Abstract
Tumor relapse and tumor cell repopulation has been explained partially by the drug-free break period between successive conventional treatments. Strategies to overcome tumor relapse have been proposed, such as the use of chemotherapeutic drugs or radiation in small, frequent fractionated doses without an extended break period between treatment intervals. Yet, tumors usually acquire resistance and eventually escape the therapy. Several mechanisms have been proposed to explain the resistance of tumors to therapy, one of which involves the cancer stem cell or tumor-initiating cell (TIC) concept. TICs are believed to resist many conventional therapies, in part due to their slow proliferation and self-renewal capacities. Therefore, emerging efforts to eradicate TICs are being undertaken. Here we show that treatment with Photofrin II, among the most frequently used photosensitizers, sensitized a TIC-enriched U-87MG human glioblastoma cell to radiation, and improve treatment outcome when used in combination with radiotherapy. A U-87MG tumor cell population enriched with radiation-resistant TICs becomes radio-sensitive, and an inhibition of cell proliferation and an increase in apoptosis are found in the presence of Photofrin II. Furthermore, U-87MG tumors implanted in mice treated with Photofrin II and radiation exhibit a significant reduction in angiogenesis and vasculogenesis, and an increased percentage of apoptotic TICs when compared with tumors grown in mice treated with radiation alone. Collectively, our results offer a new possible explanation for the therapeutic effects of radiosensitizing agents, and suggest that combinatorial treatment modalities can effectively prolong treatment outcome of glioblastoma tumors by inhibiting tumor growth mediated by TICs.
Introduction
Glioblastoma multiforme (GBM) comprises 70% of all brain tumors, and is considered one of the most aggressive forms of brain malignancies. Despite recent advances in diagnosis and treatment approaches, GBM tumors are aggressive and considered incurable. Standard therapy consists of radical resection and postoperative irradiation. The median survival rates are in the range of 6–13 mo.Citation1 A recent study showed that following surgery, adjuvant treatment with temozolomide chemotherapy in combination with radiotherapy, extended the median overall survival by approximately 2 mo.Citation2 Gene silencing represents one possible mechanism that determines resistance to therapy. For example, methylation of the promoter of the MGMT gene leads to increased sensitivity to temozolomide in GBM.Citation3 Yet, treatment options are still limited, necessitating extensive research into new therapeutic modalities to overcome GBM drug resistance.
One possible reason for the resistance of many, if not all, solid tumors to radiation and/or chemotherapy can be explained by the cancer stem cell concept.Citation4,Citation5 These cells are thought to be the sole cells within the tumor that contribute to tumor initiation and growth and are therefore also termed tumor-initiating cells (TICs).Citation5,Citation6 Such cells are pluripotent and they possess many stem cell characteristics, such as self-renewal and multi-lineage differentiation capabilities and the ability to divide limitlessly.Citation5,Citation6 The initial identification of TICs was reported in AML, in which tumor cells expressing the surface marker CD34+ but not CD38-, were able to initiate leukemic growth.Citation4 Additional studies have shown that TICs have been identified in a variety of solid tumors including melanomas, gliomas, breast, colon, pancreas, prostate, lung and head and neck tumors.Citation7 Like “normal” stem cells, TICs are thought to acquire resistance to chemotherapy and radiation more than “differentiated” tumor cells.Citation8,Citation9
A large number of mechanisms explaining how cancer cells acquire resistance to chemotherapy have been proposed. A variety of proteins, for which targeted drugs have been developed, e.g., Her2, EGFR monoclonal antibodies or small molecule drugs, are mutated and overexpressed in resistant tumor cells.Citation10 Therefore, selective proliferation of the more resistant tumor cells during the course of the therapy promotes tumor cell repopulation and relapse.Citation11 According to the TIC concept, a growing body of evidence suggests that the slow proliferation of such cells, and the overexpression of p21 and other cell cycle-regulating molecules, may provide an alternative explanation for multidrug resistance of cancer cells.Citation8,Citation9,Citation12,Citation13
One possible way to overcome treatment resistance is to combine the drug of choice with sensitizing components that increase the specificity and efficacy of the drug used. Studies from the last few decades have shown that the effect of radiation on tumors can be optimized by the addition of radiosensitizing agents, in order to achieve greater damage to the neoplastic tissue than the expected damage from radiation alone.Citation14 Porfimer-sodium (Photofrin-II®) or its derivatives have been studied as photodynamic therapy agents for the treatment of several malignancies including esophageal, intraductal papillary mucinous and glioblastoma cancers.Citation15-Citation17 Photofrin-II has also been considered as a radiosensitizing agent, and was evaluated in several preclinical tumor modelsCitation18-Citation20 as well as in the clinic.Citation21,Citation22 This substance constitutes of a complex mixture of porphyrins produced by acid treatment of hematoporphyrin (Hp), which along with irradiation, amplifies cellular damage.Citation23 Photofrin-II, as well as other porphyrins and their analogs, are known to possess a degree of selectivity to several types of tumors. They tend to concentrate in the tumor tissue when compared with skin, muscle and a variety of other tissues. The maximum concentration differentials for Photofrin-II range between two and five. As porphyrins are unable to cross the blood-brain barrier, the degree of tumor selectivity of Photofrin-II is especially high in the brain.Citation23-Citation25
Based on the fact that TICs are usually resistant to therapy, it would appear that TICs of glioblastoma tumors are probably abundant in the tumor microenvironment following radiotherapy or chemotherapy, and therefore contribute to the rapid proliferation of tumor cells at the drug-free break periods,Citation11 contributing to the aggressiveness and drug resistance of glioblastoma. We hypothesized that the combination of radiosensitizing agents along with radiation targets both “differentiated” tumor cells and resistant TICs of glioblastoma tumors, and therefore may inhibit the potential of TICs to repopulate at the treatment-free periods, hence it can inhibit glioblastoma re-growth or resistance to therapy. We used the U-87MG—a human glioblastoma cell line, which has been shown to grow aggressively, and effectively enrich for TICs in stem-like cell conditions.Citation13 Here we show that the combination of high-dosage radiation and Photofrin-II can induce TIC apoptosis, and subsequently inhibit angiogenesis and vasculogenesis. Tumors grown in mice that were treated with a radiation dose of 10 Gy and Photofrin-II were smaller and avascularized when compared with tumors grown in mice treated with radiation monotherapy. These results further imply a possible mechanism by which the combinatorial treatment strategy reduces tumor relapse and maintains radiation treatment responsiveness in glioblastoma cancer patients.
Results
TICs of glioblastoma tumor cells exhibit increased resistance to radiotherapy, and are sensitized by Photofrin-II
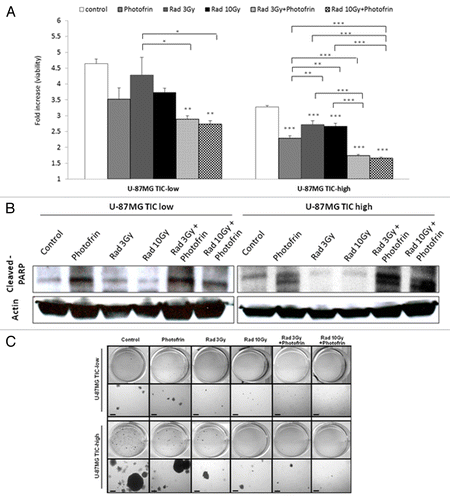
In order to test the impact of the combination of Photofrin-II and radiation on TICs of U-87 MG glioblastoma cells, we enriched for TICs by culturing the cells in stem cell culture conditions as described in Materials and Methods. We found that U-87 MG glioblastoma TIC-high fractions can grow in spheres, and are enriched for TICs based on increased expression of CD133+, a stem cell surface marker, on cancer cells in TIC-high when compared with TIC-low fractions (Fig. S1). Next, TIC-high and TIC-low cultured cells were exposed to 3 Gy or 10 Gy radiation in the presence or absence of Photofrin-II. Cell viability was measured using AlamarBlue. The results in demonstrate that U-87 MG cells from the TIC-high fraction were more resistant to both 3 Gy and 10 Gy radiation than cells from the TIC-low fractions. Furthermore, cells treated with both radiation and Photofrin-II exhibited a significant and remarkable reduction in cell viability. This effect was evident in both TIC-high and TIC-low fractions. We also noted that Photofrin-II, on its own, inhibited cell viability of U-87MG cells. Notably, cell viability was lower in TIC-high than in TIC-low fraction, probably due to reduced proliferation rate of TICs.Citation26 Furthermore, the radiosensitivity effects by means of cell viability were also observed in TICs of MCF7 breast carcinoma, but not of HT29, nor of A549 or PANC1 (Fig. S2).
Figure 1. Cell viability and dealth in TIC-high and TIC-low U-87 MG cells following treatment with radiation and Photofrin-II. TIC-high and TIC-low U-87MG cells were exposed to 1µg/ml Photofrin-II (Photofrin) and/or 3 Gy or 10 Gy radiation (Rad). After 48 h, cell viability was evaluated by the AlamarBlue assay (A), and the levels of cleaved PARP and Actin (loading control) were assessed by western blot (B). In a parallel experiment, TIC-high and TIC-low U-87 MG cells were exposed to the same treatment combination as above. Eight hours later, cells were mixed with soft agar and cultured for 3 wk in agar colony formation medium as described in Materials and Methods. Colonies are presented in two magnifications (scale bar = 200 µm, where indicated) (C). In (A), results were plotted as fold-increase from background values (blank). *, 0.05 > p > 0.01; **, 0.01 > p > 0.001; ***, p < 0.001 from control group, unless indicated otherwise.
The combination of radiation and Photofrin-II increases TIC death, and decreases the number of TIC colonies
The reduced cell viability in TIC-high and TIC-low cells in response to radiation and/or Photofrin-II prompted us to determine whether these treatments induce cell death. Recent studies showed that Photofrin-II can act as an effective radiosensitizing agent, if applied under appropriate conditions.Citation19,Citation20,Citation22 We therefore assessed cell death using PARP activity as an apoptosis marker. TIC-high and TIC-low cells were placed in serum-free medium and were subsequently exposed to 3 Gy or 10 Gy radiation in the presence or absence of Photofrin-II. After 8 h, cells were harvested for the analysis of active PARP expression by western blot. The results in show that PARP activity was markedly enhanced following 3 Gy or 10 Gy radiation in the presence of Photofrin-II. Again, Photofrin-II on its own also induced apoptosis, but not to the same degree as the combination therapy. Comparable results were observed when the cells from each fraction were evaluated for Annexin V expression as another marker for apoptosis using flow cytometry (Fig. S3).
Next, in order to test the capacity of cells from both TIC-high and TIC-low fractions following their exposure to radiation and/or Photodrin-II to form tumorigenic colonies, a soft agar colony formation assay was performed. To this end, TIC-high and TIC-low cells were exposed to radiation and/or Photofrin-II. Cells were then cultured in soft agar, and 3 wk later, colonies were visualized. The results in show that similar to cell viability, PARP expression and Annexin V positivity, the combination of Photofrin-II and radiation (in both doses) reduced the number of colony formed by TIC and non-TIC populations. Taken together, Photofrin-II, in combination with radiation, induces tumor cell apoptosis of both TIC-high and TIC-low cell fractions, in clear contrast to radiation alone, to which TICs were resistant.
The combination of Photofrin-II and radiation delayed tumor growth
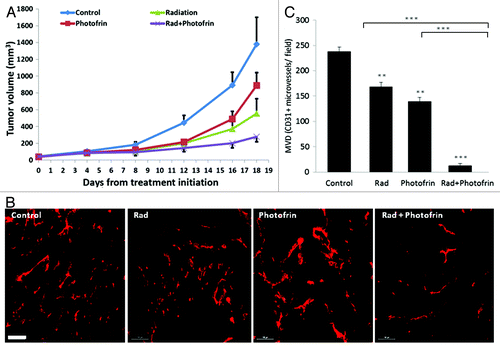
Based on our in vitro results suggesting that Photofin-II in combination with radiation can induce TIC killing, we next assessed the in vivo effects of this combination therapy. In particular, we were interested in evaluating whether a single dose of the different types of therapies would increase treatment benefit by means of delayed tumor growth. To this end, 8–10-wk-old athymic nude mice were implanted with 5 × 106 U-87 MG tumor cells. When tumors reached a size of 50 mm3, mice were treated with 10 mg/kg Photofrin-II, a dose of which was previously determined,Citation19 in combination with a single dose of 10 Gy ionizing radiation. Control mice received PBS as the vehicle of Photofrin-II. The results in show that tumor size was reduced in mice exposed to a single dose of radiation and Photofrin-II in comparison to all other treatment groups. Most notably, Photofrin-II on its own also had some antitumor activity, with no overt toxicity, as measured by body weight (Fig. S4). Furthermore, since the therapy was administered only once, we were able to monitor tumor growth over time. As shown in , over the next 18 d, tumor growth was faster in mice undergoing radiation or Photofrin-II monotherapies, as compared with the combination of the two therapies, suggesting that such treatment combination delays tumor growth.
Figure 2. Evaluation of tumor growth and angiogenesis in mice bearing U-87 MG tumors following a single treatment with Photofrin-II and radiation. U-87 MG human glioblastoma cells (5 × 106) were subcutaneously injected into the flanks of 8–10-wk-old athymic nude mice (n = four to five mice/group). When tumors reached a size of 50 mm3, mice were treated with 10 mg/kg Photofrin-II (Photofrin) and/or were exposed to 10 Gy radiation (Rad). Tumors were measured periodically, and tumor volume was plotted using the formula widthCitation2 × length × 0.5 (A). At end point, tumors were removed and evaluated for microvessel density (MVD) by staining for the endothelial cell marker, CD31 (in red) and visualized by Leica CTR 6000 (scale bar = 100 µm) (B). Quantification of the number of vessels per field was plotted (C). **, 0.01 > p > 0.001; ***, p < 0.001 from control group, unless indicated otherwise.
Photofrin-II therapy reduces angiogenesis and BMDC colonization in radiated tumors
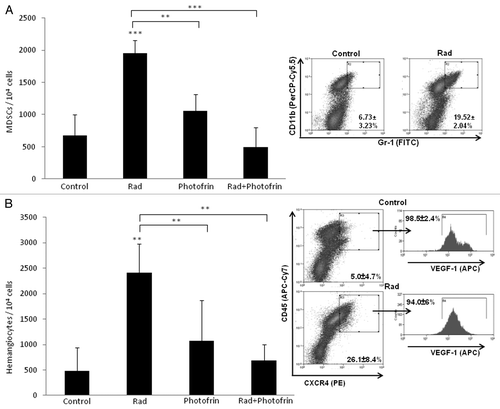
Recent studies have shown that radiotherapy can induce mobilization of several types of BMDCs known to promote angiogenesis. For example, Kioi et al. showed that glioblastoma tumors treated with five daily 2 Gy or 4 Gy local radiation to the brain induced Gr1/CD11b monocytic cell recruitment into the treated tumors.Citation27 Such bone marrow cells are known to contribute to tumor angiogenesis or to promote refractoriness of tumors following antiangiogenic therapy.Citation28 We therefore sought to determine the angiogenesis profile of tumors grown in mice treated with radiation and Photofrin-II. To do so, tumors implanted in mice were harvested at endpoint and tumor sections were analyzed for microvessel density (MVD) using the endothelial cell marker, CD31. The results in show that radiation and Photofrin-II monotherapies exhibited a mild and similar antiangiogenic effect when compared with control. Notably, MVD was substantially reduced in mice treated with the combination therapy as compared with radiation and Photofrin-II monotherapies. To assess the systemic angiogenesis effects mediated by BMDCs of the combination therapy, tumors were prepared as single-cell suspensions, and subsequently were stained for hemangiocytes and MDSCs, both of which are known to promote angiogenesis.Citation28,Citation29 Using flow cytometry, we found that the percentage of both MDSCs and hemangiocytes were significantly and profoundly increased following radiotherapy. However, the combination therapy significantly reduced the percentage of MDSCs and hemangiocytes in treated tumors when compared with radiation-treated tumors (). Taken together, these results indicate that the combination of Photofrin-II and radiation significantly decreases tumor angiogenesis and vasculogenesis mediated by different types of BMDCs found in tumors following radiotherapy.
Figure 3. Evaluation of the percentage of myeloid-derived suppressor cells and hemangiocytes in U-87 MG tumors treated with radiation and Photofrin-II. U-87 MG tumors (described in ), were harvested and prepared as single-cell suspensions. Cells were analyzed for myeloid-derived suppressor cells (MDSCs) (Gr1+/CD11b+) (A) and hemangiocytes (CXCR4+/VEGFR1+/CD45+) (B), using flow cytometry. Only control and radiation (Rad) groups are presented in the flowchart. **, 0.01 > p > 0.001; ***, p < 0.001 from control group, unless indicated otherwise.
The combination of radiotherapy and Photofrin-II decreases the proximity of U-87 MG TICs to blood vessels
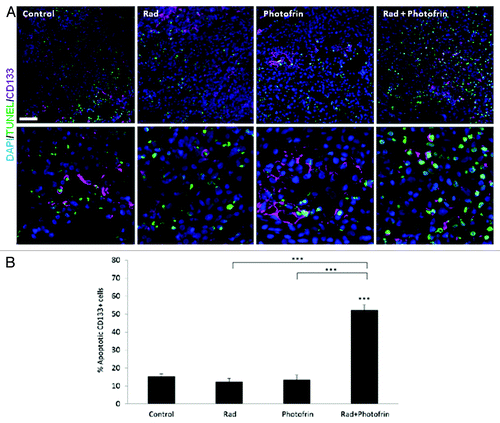
Previous studies demonstrated that TICs of brain tumors are found at the perivascular site of blood vessels, providing an additional basis for the proangiogenic properties of TICs.Citation30 We therefore investigated the pro-angiogenic effect of TICs in U-87 MG tumors treated with the combination of Photofrin-II and radiation. To do so, we analyzed the percentage of CD133+ cells (as a marker for glioblastoma stem cellCitation31) in tumors from all treated groups. The results in show that the percentage of CD133+ cells at the tumor site was significantly increased following radiotherapy when compared with their percentage in untreated tumors. However, the percentage of such cells was markedly and significantly reduced following the combination of radiation and Photofrin-II when compared with the radiotherapy group.
Figure 4. Evaluation of TICs and blood vessels in U-87 MG tumors treated with Photofrin-II and radiation. U-87MG cells (5 × 106) were subcutaneously injected into flanks of 8–10-wk-old athymic nude mice (n = four to five mice/group). When tumors reached a size of 50 mm3, mice were treated with 10 mg/kg Photofrin-II (Photofrin) and/or were exposed to 10 Gy radiation (Rad). At end point, tumors were removed and either prepared as single-cell suspensions and evaluated for the percentage of CD133+ cells (as a marker for glioblastoma TICs) using flow cytometry (A), or tumor sections were immunostained for murine-CD31 as a marker for endothelial cells (red), human-CD133+ as a marker for TICs (violet) and murine-CD45+ as a marker for hematopoietic cells (green).Nuclei were stained with DAPI (blue). Images were captured using Leica TCS SP5 confocal imaging systems (scale bar = 50 µm). High magnification images are shown in the lower panel (zoom factor X4). Filled arrows indicate endothelial cells, and empty arrows indicate TICs (B). A summary of the quantification of the distance between CD133+ and CD31+ cells is provided in Figure S5. *, 0.05 > p > 0.01; **, 0.01 > p > 0.001.
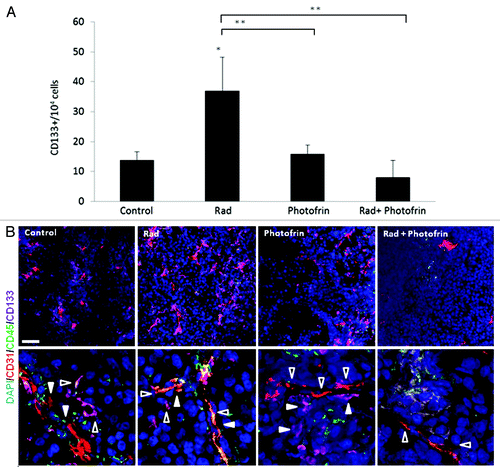
Next, we analyzed the proximity of TICs to blood vessels in all treated tumors, by staining tumor slides for murine-CD31, human-CD133 and murine-CD45 representing endothelial cells, U-87MG TICs and hematopoietic cells, respectively. The results in and Figure S5 show that TICs of U-87 MG tumors were found in close proximity to blood vessels following radiation or Photofrin-II monotherapies. However, the distance was markedly and significantly increased in tumors treated with Photofrin-II and radiation.
The combination of radiation and Photofrin-II induces TIC death
The low percentage of TICs as well as the dramatically reduced angiogenesis and vasculogenesis in tumors treated with the combination of Photofrin-II and radiation, prompted us to evaluate the viability of TICs in the re-growing tumors. To test this, sections from tumors obtained at endpoint were stained for human-CD133 and TUNEL to detect apoptotic TICs. Of note, we verified that the human CD133 does not cross-react with mouse tissue, using bone marrow cells expressing CD133 (data not shown). The results in and Figure S6 demonstrate a marked increase in the percentage and the number of CD133+TUNEL+ cells in the combination treatment group as compared with all other groups. Notably, increased number of TICs in radiotherapy-treated groups was also observed implicating the TIC resistance to such therapy (Fig. S6). These results further suggest that the combination of radiation and Photofrin-II targets tumor cells and their TIC subpopulation in U-87 MG tumors, resulting in induced tumor cell apoptosis and delayed tumor relapse.
Figure 5. Analysis of TUNEL+TICs in U-87 MG tumors following Photofrin-II and/or radiation treatment. U-87 MG tumors (described in ) were (A) sectioned and subsequently immnostained for CD133+ as a marker for TICs (violet), and TUNEL as a marker for apoptosis (green). Nuclei were stained with DAPI (blue). Images were captured using Leica TCS SP5 confocal imaging systems (scale bar = 50 µm). High magnification images are shown in the lower panel (zoom factor X3). (B) A summary of the percentage of CD133+ and TUNEL+ cells is provided. ***, p < 0.001 from control group, unless indicated otherwise.
Discussion
A growing body of evidence suggests that TICs of glioblastoma tumors can contribute to tumor growth, in part by the induction of angiogenesis. TICs were shown to be located at the perivascular site, secreting proangiogenic factors to attract endothelial cells.Citation30 In addition, C6 glioblastoma cells have been shown to promote not only angiogenesis, but also vasculogenesis mediated by different types of BMDCs.Citation32 This study revealed that targeted antiangiogenic therapy or chemotherapy administered as monotheapies did not affect the number of TICs in treated tumors, whereas the combination of an antiangiogenic drug with chemotherapy significantly reduced the number of TICs in treated tumors. These findings suggest that eradication of TICs can be achieved not only by direct TIC killing, but also by disrupting the angiogenic TIC microenvironment.Citation33 In the current study, we evaluated the impact of radiation in combination with the radiosensitizing agent, Photofrin-II, on tumor growth. To this end, we grew U-87 MG gliobalstoma tumor cells in either TIC-high (stem cell) or TIC-low (“normal”) conditions. We found that the TIC-enriched population was more resistant to both 3 Gy and 10 Gy radiation than U-87 MG cells from the TIC-low culture. This is consistent with the findings of Bao et al. who have shown that TICs of glioma tumors contribute to tumor radio-resistance via the high activation of DNA damage checkpoint kinases.Citation34 Importantly, we found that the addition of Photofrin-II to TIC-high and TIC-low radiated cells resulted in reduced cell proliferation and increased cell death, as shown by high activity of PARP in treated cells, increased number of Annexin V positive cells, and reduced number of tumor cell colonies using the soft agar colony formation assay. In this regard, it would be of interest to test a combination of Photofrin-II and PARP inhibitor since PARP was found to be highly expressed in cells following exposure to Photofrin-II. We should note that cells from TIC-high conditions were also slightly sensitive to radiation (in both radiation doses) probably due to the cultured cell methodology which only enriches for TICs but does not isolate pure TICs. Thus, it is more likely that the non-TICs in the TIC-high fraction were sensitive to the radiotherapy. Overall, the use of Photofrin-II as a radiosensitizing agent targets not only “differentiated” tumor cells, but also TICs, providing a possible mechanism to explain the clinical benefits of the combination therapy for the treatment of brain tumors (M. Schaffer, unpublished observations).
Our in vitro results prompted us to test the activity of Photofrin-II in vivo in mice bearing U-87 MG glioblastomas. We found that both radiation and Photofrin-II monotherapy groups inhibited glioblastoma tumor growth. However the combination of the two therapies increased treatment activity, and further delayed tumor growth. Although the enhanced antitumor activity of Photofrin-II has already been documented,Citation22,Citation35 in the current study we demonstrate that this treatment benefit may also be a result of TIC killing, as it markedly inhibited tumor growth over a long period of time, even after a single therapy. Consistently, TUNEL staining revealed a substantial increase in the number of apoptotic TICs in the combinatorial treatment group, providing an explanation for the delayed tumor growth. We also found that tumors treated with Photofrin-II and radiation exhibit lack of angiogenesis by means of decreased microvessel density, and decreased numbers of proangiogenic BMDCs colonizing-treated tumors when compared with radiation-treated tumors. We postulate that the elimination of TICs, which are known to induce angiogenesis in brain tumors,Citation30 accounts for the antiangiogenic effects as a result of the combination therapy. Indeed, several studies have indicated that tumor cells derived from TIC populations contain more pro-angiogenic factors than tumor cells derived from non-TIC populations.Citation33,Citation36 The proangiogenic effects of TIC-derived tumors can be abolished by anti-VEGF neutralizing antibody, suggesting that TICs are a crucial source of key angiogenic factors.Citation36 Furthermore, we found that the number of TICs increases in tumors undergoing radiotherapy, but is dramatically and significantly reduced in tumors treated with Photofrin-II and radiotherapy. Taken together, although we have based our in vivo experiments on glioblastoma tumors implanted subcutaneously and not orthotopically, our results suggest that the antitumor effect of Photofrin-II and radiation is manifested by both direct tumor cell killing and the inhibition of the TIC microenvironment that supports angiogenesis and BMDCs in treated tumors. Of note, the use of a single dose of radiation is performed in order to minimally expose mice to light during the course of the therapy, since Photofrin-II serves not only as a radiosensitizing agent but also as a photosensitizing agent. In addition, the current clinical practice using the new radiation systems allows the exposure of high-dose radiation in hypofractionation setting, which was found to increase treatment outcome in relatively short periods of treatment time, with minimal toxicity. Furthermore, the antiangiogenic effect of the combination therapy can lead to the resistance of treated tumors to additional doses of radiation during the course of the therapy. Thus, daily fractionated radiotherapy may not be the preferred treatment strategy in this setting.
The mechanism of action of Photofrin-II as a radiosensitizer is not yet clear.Citation19-Citation21 One assumption is that the radiosensitizing process of Photofrin-II may involve the oligomeric porphyrin constituents which are enriched in Photofrin-II.Citation20 Cytotoxic molecules, such as hydroxyl radicals that are generated as a result of the primary interaction of irradiation with water, may enhance the overall biological cytotoxic effects of Photofrin-II through the production of radical derivatives. These derivatives act as amplifying factors propagating the extent of the cellular damage. Additional findings from a recent study suggested that Photofrin-II reacts with OH and O2 radicals, which are generated intracellularly via the radiolysis of water.Citation21 Photofrin-II was also found to reduce the repair process which often limits the radiation-induced cellular damage, via sublethal damage. The partial damage can subsequently lead to stimulated repair mechanisms, especially in the context of TICs, which consist of increased DNA repair machinery.Citation8Thus, Photofrin-II, in addition to its tumor cell killing activity, may also reduce the repair process in TICs when combined with ionizing radiation. It therefore would be of interest to characterize and isolate additional radiosensitizing agents such as specific potent derivatives of Photofrin-II which may have antitumor/radiosensitizing activities against different tumor types, and evaluate their potency in TIC killing.
Although we have not uncovered the mechanisms by which Photofrin-II sensitizes TICs to radiotherapy, several preclinical studies on murine tumor models have shown that this treatment modality inhibits tumor re-growth. Such studies measured the efficacy of Photofrin-II as both a specific and selective radiosensitizing agent, in terms of tumor-doubling time (Δt), i.e., the time interval for the tumor volume to double when compared with the volume at the time of irradiation, the value of which was found to be substantially higher in mice exposed to radiation and Photofrin-II.Citation19-Citation21 In our study, we found that the combination of Photofrin-II and radiation increased the Δt of tumor growth, suggesting that the therapeutic impact is manifested by TIC killing. It would be of interest to test the impact of radiosensitizing agents in combination with radiation on different tumor types, and their potential in TIC eradication. Our in vitro results, using a variety of cell lines, showed that at least breast cancer may benefit from this combinatorial treatment modality by reducing the viability of both TIC and non-TIC populations. However, in our study, these possible therapeutic effects were not observed in pancreatic, lung and colon cancers, although we must stress that only a single cell line for each tumor type was evaluated. Interestingly, Photofrin-II, as a monotherapy, also inhibited tumor cell viability and delayed tumor growth. Although the reasons for these antitumor effects are not yet clear, we cannot rule out that the mice and/or tumor cells were slightly exposed to light during the experimental procedures. In such cases, the activity of Photofrin-II as a photosensitizing agent may explain, at least in part, the antitumor and anti-proliferative activity of Photofrin-II when administered as a single agent.
In summary, although the mechanisms of action of Photofrin-II and probably its derivatives in sensitizing tumor cells to radiotherapy are not yet understood, it seems that they may provide a therapeutic impact on commonly resistant tumor cells displaying TIC characteristics. Treatment of glioblastoma with a radiosensitizing agent in combination with high-dose radiation resulted in the killing of TICs, cells that usually induce tumor cell repopulation, in part, via the induction of angiogenesis. We should note, however, that our study described results only of a single cell line for each tumor type tested in vitro including a glioblastoma tumor tested in vivo. Therefore, extended studies to test the therapeutic benefit of Photofrin-II in combination with the standard treatment for glioblastoma, e.g., tamozolomide and radiation are worthy. Nevertheless, our study suggests a mechanism by which such a combinatorial treatment delays glioblastoma growth.
Materials and Methods
Cell culture
U-87 MG glioblastoma, HT29 colorectal adenocarcinoma, A549 lung carcinoma, PANC1 pancreatic carcinoma and MCF7 breast adenocarcinoma cell lines (from human origin) were purchased from the American Type Culture Collection (ATCC), and were passaged in our laboratory for fewer than 6 mo in culture. Cell lines were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 1% L-glutamine, 1% sodium-pyruvate and 1% streptomycin. All cells were cultured for no more than 4 mo after being thawed from authentic stocks.
Generation of TIC-high and TIC-low cells
All cell lines were maintained either as monolayers in DMEM with 10% fetal calf serum (FCS) (TIC-low fraction), or as non-adherent tumor spheres in serum-free DMEM (TIC-high fraction), supplemented with 1% L-glutamine, 1% sodium-pyruvate and 1% streptomycin. To obtain a TIC-enriched population (TIC-high), monolayer cells were harvested with 0.25% trypsin-EDTA and washed twice with phosphate buffered saline (PBS) to remove all FCS. Subsequently, cells were resuspended in sphere-media. For U-87 MG spheres, DMEM/F-12 containing 10 µg/ml insulin, 100 µg/ml transferrin, 40 ng/ml sodium selenite solution, 100 µg/ml BSA, 60 ng/ml progesterone, 16 µg/ml putrescine, 63 µg/ml N-acetyl cysteine, 5 µM forskolin, 10 ng/ml PDGF and 10 ng/ml bFGF was used in accordance with.Citation37 For HT29 spheres, DMEM/F-12 containing: 10 µg/ml insulin, 100 µg/ml transferrin, 40 ng/ml sodium selenite solution, 0.4% BSA, 60 ng/ml progesterone, 16 µg/ml putrescine, 20 ng/ml EGF and 10 ng/ml bFGF was used in accordance with [AUTHOR’S NOTE: Missing word?].Citation13 For A549 spheres, DMEM/F-12 containing 10 µg/ml insulin, 100 µg/ml transferrin, 40 ng/ml sodium selenite solution, 0.4% BSA, 20 ng/ml EGF and 10 ng/ml bFGF was used in accordance with [AUTHOR’S NOTE: Missing word?].Citation13 For PANC1 spheres, DMEM/F-12 containing 10 µg/ml insulin, 100 µg/ml transferrin, 40 ng/ml sodium selenite solution, 0.4% BSA, 20 ng/ml EGF and 10 ng/ml bFGF was used in accordance with [AUTHOR’S NOTE: Missing word?].Citation38 For MCF7 spheres, DMEM/F-12 containing 10 µg/ml insulin, 0.4% BSA, 20 ng/ml EGF and 10 ng/ml bFGF was used in accordance with [AUTHOR’S NOTE: Missing word?].Citation39 After approximately 10 d (depending on the cell line and growth kinetics), all the five cell lines formed spherical colonies. To propagate TICs, spheres floating in the supernatant of a week-old culture were collected and subsequently resuspended in the appropriate sphere media mentioned above. Supplemental growth factors were added to the cultured spheres every other day in order to maintain TIC enrichment for the duration of the experimental period. TIC-low cells were obtained by culturing tumor cells with DMEM and 10% FCS. In all experiments, cells were maintained in 100-mm culture dishes (Nunc) in a humidified incubator at 37°C in 5% CO2. Of note, using this method we only enriched for TICs in the TIC-high fraction but did not necessarily isolate pure TIC population. An illustration for the generation of TIC-high and TIC-low cultured cells is provided in Figure S7.
Evaluation of cell viability following radiation and Photofrin-II therapy
Cell viability was evaluated quantitatively with the metabolic indicator dye AlamarBlue (Serotec Ltd.), which determines the metabolic activity of cells and is used for cell viability and proliferation as previously described.Citation40 Cells were harvested from sub-confluent cultures and re-plated (usually 500–1,000 cells/well in a 96-well plate) in their designated medium and 10% AlamarBlue (AB) solution, in the presence of 1 µg/ml (0.166 µM) Photofrin-II, the dose of which was determined as described in ref. Citation19. In parallel experiments, tumor cells either from TIC‐high or TIC‐low conditions were irradiated with a collimated 6 MeV electron beam using Elekta Precise (Elekta Oncology Systems) linear accelerator at a dose rate of 5 Gy per minute for a total dose of either 3 Gy or 10 Gy, at room temperature (Department of Radiation Therapy, Rambam Medical Center). Results were presented as the percentage of AB reduction after 48 h, and were corrected to background values of negative controls. Results are presented as fold increase from background values. Cells were kept in the dark at all times, in order to avoid the activation of Photofrin-II as a photosensitizer agent when exposed to light. All experiments were performed in triplicate, and data were presented as means ± standard error.
Colony formation in soft agar
Two milliliters of DMEM containing 0.5% Low Melt Agarose (Bio-Rad) and 10% FCS were poured into 6-well plate to form a basic layer. The layer was covered with 0.6 mL DMEM containing 0.3% Low Melt Agarose, 10% FCS, and 2.5 × 103 of either U-87 MG TIC-low or TIC-high fractions after they underwent therapy. Medium (DMEM + 10% FCS) was exchanged every 3 d. Colonies were visualized using light microscope system after 3 wk in culture.
Evaluation of surface markers and BMDCs by flow cytometry
TIC-high or TIC-low cultures were evaluated for the presence of TICs using several surface markers. For glioblastoma, TICs were defined as CD133+. In order to identify host bone marrow-derived cells (BMDCs) colonizing tumors, tumors were prepared as single-cell suspensions and cells were stained with CXCR4+/CD11b+/VEGFR1+/CD45+ and CD11b+/Gr-1+ surface markers to identify hemangiocytes and myeloid-derived suppressor cells (MDSCs), respectively. In order to evaluate apoptosis, tumor cells from TIC-high and TIC-low fractions were immunostained with Annexin V antibody, followed by the manufacturer’s instructions (BD Biosciences). All monoclonal antibodies were purchased from BD Biosciences, R&D Systems and Macs MilitenyiBiotec, and used in accordance with the manufacturer’s instructions. At least 100,000 events were acquired using a Cyan ADP flow cytometer and analyzed with Summit software (Beckman Coulter).
Animal tumor models and drug concentrations
Five million U-87 MG cells were injected subcutaneously into the flanks of 8–10-wk-old athymic-nude mice. Tumor size was assessed regularly with Vernier calipers using the formula, widthCitation2 × length × 0.5. When tumors reached 50 mm3, mice were treated with 10 mg/kg Photofrin-II (AxcanPharma), a previously determined dosage.Citation19,Citation22 Control mice were treated with the appropriate vehicles (PBS). Mice were locally irradiated to the tumor area with a linear accelerator 6 MeV electron beam radiation using Elekta Precise (ElektaOncology Systems) at a dose rate of 5 Gy per minute for a total dose of 10 Gy at room temperature (Department of Radiation Therapy, Rambam Medical Center). Animals were kept in the dark for 48 h following the administration of Photofrin-II in order to avoid the therapeutic activation of Photofrin-II as a photosensitizer agent when exposed to light. All animal studies and experimental protocols were approved by the Animal Care and Use Committee of the Technion.
Immunostaining and immunoblotting
Tissue processing and immunohistochemistry were performed as described previously.Citation40,Citation41 Briefly, tumor cryosections (5–10 μm) were used for the analysis of microvessel density (MVD), using anti-CD31 endothelial cell specific antibody (1:200, BD Biosciences) and Cy3-conjugated secondary antibody (1:500, Jackson immunoresearch laboratories). The number of vessel structures per field were counted and plotted (approximately 5 fields/tumor, n > 20 fields/group). For the analysis of BMDC colonization of tumors, a FITC-conjugated antibody against mouse-CD45, a panhematopoietic marker was used (1:150, BD Biosciences). A primary antibody against CD133 to identify prominin-1 (1:250, Macs MiltenyiBiotec), and a Cy5-conjugated secondary antibody (1:500, Jackson immunoresearch laboratories) were used for identifying TICs of U-87 MG. Apoptotic cells were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining (Roche Diagnostics). Nuclei were stained with 6-diamidino-2-phenylindole (DAPI). Tumor sections were visualized by either Leica CTR 6000 microscope or Leica TCS SP5 confocal imaging systems. All images were taken using a multi-track channel acquisition to prevent emission cross-talk between fluorescent dyes. Single XY plane-images were acquired in 1,024 × 1,024 resolution. In order to evaluate the proximity between blood vessels (CD31+) and TICs (CD133+), the Leica application suite V3 software was used. Using micrographs of X400 magnification, the shortest distance between the center of a TIC (CD133+) and the center of a blood vessel (CD31+) was calculated. Measurements were performed on at least 10 fields per group, and a summary of the distance between TICs and blood vessel was provided. For immunoblotting of active PARP and Actin, TIC-high or TIC-low cultured cell extracts were lysed and then determined for the protein concentration. Fifty μg of protein was loaded on SDS-PAGE gel. After the proteins were transferred to PVDF membrane, they were probed with cleaved-PARP (1:1,000, Abcam) and actin (1:5,000, Sigma Aldrich) antibodies, followed by HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories).
Statistical analysis
Data are presented as means ± standard deviation. Statistical significance differences in mean values was assessed by one way ANOVA, followed by Newman-Keuls ad hoc statistical test using GraphPad Prism 4 software and were considered significant at values of *p < 0.05, **p < 0.01, and ***p < 0.001.
Additional material
Download Zip (842.4 KB)Acknowledgments
This work was supported by research grants from the Israeli Ministry of Health, Israel Science Foundation, European Commission under FP7 program (Marie Curie, no. 239212). L.B. was supported by Israel Student Education Foundation (ISEF), Fine and Jacobs studentships.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008; 359:492 - 507; http://dx.doi.org/10.1056/NEJMra0708126; PMID: 18669428
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987 - 96; http://dx.doi.org/10.1056/NEJMoa043330; PMID: 15758009
- Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol 2008; 26:2192 - 7; http://dx.doi.org/10.1200/JCO.2007.14.8163; PMID: 18445844
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645 - 8; http://dx.doi.org/10.1038/367645a0; PMID: 7509044
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105 - 11; http://dx.doi.org/10.1038/35102167; PMID: 11689955
- Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 2003; 3:895 - 902; http://dx.doi.org/10.1038/nrc1232; PMID: 14737120
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755 - 68; http://dx.doi.org/10.1038/nrc2499; PMID: 18784658
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275 - 84; http://dx.doi.org/10.1038/nrc1590; PMID: 15803154
- Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov 2009; 8:806 - 23; http://dx.doi.org/10.1038/nrd2137; PMID: 19794444
- Press MF, Lenz HJ. EGFR, HER2 and VEGF pathways: validated targets for cancer treatment. Drugs 2007; 67:2045 - 75; http://dx.doi.org/10.2165/00003495-200767140-00006; PMID: 17883287
- Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 2005; 5:516 - 25; http://dx.doi.org/10.1038/nrc1650; PMID: 15965493
- Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009; 457:51 - 6; http://dx.doi.org/10.1038/nature07618; PMID: 19122635
- Benayoun L, Gingis-Velitski S, Voloshin T, Segal E, Segev R, Munster M, et al. Tumor-initiating cells of various tumor types exhibit differential angiogenic properties and react differently to antiangiogenic drugs. Stem Cells 2012; 30:1831 - 41; http://dx.doi.org/10.1002/stem.1170; PMID: 22782858
- Edward CH, Carlos AP, Luther WB. Chemical Modifiers of Radiation Response. 2008;
- Kostron H, Fritsch E, Grunert V. Photodynamic therapy of malignant brain tumours: a phase I/II trial. Br J Neurosurg 1988; 2:241 - 8; http://dx.doi.org/10.3109/02688698808992675; PMID: 2855780
- Overholt BF, Panjehpour M. Photodynamic therapy in Barrett’s esophagus. J Clin Laser Med Surg 1996; 14:245 - 9; PMID: 9612190
- Topazian M, Zhong N, Baron TH, Vege SS, Wang KK. Photodynamic therapy of intraductal papillary mucinous neoplasm. Endoscopy 2012; 44:213 - 5; http://dx.doi.org/10.1055/s-0031-1291539; PMID: 22271032
- Luksiene Z, Juzenas P, Moan J. Radiosensitization of tumours by porphyrins. Cancer Lett 2006; 235:40 - 7; http://dx.doi.org/10.1016/j.canlet.2005.03.041; PMID: 15946797
- Schaffer M, Schaffer PM, Corti L, Gardiman M, Sotti G, Hofstetter A, et al. Photofrin as a specific radiosensitizing agent for tumors: studies in comparison to other porphyrins, in an experimental in vivo model. J Photochem Photobiol B 2002; 66:157 - 64; http://dx.doi.org/10.1016/S1011-1344(02)00237-3; PMID: 11960724
- Kulka U, Schaffer M, Siefert A, Schaffer PM, Olsner A, Kasseb K, et al. Photofrin as a radiosensitizer in an in vitro cell survival assay. Biochem Biophys Res Commun 2003; 311:98 - 103; http://dx.doi.org/10.1016/j.bbrc.2003.09.170; PMID: 14575700
- Schaffer M, Ertl-Wagner B, Schaffer PM, Kulka U, Jori G, Wilkowski R, et al. Feasibility of photofrin II as a radiosensitizing agent in solid tumors--preliminary results. Onkologie 2006; 29:514 - 9; http://dx.doi.org/10.1159/000095979; PMID: 17068386
- Schaffer M, Ertl-Wagner B, Schaffer PM, Kulka U, Jori G, Dühmke E, et al. The Application of Photofrin II as a sensitizing agent for ionizing radiation--a new approach in tumor therapy?. Curr Med Chem 2005; 12:1209 - 15; http://dx.doi.org/10.2174/0929867053764653; PMID: 15892632
- Orenstein A, Kostenich G, Roitman L, Shechtman Y, Kopolovic Y, Ehrenberg B, et al. A comparative study of tissue distribution and photodynamic therapy selectivity of chlorin e6, Photofrin II and ALA-induced protoporphyrin IX in a colon carcinoma model. Br J Cancer 1996; 73:937 - 44; http://dx.doi.org/10.1038/bjc.1996.185; PMID: 8611429
- Bellnier DA, Ho YK, Pandey RK, Missert JR, Dougherty TJ. Distribution and elimination of Photofrin II in mice. Photochem Photobiol 1989; 50:221 - 8; http://dx.doi.org/10.1111/j.1751-1097.1989.tb04152.x; PMID: 2528753
- Gomer CJ, Ferrario A. Tissue distribution and photosensitizing properties of mono-L-aspartyl chlorin e6 in a mouse tumor model. Cancer Res 1990; 50:3985 - 90; PMID: 2354446
- Karimi-Busheri F, Rasouli-Nia A, Mackey JR, Weinfeld M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res 2010; 12:R31; http://dx.doi.org/10.1186/bcr2583; PMID: 20525204
- Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 2010; 120:694 - 705; http://dx.doi.org/10.1172/JCI40283; PMID: 20179352
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol 2007; 25:911 - 20; http://dx.doi.org/10.1038/nbt1323; PMID: 17664940
- Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 2006; 12:557 - 67; http://dx.doi.org/10.1038/nm1400; PMID: 16648859
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11:69 - 82; http://dx.doi.org/10.1016/j.ccr.2006.11.020; PMID: 17222791
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004; 432:396 - 401; http://dx.doi.org/10.1038/nature03128; PMID: 15549107
- Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, et al. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res 2009; 69:7243 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-09-0167; PMID: 19738068
- Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res 2007; 67:3560 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-06-4238; PMID: 17440065
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756 - 60; http://dx.doi.org/10.1038/nature05236; PMID: 17051156
- Schaffer M, Schaffer PM, Corti L, Gardiman M, Sotti G, Hofstetter A, et al. Photofrin as a specific radiosensitizing agent for tumors: studies in comparison to other porphyrins, in an experimental in vivo model. J Photochem Photobiol B 2002; 66:157 - 64; http://dx.doi.org/10.1016/S1011-1344(02)00237-3; PMID: 11960724
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 2006; 66:7843 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-1010; PMID: 16912155
- Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA 2004; 101:781 - 6; http://dx.doi.org/10.1073/pnas.0307618100; PMID: 14711994
- Gou S, Liu T, Wang C, Yin T, Li K, Yang M, et al. Establishment of clonal colony-forming assay for propagation of pancreatic cancer cells with stem cell properties. Pancreas 2007; 34:429 - 35; http://dx.doi.org/10.1097/MPA.0b013e318033f9f4; PMID: 17446842
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 2005; 65:5506 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-05-0626; PMID: 15994920
- Voloshin T, Gingis-Velitski S, Bril R, Benayoun L, Munster M, Milsom C, et al. G-CSF supplementation with chemotherapy can promote revascularization and subsequent tumor regrowth: prevention by a CXCR4 antagonist. Blood 2011; 118:3426 - 35; http://dx.doi.org/10.1182/blood-2010-11-320812; PMID: 21685373
- Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science 2006; 313:1785 - 7; http://dx.doi.org/10.1126/science.1127592; PMID: 16990548