enhanced FAK activation has been reported previously,Citation28-Citation30 we investigated the relationship between CLCA2 and FAK expression in a large published data set that includes 251 clinically annotated cases of breast cancer using a cDNA microarray. Importantly, we found an inverse correlation between CLCA2 and FAK expression ( = 4.2 × 10−6), which is consistent with the ability of CLCA2 to strongly suppress endogenous FAK. These results suggest that CLCA2 is involved in FAK regulation and thus possesses a potential suppressive role in carcinogenesis.
Figure 7. A negative correlation between CLCA2 and FAK expression in human breast cancer tissues. An expression microarray experiment conducted by Miller, et al.Citation37 and available at the GEO database (data set GSE3494) was queried for levels of CLCA2 and FAK. Normalized mRNA signals (lowest = 0, highest = 1) from 251 breast cancer samples were plotted. The Pearson correlation coefficient is shown.
Discussion
Initially, CLCA2 was reported as a p53 target that regulates p53-mediated apoptosis.Citation14 Recently, CLCA2 was also shown to be involved in the p53-mediated senescence pathway.Citation31 In this study, we identified CLCA2 as a common p53 family target. We found that regulation of the CLCA2 gene by p53 is evolutionally conserved between humans and rodents. We also identified a specific binding site for p53 family proteins, RE-CLCA2, that is highly conserved between humans and rodents. A reporter assay demonstrated that RE-CLCA2 is responsive to p53 family proteins (). When the critical nucleotides in RE-CLCA2 were substituted, the mutant response element became inactive.
Although many p53 family target genes have been identified, CLCA2 is unique in several ways. CLCA2 knockdown promotes cancer cell migration and invasion, whereas its overexpression inhibits cancer cell migration. Interestingly, we found that the knockdown of CLCA2 leads to an increased expression of FAK, which is known as an integrin- and growth-factor-associated tyrosine kinase that promotes cell motility. Indeed, CLCA2 expression negatively correlates with FAK expression in a large published breast cancer data set (). Additionally, knockdown of CLCA2 in HEK293 cells activates the FAK promoter, while CLCA2 overexpression represses it in HCT116 and Saos2 cells. These results suggest that CLCA2 negatively regulates FAK expression though transcriptional inactivation of the FAK gene. Importantly, we found that cell migration and invasion mediated by CLCA2 knockdown is FAK dependent, as FAK inhibitor rescues the effect induced by silencing CLCA2. In addition, through searching the Oncomine database, CLCA2 expression was found to be markedly decreased in primary tumor tissues of lung, prostate and bladder but decreased in glioma and leukemia (data not shown). Taken together, these data suggest that CLCA2 is an important mediator of the p53 family in the regulation of cell motility.
Previous works reported that p53 directly binds to its consensus binding site on the promoter of the FAK gene and inhibits its transcription.Citation27 Moreover, FAK overexpression and p53 mutation correlate strongly in human breast cancers.Citation32 In the present study, we found that wild-type p53-mediated FAK downregulation was partially inhibited in CLCA2-silencing cells. Therefore, the p53 family might play a role in the negative regulation of FAK expression both directly and indirectly via CLCA2 induction. To identify the promoter region responsible for CLCA2-mediated suppression of FAK promoter activity, additional experiments with a series of deletion constructs of the FAK promoter are needed in a future study. FAK can, in turn, suppress the transcriptional activity of p53 or promote the ubiquitin-proteasome-mediated degradation of p53 by Mdm2 through its binding to the p53 protein in the nucleus, suggesting a feedback mechanism in which FAK regulates p53.Citation33 The results presented here support a novel p53-CLCA2-FAK signaling pathway.
Overall, our demonstration that CLCA2 is a p53 target provides additional insight into p53-mediated tumor suppression, suggesting a novel network in which CLCA2 is important for regulating FAK expression downstream of p53. Thus, it is plausible that impaired CLCA2 induction by inactivation of the p53 pathway, and the subsequent failure of consequent downstream signaling events, confers a survival advantage to tumor-prone cells. In support of this view, CLCA2 expression has been shown to be downregulated in various types of tumors.Citation17,Citation18,Citation34 For instance, CLCA2 is silenced by DNA methylation in breast cancers,Citation18 suggesting that a loss of CLCA2 function may be a genetic event that promotes tumor progression. In summary, identifying this unrecognized pathway from p53-CLCA2-FAK provides an opportunity for therapeutic intervention, as it reveals a novel target for drug design that could potentially maximize the p53 response and sensitize tumor cells to cancer therapy.
Materials and Methods
Cell culture, recombinant adenovirus and plasmids
The cell lines used in this study were purchased from the American Type Culture Collection or the Japanese Collection of Research Bioresources. HCT116 (p53 wild-type) and its derivative HCT116-p53(−/−) cells lacking p53 were kindly provided by Dr. Bert Vogelstein (Howard Hughes Medical Institute, Johns Hopkins University). The construction, purification and infection procedures of replication-deficient recombinant adenoviruses encoding the human p53 family proteins with an N-terminal FLAG epitope (Ad-p53, Ad-p73α, Ad-p73β, Ad-p63γ and Ad-p63α) or the bacterial lacZ gene (Ad-lacZ) were described previously.Citation13,Citation35,Citation36 The relative efficiency of adenovirus infection was determined by X-gal staining of cells infected with a control Ad-lacZ; 90–100% of the cells can be infected at an MOI of 25 to 100.Citation35 The vector encoding FLAG-tagged human CLCA2 (pRerceiver-M11-CLCA2) was purchased from GeneCopoeia. PF-573228 was purchased from Sigma.
cDNA microarray
In an effort to identify specific targets regulated by the p53 family, an Affymetrix Genechip analysis was performed and expression patterns compared in a human osteosarcoma cell line, Saos-2, transfected separately with Ad-p53 or Ad-p63γ. The microarray data have been deposited into the NCBI Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) and are accessible under GEO series accession number GSE13504.Citation37
Real-time RT-PCR
Total RNA was reverse transcribed using SuperScript III (Invitrogen) with random primers. Quantitative RT-PCR was performed using TaqMan Gene Expression assays, and 7900HT Real-time PCR System (Applied Biosystems) was performed according to the protocols of the manufacturer. Relative levels of gene expression were quantified using the ΔΔCt method, which results in a ratio of target gene expression to an equally expressed housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase. The primer/probe sets used were as follows: human CLCA2, Hs00998916_m1; human p21, Hs00355782_m1; GAPDH, Hs99999905_m1. Values shown are the means ± standard errors of three independent experiments and normalized to their respective controls as 1.
Immunoblot analysis
The primary antibodies used for immunoblotting in this study were as follows: mouse anti-FLAG mAb (M2, Sigma), mouse anti-human p53 monoclonal antibody (DO-1, Santa Cruz Biotechnology), goat anti-human CLCA2 antibody (C-18, Santa Cruz Biotechnology), mouse anti-human p21 mAb (F-5, Santa Cruz Biotechnology), mouse anti-human MDM2 mAb (SPM14, Santa Cruz Biotechnology) and mouse anti-β-actin mAb (C4, Chemicon). Proteins were transferred onto Immobilon P membranes (Millipore) using electroblotting, and immunoblot analysis was performed as described previously.Citation16
Chromatin immunoprecipitation assay (ChIP)
The ChIP assay was performed as described previouslyCitation16 using the ChIP Assay Kit (Upstate Biotechnology) and antibodies against FLAG (M2). A total of 2 × 106 cells were cross-linked with a 1% formaldehyde solution for 15 min at 37°C. The cells were then lysed in 200 µl of SDS lysis buffer and sonicated to generate 300–800 bp DNA fragments. After centrifugation, the cleared supernatant was diluted 10-fold with ChIP dilution buffer and split into two equal portions; one was incubated with anti-FLAG antibody (5 μg) at 4°C for 16 h, and the other portion was used as a negative control (no antibody). A volume of 1/50 of total extract was used for PCR amplification as the input control. Immune complexes were precipitated, washed and eluted, and DNA-protein cross-links were reversed by heating at 65°C for 4 h. DNA fragments were purified and dissolved in 40 ml of TE. Two microliters of each sample were used as a template. PCR amplification of the consensus p53-binding sequence upstream of the human CLCA2 gene (-73, named the RE-CLCA2) were performed on immunoprecipitated chromatin using the specific primers 5′-GGCAGAATGTTTCTTGCCCAACCAT-3′ (forward) and 5′-AGAGAAGCCTCCAGTTCACTCACAC-3′ (reverse). Amplifications were also performed on immunoprecipitated chromatin using the specific primer 5′-GTTCAGTGGGCAGGTTGACT-3′ and 5′-GCTACAAGCAAGTCGGTGCT-3′ for the MDM2 promoter containing a p53-binding sequence. To ensure that PCR amplification was performed in the linear range, template DNA was amplified for a maximum of 35 cycles. PCR products generated from the ChIP template were directly sequenced to verify the identity of the amplified DNA.
Luciferase assay
A 20-bp fragment of the response element for p53 family, RE-CLCA2 (5′-GGACTTGTAGAGGCAGGTCC-3′), and its mutant form RE-CLCA2-mut (5′-GGAATTTTAGAGGAAGTTCC-3′) were synthesized and inserted upstream of a minimal promoter in the pGL3-promoter vector (Promega), and the resulting constructs were designated pGL3-RE-CLCA2 and pGL3-RE-CLCA2-mut, respectively. A luciferase assay was subsequently conducted as previously described.Citation13,Citation16 The P1020 FAK promoter-luciferase reporter construct was a kind gift from Dr. V. Golubovskaya (Roswell Park Cancer Institute). The luciferase assay was performed as described previously.Citation13,Citation16 All experiments were performed in triplicate and repeated at least three times.
Knockdown of CLCA2 by RNA interference
For CLCA2 knockdown studies, a small interfering RNA (siRNA) vector against human CLCA2-containing H1 promoter (iLenti-GFP-si-CLCA2) was purchased from Applied Biological Materials. This siRNA vector has an independent open reading frame of green fluorescent protein (GFP). The GFP-positive cells were monitored as a marker of the transfection. The target sequence is 5′-CTACAGAATATCCAAGATGACTTTAACAA-3′. As controls, we used siRNA vector without the insert synthesized by Applied Biological Materials. To establish stable cell clones, transfected CHC-Y1 cells were seeded at clonal densities and selected in medium containing 0.8 µg/ml geneticin. Two weeks later, single clones were isolated and expanded.
Cell migration and invasion assays
Invasiveness was determined using Matrigel invasion chambers with an 8-µm pore membrane (24-well plate, BD Biosciences). Cells were suspended in serum-free medium and loaded at a density of 1 × 105 cells/mL to an insert of a transwell chamber as indicated. After 24 h of incubation, the uninvaded cells that remained on top of the filter were removed. The invaded cells were fixed, stained with hematoxylin and photographed. The number of invaded cells was quantified by counting cells from five microscopic fields. For the cell migration assay, the stably transfected cells were seeded at a high density, 1 × 106 cells, on poly-d-lysine-coated coverslips and wounded 1 d later when the cells formed a confluent monolayer. Scraping a pipette tip through the cell monolayer created the wound. Wound healing was monitored with an inverted phase-contrast microscope. Three randomly chosen regions of a single wound (each 2 cm long) were photographed at 4x magnification.
Clinical samples
FAK and CLCA2 mRNA levels in a publicly available breast cancer data set, GSE3494, which includes gene-expression data from 251 cases, were investigated.Citation37 Statistical analyses were performed using two-sided Student's t tests.
Additional material
Download Zip (5.3 MB)Acknowledgments
We thank Kanae Ohori and Masami Ashida for excellent technical support and Kohzoh Imai, Nobuyuki Yokoyama and Shota Sato for critical comments about this manuscript. This research was supported in part by Grants-in-Aid for Cancer Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- el-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol 1998; 8:345 - 57; http://dx.doi.org/10.1006/scbi.1998.0097; PMID: 10101800
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 10; http://dx.doi.org/10.1038/35042675; PMID: 11099028
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997; 90:809 - 19; http://dx.doi.org/10.1016/S0092-8674(00)80540-1; PMID: 9288759
- Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 1998; 4:839 - 43; http://dx.doi.org/10.1038/nm0798-839; PMID: 9662378
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2:305 - 16; http://dx.doi.org/10.1016/S1097-2765(00)80275-0; PMID: 9774969
- Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010; 2:a004887; http://dx.doi.org/10.1101/cshperspect.a004887; PMID: 20484388
- Barbieri CE, Pietenpol JA. p53 family members: similar biochemistry, different biology. Cancer Biol Ther 2005; 4:419 - 20; http://dx.doi.org/10.4161/cbt.4.4.1760; PMID: 15908776
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011; 333:1157 - 60; http://dx.doi.org/10.1126/science.1208130; PMID: 21798893
- Zhang J, Chen X. The p53 family: prospect for cancer gene therapy. Cancer Biol Ther 2007; 6:116 - 8; http://dx.doi.org/10.4161/cbt.6.1.3790; PMID: 17612008
- Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol 2010; 80:724 - 30; http://dx.doi.org/10.1016/j.bcp.2010.04.031; PMID: 20450892
- Leslie M. Brothers in arms against cancer. Science 2011; 331:1551 - 2; http://dx.doi.org/10.1126/science.331.6024.1551; PMID: 21436441
- Lu C, Wang W, El-Deiry WS. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol Ther 2008; 7:2039 - 46; http://dx.doi.org/10.4161/cbt.7.12.7461; PMID: 19106635
- Sasaki Y, Negishi H, Koyama R, Anbo N, Ohori K, Idogawa M, et al. p53 family members regulate the expression of the apolipoprotein D gene. J Biol Chem 2009; 284:872 - 83; http://dx.doi.org/10.1074/jbc.M807185200; PMID: 19001418
- Walia V, Ding M, Kumar S, Nie D, Premkumar LS, Elble RC. hCLCA2 Is a p53-Inducible Inhibitor of Breast Cancer Cell Proliferation. Cancer Res 2009; 69:6624 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-08-4101; PMID: 19654313
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet 1992; 1:45 - 9; http://dx.doi.org/10.1038/ng0492-45; PMID: 1301998
- Sasaki Y, Ishida S, Morimoto I, Yamashita T, Kojima T, Kihara C, et al. The p53 family member genes are involved in the Notch signal pathway. J Biol Chem 2002; 277:719 - 24; http://dx.doi.org/10.1074/jbc.M108080200; PMID: 11641404
- Bustin SA, Li SR, Dorudi S. Expression of the Ca2+-activated chloride channel genes CLCA1 and CLCA2 is downregulated in human colorectal cancer. DNA Cell Biol 2001; 20:331 - 8; http://dx.doi.org/10.1089/10445490152122442; PMID: 11445004
- Li X, Cowell JK, Sossey-Alaoui K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene 2004; 23:1474 - 80; http://dx.doi.org/10.1038/sj.onc.1207249; PMID: 14973555
- Hoffmann EK. Ion channels involved in cell volume regulation: effects on migration, proliferation, and programmed cell death in non adherent EAT cells and adherent ELA cells. Cell Physiol Biochem 2011; 28:1061 - 78; http://dx.doi.org/10.1159/000335843; PMID: 22178996
- Lang F, Föller M, Lang K, Lang P, Ritter M, Vereninov A, et al. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol 2007; 428:209 - 25; http://dx.doi.org/10.1016/S0076-6879(07)28011-5; PMID: 17875419
- Ransom CB, O’Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci 2001; 21:7674 - 83; PMID: 11567057
- Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. Focal adhesion kinase activated by beta(4) integrin ligation to mCLCA1 mediates early metastatic growth. J Biol Chem 2002; 277:34391 - 400; http://dx.doi.org/10.1074/jbc.M205307200; PMID: 12110680
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci 2003; 116:1409 - 16; http://dx.doi.org/10.1242/jcs.00373; PMID: 12640026
- Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Rondé P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci 2005; 118:4415 - 25; http://dx.doi.org/10.1242/jcs.02565; PMID: 16159962
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci 1999; 112:2677 - 91; PMID: 10413676
- Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem 2007; 282:14845 - 52; http://dx.doi.org/10.1074/jbc.M606695200; PMID: 17395594
- Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, et al. p53 regulates FAK expression in human tumor cells. Mol Carcinog 2008; 47:373 - 82; http://dx.doi.org/10.1002/mc.20395; PMID: 17999388
- Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev 2004; 14:92 - 101; http://dx.doi.org/10.1016/j.gde.2003.12.002; PMID: 15108811
- Siesser PM, Hanks SK. The signaling and biological implications of FAK overexpression in cancer. Clin Cancer Res 2006; 12:3233 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-06-0456; PMID: 16740741
- Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histol Histopathol 2009; 24:503 - 10; PMID: 19224453
- Tanikawa C, Nakagawa H, Furukawa Y, Nakamura Y, Matsuda K. CLCA2 as a p53-inducible senescence mediator. Neoplasia 2012; 14:141 - 9; PMID: 22431922
- Golubovskaya VM, Conway-Dorsey K, Edmiston SN, Tse CK, Lark AA, Livasy CA, et al. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int J Cancer 2009; 125:1735 - 8; http://dx.doi.org/10.1002/ijc.24486; PMID: 19521985
- Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell 2008; 29:9 - 22; http://dx.doi.org/10.1016/j.molcel.2007.11.031; PMID: 18206965
- Elble RC, Pauli BU. Tumor suppression by a proapoptotic calcium-activated chloride channel in mammary epithelium. J Biol Chem 2001; 276:40510 - 7; http://dx.doi.org/10.1074/jbc.M104821200; PMID: 11483609
- Oshima Y, Sasaki Y, Negishi H, Idogawa M, Toyota M, Yamashita T, et al. Antitumor effect of adenovirus-mediated p53 family gene transfer on osteosarcoma cell lines. Cancer Biol Ther 2007; 6:1058 - 66; http://dx.doi.org/10.4161/cbt.6.7.4320; PMID: 17568187
- Sasaki Y, Negishi H, Idogawa M, Yokota I, Koyama R, Kusano M, et al. p53 negatively regulates the hepatoma growth factor HDGF. Cancer Res 2011; 71:7038 - 47; http://dx.doi.org/10.1158/0008-5472.CAN-11-1053; PMID: 22006999
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A 2005; 102:13550 - 5; http://dx.doi.org/10.1073/pnas.0506230102; PMID: 16141321