Abstract
Primitive neuroectodermal tumors (PNET) arising directly from the lung are very rare but particularly aggressive neoplasms. We report a case of a 31-y-old man with primary pulmonary neuroectodermal tumor. We review the clinical as well as pathological features. As typical for these tumors, the diagnosis was initially delayed in our patient and prognosis was poor despite aggressive surgical resection, postoperative chemotherapy and local irradiation. Recent biological insights have revealed unique chromosomal translocations crucial to the pathogenesis of these tumors, most notably the EWS-FLI-1 translocation. We provide an overview of the molecular features of the Ewing Sarcoma Family of Tumors (ESFT) including PNET and their potential implications for therapeutic targeting.
Introduction
Primitive neuroectodermal tumors (PNET) and Ewing sarcoma (EWS) belong to the same family of malignant, small, round-cell neoplasms of soft tissue or bone origin. Common locations for EWS-PNET include chest wall, pelvis and extremities. EWS-PNETs that arise in the lung parenchyma without chest wall involvement are extremely rare in adults. We report a case of EWS-PNET of the lung in an adult and review the distinct clinical, pathological and molecular features of these tumors.
Clinical Course
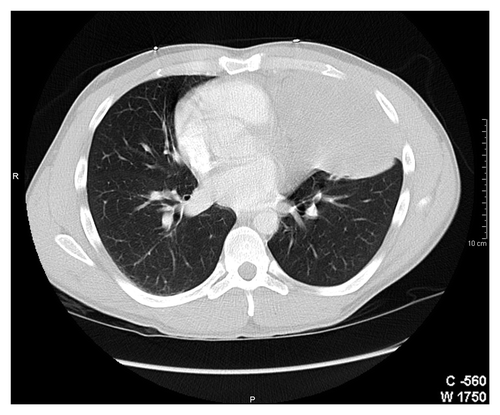
A 31-y-old Hispanic man presented with hemoptysis. His past medical history was remarkable for childhood asthma, a spontaneous pneumothorax and a remote history of smoking. At the time of his initial presentation in 2006, he was evaluated with chest radiography and a CT (CT) scan that showed a 2 cm nodule in the left lung, involving lingula. He underwent a fine needle biopsy of this nodule. However, histopathological examination was inconclusive showing only fibrosis. Fiberoptic bronchsoscopic biopsy was recommended to fully characterize this nodule. Unfortunately, the patient was lost to follow up until end of 2007 when presented with recurrent hemoptysis associated with left sided chest pain and fever. This time chest X-ray showed left lung opacity (), and a repeat CT scan of the chest revealed a well-defined dense mass in the lingula of the left lung measuring 10.7 × 7 × 11.4 cm ().
Figure 1. Chest radiogram of a 31-y-old man with primary pulmonary primitive tumor (PNET) in the left lung (lingula).
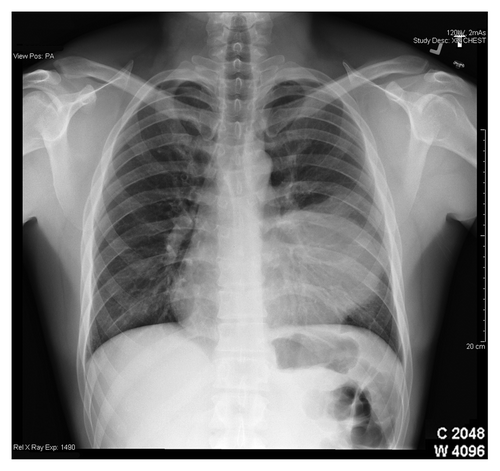
Figure 2. Computer tomographic scan of a 31-y-man with primary pulmonary primitive tumor (PNET) in the left lung.
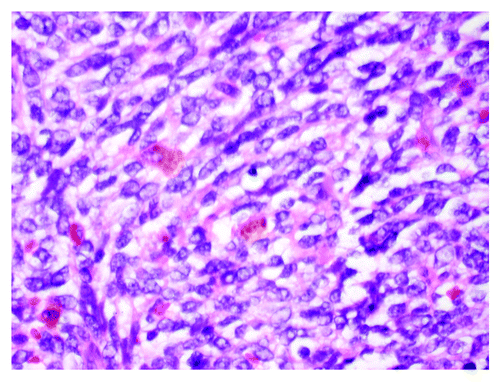
A CT-guided core needle biopsy of this mass was inconclusive, but suggestive of a neoplasm. Histopathological examination showed fibrosis, with focal small-cell keratin-negative infiltrate. Immunohistochemical staining for keratin, EMA, S100, CD34, desmin were negative, positive for NSE and synaptophysin with an equivocal CD99 staining, suspicious for malignant small blue-cell neoplasm. To establish the final diagnosis, the patient underwent posterolateral thoracotomy with resection of the mass without lobectomy. Gross pathological examination showed a large mass in the linguala, which was adherent to the chest wall anteriorly and to the pericardium medially with extensive central necrosis and hemorrhage (). The pleural surface was smooth, pink and focally congested with a few adhesions. The soft tissue overlying the mass was congested with diffuse, red adhesions. For diagnosis confirmation, the tissue was evaluated for t(11:22) translocation. Final results showed positive staining for CD99 () and a chimeric fusion translocation for EWSR1-ERG consistent with pulmonary EWS-PNET. Post-surgery follow-up CT chest showed a residual opacity that was 5.8 cm in transverse, and 4.7 cm in antero-posterior, 9.5 cm in craniocaudal dimensions. These radiographic changes were interpreted as postoperative in nature, although the persistent tumor could not have been completely excluded. There was no mediastinal lymphadenopathy. There were no lytic or blastic osseous lesions noted on chest CT scan, with the exception of posterior left seven fractures from thoracotomy and a whole-body nuclear bone scan showed no scintigraphic evidence of bone metastasis. A CT scan of the abdomen and pelvis showed no evidence of metastatic disease. Following the completion of staging studies, the decision was made to start aggressive systematic chemotherapy and adjuvant RT considering the inability to obtain clear surgical margins, as well as the high probability of recurrence. The chemotherapy regimen consisted of alternating cycles every 3 wk of cyclophosphamide, doxorobucin and vincristine, with isofosfamide and etoposide, respectively, in conjunction with growth factor support.
Figure 3. Well demarcated primary pulmonary primitive tumor (PNET) with extensive tumor necrosis inside.
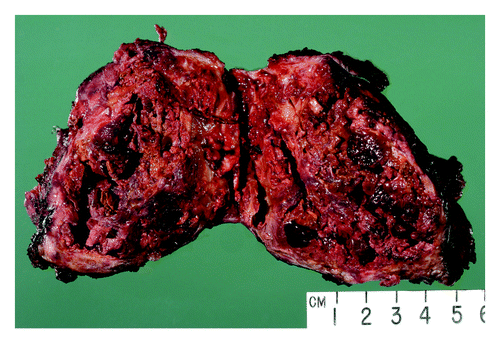
Figure 4. HH&E stained section of a small round cell lung tumor-primary pulmonary tumor (PNET).
After completing four cycles of chemotherapy, the patient started external beam irradiation therapy of the left hemi-thorax including: chest wall, pericardium and hilum, which were completed within 6 mo after surgery. He completed a total of 11 cycles of chemotherapy over the period of 1 y. He underwent serial CT chest for restaging that showed stable left lingular mass with no evidence of recurrent lung malignancy. Hence, during this time, he continued to have progressive worsening of chest pain at the thoracotomy site, and recurrent lung infections. Two months later following chemotherapy, CT of the chest showed a left-side pleural mass that was increasing in size, a left pleural effusion and a new right middle lung nodule. A core needle biopsy of the pleural mass confirmed that tumor relapsed.
He had progression of his disease with development of lesions at both the chest wall and within the lung parenchyma despite trying multiple palliative chemotherapy regimens that included cyclophosphamide, topotecan, carboplatin and gemcitabine, radiotherapy and docetaxel and patient succumbed to his disease after 3 y.
Discussion
In 1918, Stout et al.Citation1 reported an extracranial PNET originating in the ulnar nerve of an adult patient. Ewing sarcoma (EWS), initially named diffuse endothelioma, was first described in 1921 by James Ewing, who reported a case of highly malignant tumor in the diaphysis of long bones in a 14-y-old girl.Citation2 In 1979, Askin et al.Citation3 described a similar rare malignant small-cell tumor of the thoracopulmonary region in 20 children and adolescents with a mean age of 14 y, referred to as Askin’s tumor. ES, extra-osseous Ewing tumors (EES), PNET and Askin’s tumors (PNET of the chest wall) have been categorized as members of the Ewing’s sarcoma family of tumors (ESFT). Most commonly seen in children and young adults, these highly malignant tumors of bone and soft tissue are histopathologically described as poorly differentiated “small round cell tumors.” ESFT are tumors of neural crest derivation showing varying degrees of neuroectodermal differentiation.Citation4 Current evidence indicates that ES and PNET represent a single neoplastic entity, differing only in their degree of neural differentiation. Tumors that demonstrate neural differentiation by these analyses have been traditionally named PNETs, and those that are undifferentiated have been called Ewing's sarcoma.Citation5
Clinical symptoms depend on the site of presentation but invariably include pain and swelling of the surrounding structures due to mass effect. Other reported symptoms and signs are site specific, such as hemoptysis with lung involvement. Definitive diagnosis of primary pulmonary EWS-PNET is based on a broad combination of imaging tests and information obtained with light microscopy, immunohistochemical staining and cytogenetic analysis. Imaging tests include chest radiographs, CT scans and MRI to determine the extent of tumor involvement. On CT scans, EWS-PNETs appear as heterogeneous masses, often invading surrounding tissues, including bone. MRI typically reveals a mass isointense to muscle on T1-weighted images, while hyperintense on T2-weighted images. Most of the tumors located in the chest show necrosis and multilocular cysts, while some of the tumors appear as homogeneous solid masses.Citation6 Imaging tests can be used to guide biopsy of identified masses. On light microscopy, EWS-PNETs appear as small round blue cells with hyperchromatic nuclei and scant cytoplasm, associated with extensive necrosis. Immunohistochemically, EWS-PNETs typically co-express CD99 (the glycoprotein MIC2) and vimentin. MIC2 expression has a high sensitivity for diagnosis of EWS-PNET family of tumors, but it lacks specificity in that many other tumors express MIC2.Citation7 Other nonspecific markers include S-100, neuron-specific enolase, CD75 and synaptophysin. Because PNETs are uncommon yet highly aggressive, a high index of suspicion is required for timely diagnosis and prompt initiation of treatment. The treatment of PNETs should be aggressive consisting of early surgical resection followed by a combination of intense chemotherapy and high-dose radiation therapy. The most compelling evidence suggests chemotherapeutic five-drug regimens that include alternating cycles of vincristine, doxorubicin and cyclophosphamide with ifosfamide and etoposide. It has been shown that patients with loco-regional disease have an improved survival with dose-intensified chemotherapy.Citation8 Prognosis is mainly related to the ability to achieve disease-free surgical margins and the extent of anatomical spread to surrounding structures such as bone, pleura, and the epidural space. Our patient had pleural involvement without bone extension. Historically, outcome has been poor because of the limitations of traditional chemotherapeutic approaches.Citation9 This case highlights the importance of early diagnosis and treatment of primary pulmonary PNETs. It also demonstrates the shortcomings of traditional chemotherapeutic agents in altering the aggressive natural history of these tumors. In the last few years, emerging insights into the molecular biology of these tumors have opened the door to the development of targeted therapies needed to improve the prognosis of these rare aggressive tumors.
Molecular biology of ESFTs
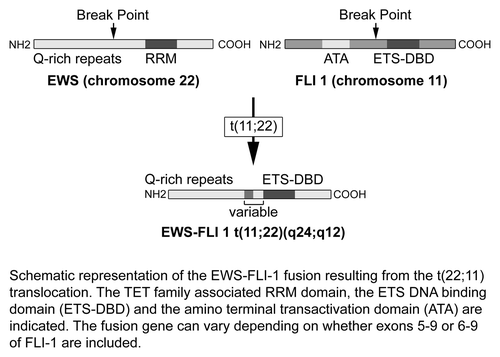
ESFTs are characterized by a specific balanced chromosomal translocation which leads to classification of these tumors as a single entity with a spectrum of phenotypic differentiation. The most common and studied fusion genes are EWS and FLI1. These translocations interrupt specific genes and recombine them to create novel fusion genes that encode tumor-specific proteins ().Citation10
Figure 5. Schematic diagram of EWS-FLI1 gene fusion resulting from t(22;11) translocation. The TET family associated RRM domain, the ETS DNA-binding domain (ETS-DBD) and the N-terminal transactivation domain (ATA) are shown. The fusion gene can vary depending on whether exons 5–9 or 6–9 of FLI1 are included. Adapted with permission from ref. Citation10.
Ewing sarcoma (EWS) gene and protein
The EWS gene is located on band q12 of human chromosome 22, it spans about 40 kb of DNA encoding 17 exon.Citation11,Citation12 The EWS protein is part of the TET family of proteins, together with the Translocated in liposarcoma (TLS) and TATA-binding protein-associated factor 15.Citation13 TET proteins share a characteristic 87-amino acid ribonucleoprotein consensus sequence (RNP-CS)/RNA recognition motif (RRM) domain that plays a key role in protein-RNA binding.Citation14 On one end, TET proteins possess a glutamine rich N-terminal region that fuses to ETS genes in ESFT. On the other end, at the C terminal, TET proteins have a distinctive region with a variable number of RGG (arginine-glycine-glycine) residues thought to mediate interactions with RNA or single-stranded DNA.Citation15 Functionally TET proteins are involved in transcription and mRNA processing by binding components of both transcription and splicing regulators. EWS is ubiquitously expressed in mammalian cells and binds to transcription products of RNA polymerase II, thus having a pivotal role in basic transcriptional regulation.Citation16
Friend leukemia integration 1 (FLI-1) gene and protein
FLI-1 gene is a member of the ets family of genes. FLI-1 maps to the mouse chromosome region syntonic with human chromosome 11q23.Citation17 This is mainly expressed in hematopoietic cells, both during development and in adult tissues with a lower expression levels in non-hematopoietic adult tissues, such as lung, heart and ovaries. It was identified proximal to the insertion of Friend’s murine leukemia virus as well as other viruses.Citation18,Citation19 The human FLI-1 gene codes for a 452-residue protein consisting of two domains separated by a FLI-1-specific domain (FLS); the 3′ region encodes for an 89-amino acid carboxyterminal trans-activation (CTA) domain involved in DNA binding; the 5′ ets and FLS regions encode for N-terminal transcriptional activation (ATA) domain.Citation20,Citation21 FLI-1 plays a pivotal role in the regulation of hematopoiesis and vasculogenesis. FLI-1 affects the normal balance between differentiations and proliferation in primary erythroblasts, inducing malignant transformation; the main mechanism is induction of Bcl2 expression and further enhancement of cell survival. In addition, FLI-1 inhibits the expression of retinoblastoma protein at the transcriptional level, further promoting the G1 and S transitions of the cell cycle.Citation22
Unique genetic translocations
A common genetic abnormalities involving fusion of the EWS gene on chromosome 22 and one of the several members of the ETS family genes constitute the genetic hallmark of ESFTs. In 85% of cases, the gene fusion is the result of a balanced chromosomal translocation between EWS gene and FLI-1 t(11;22)(q24;q12).Citation23 In 5–10% of cases 10% of EWS-PNETs, there is a gene fusion resulting from translocation between EWS and ERG (21q22), which is another member of the ETS family of transcription factors closely related to FLI1.Citation24 Several other translocations are rarely seen. EWS-ERG was initially thought to be a simple balanced reciprocal translocation; however, further investigation using systematic analysis with locus-specific probes identified the breakpoints within chromosome 21 and 22. These breakpoints are required for the excision and exchange of both 3′ERG and 3′EWS, that results in the formation of the EWS-ERG fusion gene present on the der(22).Citation25 Additional acquired chromosomal and genomic abnormalities were observed also, highlighting the complex chromosomal rearrangements of EWS-ERG Ewing sarcoma (ES) gene. Combinations of EWS and ETS family drive ontogenesis in ESFTs. EWS rearrangements with other genes result in different types of sarcomas emphasizing that it is the partner gene, not EWS that drives the final tumor pathology. In fact, rare fusions that do not even involve EWS (CIC/DUX4, BRD4/NUT) were found to induce similar molecular changes to EWS/ETS fusions. A new rare translocation that fuses EWS to NFATc2 was also identified.Citation26
EWS-FLI-1 fusion protein
The EWS-FLI1 fusion transcript encodes an oncoprotein with two primary domains, the N-terminal domain of EWS, which is a potent transcriptional activator and the C-terminal region of FLI1, which harbors the DNA binding domain.Citation27 EWS-FLI1 and other EWS-ETS fusions are multifunctional: activating or repressing multiple target genes.Citation28 The list of both direct and indirect genomic and proteomic targets for EWS-FLI1 is growing; one of the most critical targets is transcription factor NKX2–2, which acts as a transcriptional repressor. EWS-FLI-1 upregulates NKX2–2 mediating a downregulated transcriptional signature that results in oncologic transformation. Both EWS-FLI1 domains are important for oncogenic transformation of target cells, as deletion of either domain results in complete loss of oncogenic activity.Citation29,Citation30
Diagnostic and prognostic implications
Many molecular techniques can be used to detect translocations associated with EWS-PNETs, including fluorescent in situ hybridization, Southern blotting, DNA-based polymerase chain reaction (PCR), RNA-based PCR, also called reverse transcriptase-PCR (RT-PCR). The mainstay in molecular diagnosis is RT-PCR, because of high sensitivity and specificity.Citation31 Better outcomes were observed in patients with localized tumors that express the most common translocation, EWS exon 7 fused to FLI1 exon 6, compared with other less common fusion types.Citation32 The heterogeneity in EWS-PNETs prognosis correlates with molecular heterogeneity that resulting from changes either in the fusion partner (FLI1, ERG, ETV1, E1A, or FEV), or the breakpoint location within the genes. TP53 mutation, del1p, homogenous deletion of CDKN2A is associated with poor prognosis.Citation33
EWS-FLI-1 as a therapeutic target
The limitations of traditional chemotherapeutic approaches have fueled the search for novel targeted therapies to improve clinical outcomes. The elucidation of the EWS-FLI1 translocation and its cellular functions presents a unique opportunity for therapeutic targeting. EWS and ETS fusions play a pivotal role in ESFTs’ pathogenesis, thus referred to as the “perfect target without a therapeutic agent.”Citation34 Different therapeutic strategies targeting EWS-FLI1 are currently under investigation. In vitro experiments show that simultaneous administration of EWS/FLI-1-targeted antisense oligonucleotides and rapamycin induces apoptosis of ES cells. In vivo experiments with the antisense/rapamycin combination show inhibition of the growth of the ESFT xenografts in mice.Citation35
Another promising strategy involves the use of small interfering RNA (siRNA). An siRNA molecule was designed with an aromatic compound at the 3′-end to target the breakpoint of EWS/Fli1 fusion gene. In vitro experiments showed that in ES cells, this siRNA molecule significantly suppresses the expression of the EWS/Fli1 protein sequence and concomitantly reduces c-Myc protein expression. The inhibition of proliferation of ES cells was further demonstrated in vivo by using Ewing sarcoma cell line subcutaneously xenografted into mice.Citation36 More research is needed on the safety and efficacy of these novel targeted approaches for treatment of ESFT in humans.
Conclusions
We present the rare case of a patient with pulmonary PNET, a tumor from the ESFTs. Based on the small number of cases (), PNETs are rare aggressive neoplasms with a very high rate of local recurrence. Despite aggressive therapy, our patient rapidly succumbed to the tumor. Improving understanding of the molecular pathogenesis underlying ESFTs especially the EWS-FLI1 translocation presents unique opportunities for therapeutic targeting. More research is needed to translate these basic discoveries from the bench to therapies that would result in clinical benefit for patients afflicted with these aggressive malignant soft tissue tumors.
Table 1. A literature summary of case reports of Primary Primitive Neuroectodermal Tumor (PNET) of the lung/chest wall
| Abbreviations: | ||
| PNET | = | primary primitive neuroectodermal tumor |
| ES | = | Ewing’s sarcoma |
| EES | = | extra-osseous Ewing tumor |
| ESFT | = | Ewing’s sarcoma family of tumors |
| CT | = | computed tomography |
| RT | = | radiotherapy |
| PCR | = | polymerase chain reaction |
| RT | = | reverse transcriptase |
| siRNA | = | small interferering RNA |
| TLS | = | translocated in liposarcoma |
| FLI-1 | = | Friend leukemia integration 1 |
Acknowledgments
The authors wish to thank Blackman J. for his assistance with figures in the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Stout AP. A tumor of the ulnar nerve. Proc NY Pathol Soc 1918; 18:2-12.
- Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J Clin 1972; 22:95 - 8; http://dx.doi.org/10.3322/canjclin.22.2.95; PMID: 4622125
- Askin FB, Rosai J, Sibley RK, Dehner LP, McAlister WH. Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinicopathologic entity of uncertain histogenesis. Cancer 1979; 43:2438 - 51; http://dx.doi.org/10.1002/1097-0142(197906)43:6<2438::AID-CNCR2820430640>3.0.CO;2-9; PMID: 222426
- Scurr M, Judson I. How to treat the Ewing’s family of sarcomas in adult patients. Oncologist 2006; 11:65 - 72; http://dx.doi.org/10.1634/theoncologist.11-1-65; PMID: 16401715
- Kumar V, Fausto N, Abbas A. Robbins & Cotran Pathologic Basis of Disease Seventh Edition, 2004.
- Zhang WD, Xie CM, Mo YX, Li JY. [CT and MRI features of peripheral primitive neuroectodermal tumor]. Ai Zheng 2007; 26:643 - 6; PMID: 17562273
- de Alava E, Gerald WL. Molecular biology of the Ewing’s sarcoma/primitive neuroectodermal tumor family. J Clin Oncol 2000; 18:204 - 13; PMID: 10623711
- Kolb EA, Kushner BH, Gorlick R, Laverdiere C, Healey JH, LaQuaglia MP, et al. Long-term event-free survival after intensive chemotherapy for Ewing’s family of tumors in children and young adults. J Clin Oncol 2003; 21:3423 - 30; http://dx.doi.org/10.1200/JCO.2003.10.033; PMID: 12972518
- Jürgens H, Bier V, Harms D, Beck J, Brandeis W, Etspüler G, et al. Malignant peripheral neuroectodermal tumors. A retrospective analysis of 42 patients. Cancer 1988; 61:349 - 57; http://dx.doi.org/10.1002/1097-0142(19880115)61:2<349::AID-CNCR2820610226>3.0.CO;2-0; PMID: 3334970
- Riggi N, Stamenkovic I. The Biology of Ewing sarcoma. Cancer Lett 2007; 254:1 - 10; http://dx.doi.org/10.1016/j.canlet.2006.12.009; PMID: 17250957
- Ohno T, Ouchida M, Lee L, Gatalica Z, Rao VN, Reddy ES. The EWS gene, involved in Ewing family of tumors, malignant melanoma of soft parts and desmoplastic small round cell tumors, codes for an RNA binding protein with novel regulatory domains. Oncogene 1994; 9:3087 - 97; PMID: 8084618
- Plougastel B, Zucman J, Peter M, Thomas G, Delattre O. Genomic structure of the EWS gene and its relationship to EWSR1, a site of tumor-associated chromosome translocation. Genomics 1993; 18:609 - 15; http://dx.doi.org/10.1016/S0888-7543(05)80363-5; PMID: 8307570
- Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol 2009; 1:82 - 92; http://dx.doi.org/10.1093/jmcb/mjp025; PMID: 19783543
- Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J 1996; 15:5022 - 31; PMID: 8890175
- Arvand A, Denny CT. Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene 2001; 20:5747 - 54; http://dx.doi.org/10.1038/sj.onc.1204598; PMID: 11607824
- Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 1998; 17:603 - 10; http://dx.doi.org/10.1038/sj.onc.1201964; PMID: 9704926
- Prasad DDK, Rao VN, Reddy ES, Reddy P. Structure and expression of human Fli-1 gene. Cancer Res 1992; 52:5833 - 7; PMID: 1394211
- Ben-David Y, Giddens EB, Letwin K, Bernstein A. Erythroleukemia induction by Friend murine leukemia virus: insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1. Genes Dev 1991; 5:908 - 18; http://dx.doi.org/10.1101/gad.5.6.908; PMID: 2044959
- Blair DG, Athanasiou M. Ets and retroviruses - transduction and activation of members of the Ets oncogene family in viral oncogenesis. Oncogene 2000; 19:6472 - 81; http://dx.doi.org/10.1038/sj.onc.1204046; PMID: 11175363
- Rao VN, Ohno T, Prasad DD, Bhattacharya G, Reddy ES. Analysis of the DNA-binding and transcriptional activation functions of human Fli-1 protein. Oncogene 1993; 8:2167 - 73; PMID: 8336942
- Truong AH, Ben-David Y. The role of Fli-1 in normal cell function and malignant transformation. Oncogene 2000; 19:6482 - 9; http://dx.doi.org/10.1038/sj.onc.1204042; PMID: 11175364
- Pereira R, Quang CT, Lesault I, Dolznig H, Beug H, Ghysdael J. FLI-1 inhibits differentiation and induces proliferation of primary erythroblasts. Oncogene 1999; 18:1597 - 608; http://dx.doi.org/10.1038/sj.onc.1202534; PMID: 10102630
- Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, et al. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet 1988; 32:229 - 38; http://dx.doi.org/10.1016/0165-4608(88)90285-3; PMID: 3163261
- Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, et al. The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331:294 - 9; http://dx.doi.org/10.1056/NEJM199408043310503; PMID: 8022439
- Maire G, Brown CW, Bayani J, Pereira C, Gravel DH, Bell JC, et al. Complex rearrangement of chromosomes 19, 21, and 22 in Ewing sarcoma involving a novel reciprocal inversion-insertion mechanism of EWS-ERG fusion gene formation: a case analysis and literature review. Cancer Genet Cytogenet 2008; 181:81 - 92; http://dx.doi.org/10.1016/j.cancergencyto.2007.11.002; PMID: 18295659
- Jedlicka P. Ewing Sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol 2010; 3:338 - 47; PMID: 20490326
- Downing JR, Head DR, Parham DM, Douglass EC, Hulshof MG, Link MP, et al. Detection of the (11;22)(q24;q12) translocation of Ewing’s sarcoma and peripheral neuroectodermal tumor by reverse transcription polymerase chain reaction. Am J Pathol 1993; 143:1294 - 300; PMID: 8238248
- Ordonez JL, Osuna D, Herrero D. Enrique de Alava, Madoz-Gurpide J. Advances in Ewing’s Sarcoma Research: Where are you now and what lies ahead?. Cancer Res 2009; 69:7140 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-08-4041; PMID: 19738075
- Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 1998; 17:603 - 10; http://dx.doi.org/10.1038/sj.onc.1201964; PMID: 9704926
- May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci USA 1993; 90:5752 - 6; http://dx.doi.org/10.1073/pnas.90.12.5752; PMID: 8516324
- Ladanyi M. The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 1995; 4:162 - 73; http://dx.doi.org/10.1097/00019606-199509000-00003; PMID: 7493135
- Lin PP, Brody RI, Hamelin AC, Bradner JE, Healey JH, Ladanyi M. Differential transactivation by alternative EWS-FLI1 fusion proteins correlates with clinical heterogeneity in Ewing’s sarcoma. Cancer Res 1999; 59:1428 - 32; PMID: 10197607
- Hattinger CM, Rumpler S, Strehl S, Ambros IM, Zoubek A, Pötschger U, et al. Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 1999; 24:243 - 54; http://dx.doi.org/10.1002/(SICI)1098-2264(199903)24:3<243::AID-GCC10>3.0.CO;2-A; PMID: 10451705
- Uren A, Toretsky JA. Ewing’s sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol 2005; 1:521 - 8; http://dx.doi.org/10.2217/14796694.1.4.521; PMID: 16556028
- Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V. Combined transcriptional and translational targeting of EWS/FLI-1 in Ewing’s sarcoma. Clin Cancer Res 2006; 12:6781 - 90; http://dx.doi.org/10.1158/1078-0432.CCR-06-0609; PMID: 17121899
- Takigami I, Ohno T, Kitade Y, Hara A, Nagano A, Kawai G, et al. Synthetic siRNA targeting the breakpoint of EWS/Fli-1 inhibits growth of Ewing sarcoma xenografts in a mouse model. Int J Cancer 2011; 128:216 - 26; http://dx.doi.org/10.1002/ijc.25564; PMID: 20648560