Abstract
The early observations by Dr Otto Warburg revealed that fundamentally metabolic differences exist between malignant tumor cells and adjacent normal cells. Many studies have further reported the relationship between altered cellular metabolism and therapeutic outcomes. These observations suggest that targeting the peculiar metabolic pathways in cancer might be an effective strategy for cancer therapy. In recent years, investigations have accelerated into how altered cellular metabolism promotes tumor survival and growth. This review highlights the current concepts of altered metabolism in cancer and the molecular targets involved in glycolysis, mitochondria and glutamine metabolism and discusses future perspective of cellular metabolism-based cancer treatment.
Introduction
The tendency of cancer research during the past decades has been focused on the gain of function of oncogenes and loss of function of tumor-suppressor genes. Although this paradigm provided major insights into cancer biology, it is now clear that there are additional factors contributing to carcinogenesis that cannot be ignored. Hanahan and Weinberg recently updated their review published in 2000 on the hallmarks of cancer by incorporating the remarkable progress in cancer research that was made over the last decade, one of which is “reprogramming of energy metabolism”.Citation1 Cancer metabolism has been arousing wide interest, not only because the large number of oncogenes and tumor-suppressor genes involved, but also because it reveals novel therapeutic targets for inhibiting tumor growth. Here, we summarize recent developments and therapeutic targets in the area of cancer metabolism and discuss the future direction of metabolism-targeted cancer therapy.
The Warburg Effect and Cancer
The earliest and best-known cancer metabolic anomaly is described by Otto Warburg in the 1920s. Dr Warburg reported that tumors showed unusually high rates of glucose uptake and lactate production compared with normal tissues, even in the presence of oxygen (Warburg effect).Citation2 In fact, the enhanced tumor uptake of 2-deoxy-2(18F)-fluoro-D-glucose in positron emission tomography (PET) scans is now exploited in the clinic for diagnostic purposes.Citation3 Glycolysis produces ATP with lower efficiency, but at a faster rate than oxidative phosphorylation. This faster rate of ATP production is thought to aid the rapid proliferation of cancer cells. In addition to providing ATP, the high glycolytic rate may favor the growth of cancer cells through increasing biosynthesis of important molecules such as lipids, nucleotides, NADPH and amino acids.Citation4 Other metabolic products of glycolysis, such as lactate and H+, cause a consistent acidification of the extracellular environmentCitation5 and favor cancer invasion.Citation6 Because of the similarity, abnormally high aerobic glycolysis is now considered a hallmark of cancer. It is therefore greatly important to understand the molecular pathways that regulate aerobic glycolysis, to exploit the altered cancer metabolism for cancer therapy.
Glycolytic Pathways as Cancer Therapeutic Targets
In the standard model, glucose is first uptaken into cells through glucose transporters (GLUTs), after which it is phosphorylated by hexokinaseII (HKII) to become glucose-6-phosphate. Glucose-6-phosphate is then converted to fructose-6-phosphate by glucose-6-phosphate isomerase to proceed into glycolysis or it can be shunted into the pentose phosphate pathway (PPP) by glucose-6-phosphate dehydrogenase (G6PDH) to participate in nucleotide synthesis. The oxidative branch of PPP also produces NADPH, which is used in combating oxidative stress.Citation7 Phosphofructokinase-1 (PFK-1), located at downstream of HKII in glycolysis, converts fructose-6-phosphate to fructose-1,6-bisphosphate. Fructose-1,6-bisphosphate is then converted either to glyceraldehyde-3-phosphate to proceed into glycolysis or converted to dihydroxyacetone phosphate which is critical for lipid synthesis. In glycolysis, glyceraldehyde-3-phosphate is then converted to glycerate-2-phosphate by G3PDH and glycerate-2-phosphate is converted to phosphoenol pyruvate by enolase. Pyruvate kinase (PK) catalyzes an ATP-producing step of glycolysis in which phosphoenol pyruvate is converted into pyruvate with the release of ATP. Further down the glycolysis pathway, lactate dehydrogenase-A (LDH-A) converts pyruvate to lactate and generates NAD+ from NADH. NAD+ is necessary for continued flux through glycolysis ().
Figure 1. Schematic representation of the glucose metabolism in cancer cells. Cancer cells increase the uptake and metabolism of glucose by regulating key transporters and enzymes (shown in red font) involved in glycolytic pathways. Key oncogenic pathways are shown in green and key tumor suppressor pathways are shown in purple. The oxidative branch of PPP is required in nucleotide synthesis and dihydroxyacetone phosphate pathway is critical for lipid synthesis.
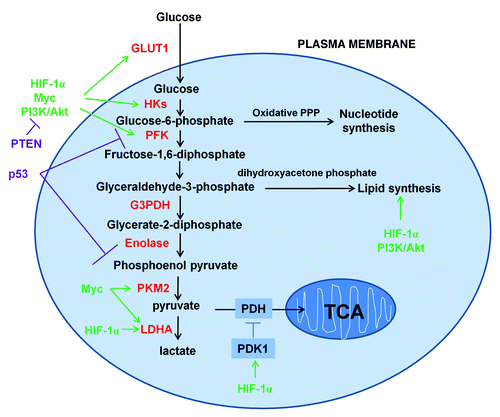
The increased dependence of cancer cells on glycolysis offers a number of attractive potential therapeutic targets for selectively killing cancer cells. Unfortunately, both cancer cells and normal proliferating cells display the Warburg effect. It is thus challenging to specifically target cancer metabolism, while sparing normal tissues. Several candidates such as GLUT1, HKII, PK, LDH-A and G6PD are currently the focus of interest for targeting, as they are overexpressed in cancer.
GLUT1 appears to be the predominant glucose transporter in many types of cancer.Citation8,Citation9 Inhibition of glucose transporter by phloretin can preferentially sensitize cancer cells to chemotherapeutic drugsCitation10 ().
Table 1. Summary table of selective potential drugs/compounds targeting cancer metabolism pathways and mode of action
HKII is a key glycolytic enzyme catalyzing the first step of glycolysis. Wolf and colleagues recently found that malignant gliomas show elevated HKII expression. Depletion of HKII in malignant glioma cells stimulates the production of ROS and increases cell death.Citation11 Another reason for considering HKII as a therapeutic target is that this enzyme is reversibly bound to the mitochondrial membrane protein named porin, which not only plays a role in maintaining HKII activity to regulate glycolysis, but also prevents cytochrome c release during the intrinsic apoptosis cascade.Citation12 Some drugs such as 3-bromopyruvate (3-BrPA) and clotrimazole can induce the detachment of HKII from mitochondria, abolish the activity of mitochondrial bound HKII and induce cancer cell death by a dramatic decrease in the level of ATPCitation13 ().
PK converts phosphoenol pyruvate to pyruvate with release of ATP. PK has two splice variants: PKM1and PKM2. PKM1 is found in many normal cells, whereas tumors express higher levels of PKM2.Citation14 Due to the high expression and activity of PKM2, this enzyme is also considered as an attractive target for cancer therapy. However, a recent study found that inhibition of PKM2 activity by cellular oxidative stress promotes accumulation of glycolytic intermediates and feeds other biosynthetic pathways (such as PPP pathway), resulting in tumor progression.Citation15 On the other hand, Goldberg and colleagues demonstrated that knockdown of PKM2 expression by RNAi increases apoptosis in vitro and causes tumor regression in vivo.Citation16 These paradoxical results may be explained by different cellular responses to varying degrees of hypoxia. Under moderate hypoxia, PKM2 is oxidized, leading to inhibition of its activity, promotion of PPP pathway and enhanced generation of cellular NADPH. This prevents accumulation of aberrantly high levels of ROS and oxidative cellular damage. Under severe hypoxia, oxidative phosphorylation is limited and cancer cells become dependent on PKM2 activity for ATP generation. Thus, PKM2 inhibitors prevent ATP production in severely hypoxic cancer cells, whereas PKM2 activators promote oxidative cellular damage in moderately hypoxic cancer cells. The therapeutic benefits of PKM2 inhibitors and activators are therefore context- and tumor type-dependent. PKM2 inhibitors may be therapeutically effective in relatively limited-oxygenated tissues such as brain, whereas PKM2 activators may be therapeutically beneficial in relatively well-oxygenated tissues such as lung.
Lactate overproduction is another important hallmark of advanced cancer. The first metabolic target for cancer therapy was LDH-A, an enzyme which converts pyruvate to lactate and converts NADH to NAD+. In addition to facilitating sustained glycolysis, the overproduction of lactate leads to an acidic tumor microenvironment associated with metastasis, tumor recurrence and poor survival.Citation17 Inhibition of LDH-A by a small-molecule inhibitor (FX11 [3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid]) reduces ATP levels, induces significant oxidative stress and inhibits the progression of sizable human lymphoma and pancreatic cancer xenograftsCitation18 ().
In addition to the above mentioned candidate targets, there are other molecules in the glycolytic pathway that are potentially implicated in cancer therapy. Fox example, 2-deoxyglucose (2-DG), a non-metabolizable glucose analog, could improve the efficacy of ABT-263 /737 (molecular antagonists of the Bcl-2 family) without harming healthy cells.Citation19 Iodoacetate, an inhibitor of G3PDH, increases cellular necrosis in pancreatic cancer.Citation20 Fluoride, an inhibitor of enolase, has been shown to induce C6 glioma cell death.Citation21 Dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase 1 (PDK1), increases mitochondrial production of ROS in cancer cells by shifting metabolism from glycolysis to glucose oxidation.Citation22 Given that NADPH is important in combating oxidative stress, inhibiting PPP could also be an attractive target. Buthionine S′R′-sulfoximine (BSO), a G6PDH inhibitor, has been shown to suppress the colony-forming efficiency of G6PDH-expressing cells.Citation23 The combination of 2DG and 6-AN (6-aminonicotinamide, another promising inhibitor of G6PDH) has been shown to greatly decrease the glutathione(GSH) content of cancer cells and enhance radiation-induced damage in human glioma and squamous carcinoma cell linesCitation24 ().
Additionally, a number of oncogenes are known to be involved in the metabolic switch from oxidative phosphorylation (OXPHOS) to glycolysis in tumor cells, such as AMP-activated protein kinase(AMPK),Citation25 hypoxia-inducible factor 1α (HIF-1α),Citation26 Myc,Citation27 PI3K/Akt/mTOR,Citation28 and rat sarcoma viral oncogene homolog (RAS).Citation29 Targeting of these oncogenes may selectively kill cancer cells by inhibiting glycolytic pathway ().
In 2008, while sequencing 22 human glioblastomas, researchers found that five of these tumors harbored mutations in the isocitrate dehydrogenase 1 (IDH1) gene.Citation30 IDH is a key player in the tricarboxylic acid cycle or Krebs cycle and catalyzes the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (α-KG). Mutations in the enzyme isocitrate dehydrogenase 1 (IDH1) have been recently identified in secondary brain glioblastomas (> 70%).Citation31 One explanation for the role of mutant IDH1 in cancer was first described in 2009. Researchers found that mutant IDH1 could reduce levels of α-KG, but lead to hypoxia-inducible factor (HIF) upregulation, a condition often seen in tumors.Citation32 Other researchers inserted mutant IDH1 into cells and found resulting high levels of a single metabolite, 2-hydroxyglutatarate (2HG). This metabolite was thought to potentially disable mitochondria and push the cancer cells toward anerobic glycolysis.Citation33 The IDH1 pathway thus represents a novel therapeutic target for cancer, particularly cancers like brain glioblastomas or acute myeloid leukemia that most commonly harbor IDH1 mutations.
Although the increased dependence of cancer cells on glycolysis offers an attractive therapeutic strategy in cancer therapy, some clinical trials have reported that utilization of glycolytic inhibitors may not be appropriate for certain types of tumors. Moreno-Sanchez and colleagues reported that some tumors located in the lung, mammary gland, cervix, ovaries and skin are not dependent on glycolysis, but are instead dependent on mitochondrial oxidative phosphorylation.Citation34 This suggests that glycolytic pathway-targeted strategies might only be useful in cancer cells with mitochondrial function defects or under hypoxia conditions when glycolysis is the major source of ATP.Citation35
Mitochondrial Oxidative Phosphorylation and Therapeutic Intervention
Mitochondria are important cellular organelles that fulfill a dual role in cell metabolism. During anabolic cell proliferation, they are the “power center” of the cell. However, under stressful circumstances, mitochondria control the intrinsic pathway of apoptosis by regulating the translocation of pro-apoptotic proteins to the cytosol.
Inspired by his initial observations, Warburg hypothesized that in tumors, mitochondrial respiration becomes defective, followed by an increasing reliance on glycolysis for ATP production.Citation2 However, this view has been disputed in recent years and may need a revision. Although glycolysis is the best-known metabolic phenotype of cancers, it is not a unique feature of all human cancers. In fact, in highly glycolytic tumor cells, defects in mitochondrial oxidative metabolism are not found.Citation34 Fogal and colleagues reported that high levels of glycolysis, in the absence of adequate OXPHOS, may not be beneficial for breast cancer growth.Citation36 Jessie and colleagues showed that introduction of activated Ras not only promotes tumorigenesis but also upregulates basal autophagy, leading to maintenance of mitochondrial function and providing energy substrates required for the tricarboxylic acid (TCA) cycle during periods of nutrient limitation.Citation37 These studies suggest that under certain circumstances, cancer cells will depend on mitochondrial oxidative phosphorylation to meet with cellular ATP demands.
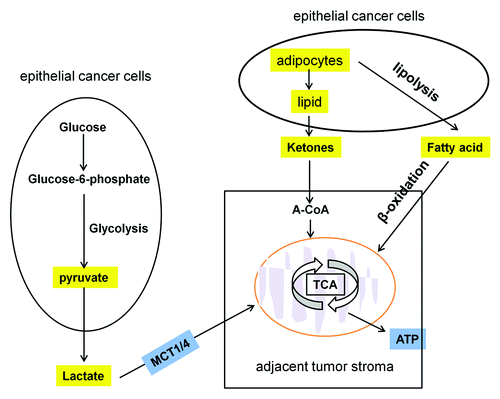
Recently, some studies have shown that hydrogen peroxide secreted by epithelial cancer cells can induce oxidative stress in adjacent tumor stroma. This oxidative stress induces autophagy, mitophagy and glycolysis in the tumor stroma.Citation38,Citation39 L-lactate derived from glycolytic tumor stroma can be transferred to the adjacent cancer cells and used for energy production via mitochondrial oxidative phosphorylationCitation38,Citation40 (). Similarly, Pierre reported that lactate generated by hypoxic tumor cells is a prominent substrate that fuels the oxidative metabolism of oxygenated tumor cells. These observations imply that glycolytic and oxidative tumor cells mutually regulate their access to energy metabolites.Citation41 It has also been reported that starvation mobilizes lipid stores from adipocytes to form ketone bodies, which can also fuel the mitochondrial oxidative phosphorylation of adjacent epithelial cancer cellsCitation38,Citation40,Citation42 (). Tumor stroma metabolites such as lactate and ketone may thus promote tumor growth by acting as high-energy metabolites. In support of this view, a recent study by Nieman and colleagues demonstrated that lipolysis in adipocytes leads to the release of free fatty acids. Ovarian cancer cells showed increased free fatty acids uptake and β-oxidation in mitochondria, which promoted ovarian cancer metastasis and provided energy for rapid tumor growth (). This study suggests that metabolic coupling between adipocytes and cancer cells may also favor tumor growth and that this can be blocked by lipid transport from adipocytes to cancer cells.Citation43 Thus, stromal-epithelial metabolic coupling and mitochondria are promising candidate targets for devising novel therapeutic intervention for cancer treatment and prevention. For instance, chloroquine inhibits autophagy and cuts off the stromal fuel supply, preventing high-energy metabolites moving from the stroma to cancer cells.Citation44 Metformin, an anti-diabetic reagent, inhibits both lipolysis in adipocytes and oxidative phosphorylation,Citation45 preventing cancer cells from using the energy-rich metabolites derived from the tumor stroma. The mitochondrial respiration complex I inhibitors, Rotenone or Bullatacin, induce apoptosis in a variety of cancer cell lines.Citation46,Citation47 The complex II inhibitor, α-TOS (α-tocopheryl succinate, Vitamine E analogs), causes electron leakage and subsequent generation of reactive oxygen species (ROS), which are cytotoxic to tumor cells.Citation48 The Complex Ш inhibitor, Benzylisothiocyanate, generates ROS and induces apoptosis in breast cancer, liver and lung solid tumor.Citation49 The complex IV and ATP synthesis inhibitors, Resveratrol or Oligmycin, show anti-tumor actions in skin, neuroblastoma and lymphoblastoid cancer modelsCitation47,Citation50 ().
Figure 2. Mitochondrial oxidative phosphorylation in cancer cells. Some energy-rich metabolites (L-lactate, ketones and fatty acids shown in yellow) derived from the tumor stroma can be transferred to the adjacent cancer cells and used for energy production via mitochondrial oxidative phosphorylation.
Target Glutamine Metabolism for Cancer Therapy
The unique role of glutamine in proliferating cells was first observed by Eagle in 1955.Citation51 Eagle observed that many cell lines consumed 10-fold higher rates of glutamine than any other amino acids and these cell lines could not proliferate or maintain their viability in the absence of glutamine. Subsequent experiments showed that glutamine is involved in oxidative mitochondrial metabolism.Citation52 In order to be oxidized, glutamine first loses its amide group to form glutamate by glutaminase (GLS), which then loses its amine group to form α-ketoglutarate by glutamate dehydrogenase, in the TCA cycle. Some cancer cells generate more than 50% ATP by oxidizing α-ketoglutarate in mitochondria. For example, in lymphoma cells, even in the absence of glucose or under hypoxia, glutamine is able to fully sustain the mitochondrial TCA cycle for ATP production.Citation53 During glutamine oxidation in the TCA cycle, glutamine metabolism contributes to mitochondrial ROS production. To combat ROS oxidative stress, glutamine can be converted directly into GSH by the enzyme glutathione cysteine ligase (GCL). The reduced form of GSH has an antioxidant role and plays a key role in controlling cellular redox homeostasis ().Citation54
Figure 3. Glutamine metabolism in cancer cells. High throughput glutamine uptake feeds cancer cell growth and proliferation with a large pool of carbon and nitrogen for the biosynthesis of the nonessential amino acids and fatty acids for membrane production. Carbon precursors derived from glutaminolysis (oxaloacetate and glutamate) serve as the carbon substrate for amino acid biosynthesis and lactate. Glutamate donates its amine group to these carbon substrates to produce non-essential amino acids (NEAA), nucleotide and α-ketoglutarate. Glutamine can be converted directly into GSH which has antioxidant role and plays a key role in controlling cellular redox homeostasis.
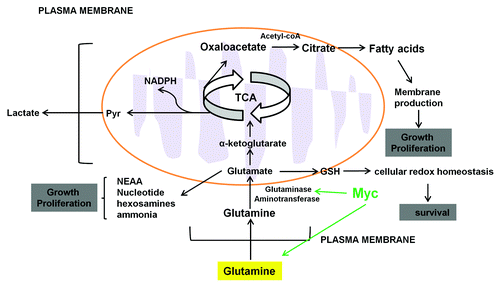
In addition to ATP production, real-time 13C NMR studies showed that glutamine provides the major source of oxaloacetate (). Oxaloacetate is the substrate that condenses with acetyl-coA to form citrate, which serves as a precursor for fatty acids synthesis.Citation55 The fatty acids are used for energy production through β-oxidation or lipids synthesis for membrane production in highly proliferating cancer cells. Fatty-acid synthesis involves one key enzyme, fatty-acid synthase (FASN), which is overexpressed in cancer. Several FASN inhibitors such as C93 and FAS31 have been tested in pre-clinical studies (). Treatment of tumor cells with these FASN inhibitors effectively suppresses the growth in small lung cancer and mouse melanoma models.Citation56,Citation57 By contributing to citrate production, the role of glutamine as a carbon source provides the mitochondria with precursors for maintaining mitochondrial membrane potential, which is critical for glutamine-dependent cancer cells. Glutamine carbon is also converted into lactate acid and secreted from cells. Conversion of glutamine into lactate acid requires malic enzyme, which decarboxylates malic acid and produces NADPH to meet the need in the proliferating cells ().Citation58
Glutamine also contributes to macromolecular synthesis by means other than production of oxaloacetate, NADPH and supply of carbon to mitochondria. Real-time 13C NMR studies showed that glutamine also serves as a nitrogen source.Citation58 The amido and amino groups of glutamine contribute to metabolic intermediates in the synthesis of non-essential amino acids, nucleotide biosynthesis, hexosamines and ammonia (). Yuneva and colleagues reported that supplementation of glutamine-starved Myc-driven cells with TCA intermediates could rescue cell viability but not proliferation.Citation59 Similarly, Meng and colleagues reported that supplementation of glutamine-starved Hep3B cells with alternate nitrogen sources such as alanine or asparagine could rescue cell proliferation.Citation60 In nucleotide biosynthesis, the role of glutamine as nitrogen donor for both purine and pyrimidine has been implicated in support of proliferation. Gaglio and colleagues showed that the K-Ras transformed fibroblasts cultured in the glutamine-deprived media exhibit reduced cellular proliferation and decreased progression into S phase. The growth potential of the transformed cells was restored by adding the four deoxyribonucleotides.Citation61 These results suggest that the role of glutamine as a nitrogen donor might explain some of the proliferation-inhibitory effects of glutamine deprivation.
Many types of cancer cells are sensitive to glutamine deprivation, including pancreatic cancer cells, lung cancer cells and glioma cells. Pre-clinical tests of L-asparaginase can significantly deplete blood glutamine levels and has some success in the treatment of pediatric acute lymphoblastic leukemia (ALL) (). However, L-asparaginase treatment has shown significant toxicity and side effects, such as increasing mental confusion and early signs of coagulopathy.Citation62 Novel glutamine metabolism-based therapies for cancer are needed which do not impair normal tissues. Glutamine-dependent cancer cells always make use of mitochondria to produce precursors from glutamine for the synthesis of lipids, proteins and nucleotides through activities of the mitochondrial electron transport chain. Thus, inhibition of mitochondrial respiration chain by mitoformin can also inhibit glutamine metabolism and slow the growth of glutamine-dependent cancer cells.Citation63 The oncogene Myc regulates several steps in the glutamine metabolism and the expression level of Myc has been demonstrated to be linked to the increased glutaminolysis through transcription program. L-γ-glutamyl-p-nitroanilide (GPNA), an inhibitor of SLC1A5 which is a direct target of Myc, can inhibit glutamine uptake and suppress glutamine-dependent mTOR activationCitation64 (). Glutamine metabolism is mediated by three types of enzymes: glutaminase (GLS), glutamate dehydrogenase and aminotransferase. Active Rho GTPases can increase GLS for transformation.Citation65 One compound 968, a small molecular inhibitor of Rho GTPase, can inhibit growth, migration and invasive activity of transformed fibroblasts and human cancer cells and shrink tumors in mouse xenograft models without obvious adverse effectsCitation66 (). Another recent study indicates that glutamine dehydrogenase may not be the rate-limiting step in glutamine metabolism. The major route of glutamine-derived carbon that enters the TCA cycle is through transamination.Citation67 Thus, aminotransferase appears to be a promising target for cancer therapy.
Amino-oxyacetic acid (AOA) (), an inhibitor of transaminase, can selectively suppress the proliferation of MDA-MB-231 cells and inhibit the growth of MDA-MB-231 breast tumor xenografts in mouse model.Citation68 These studies suggest that selective inhibition of one step of glutamine metabolism (such as glutaminase or glutamine aminotransferase) might produce an anticancer effect without the toxicity associated with inhibition of the full glutamine metabolism chain. Based on the characteristics of tumors exhibiting increased glutamine metabolism, novel glutamine-based imaging techniques have recently emerged. Glutamine PET tracers 18F-(2S,4R)4-fluoroglutamine and L-[5-11C]-glutamine have been shown to be taken up by glutaminolytic cancer cell lines and tumors in the mouse model and can be used to image glutamine metabolism both in vitro and in vivo.Citation69 This relatively recent technology will facilitate the ongoing development of drugs that target glutamine metabolism pathway.
Conclusions and Perspectives
Modulating cancer metabolism is a novel therapeutic strategy for suppression of tumor growth. The increasing interest in cancer metabolism has already generated a series of new potentially useful therapeutic agents. Nevertheless, there are potential difficulties and concerns due to the low selectivity and specificity of the current generation of agents. It is known that in addition to cancer cells, some normal tissues (brain, retina or testis), stem cells and immune cells also display glycolysis, mitochondrial oxidative phosphorylation and glutaminolysis. Therefore, most current metabolism-based therapeutic agents show some toxic effects on normal cells. The question for the next decade of metabolic cancer research is: What is the best way to specifically target the metabolism of cancer cells? First, a better understanding is necessary of the key metabolic differences between cancer cells and non-malignant cells. A better understanding of how different cancer cells adapt these processes to fulfill their energy requirements may help improve the selectivity of cancer metabolism-based therapy. These efforts may lead to generation of highly specific cancer metabolism-targeted agents that uniquely induce cancer cell death. For example, Myc addicts cancer cells to glutamine by preventing them from supplying the TCA with other nutrients.Citation59 Thus, glutamine addiction is often exploited for metabolic therapy.
Second, we need a better understanding of how oncogenic activation controls cancer cell metabolism for proliferation. The metabolic type of a given tumor may vary widely from oxidative to glycolytic due to oncogenes activated or the micro-environmental change.Citation34 For example, under stressful environments, cancer cells can take advantage of oncogene-induced autophagy or mitophagy for energy demand. Some mutations in BRAF or KRAS can sustain cancer cell growth in low glucose conditions.Citation70 In this regard, key metabolic oncogene regulators could serve as therapeutic targets without impairing normal tissues.
Third, heterogeneity of metabolic profiles of different cancer types needs to be further clarified, such as brain cancer (glycolytic type), lung cancer (OXPHOS type) and others. If the metabolic profile primarily used for energy production is better elucidated, targeting the predominant bio-energetic pathway might lead to more effective utilization of the metabolism-targeted agents. Hence, re-activation of mitochondrial oxidative metabolism in glycolytic tumors or reduced mitochondrial content and OXPHOS capacity in “OXPHOS” type of tumors may be considered as potential therapeutic strategies.
Fourth, delivering the metabolism-targeted agents to the tumor sites by specific delivery systems will enhance the anticancer efficiency of those drugs and decrease their side effects. Although the success of nanoparticle interventions has currently been primary limited to laboratory tests in human cancer cell lines and mouse xenograft models, this new approach shows promise to provide an alternative strategy for clinical cancer diagnosis and therapy.
Cancer cells display metabolic flexibility, which provides ability for a given cancer cell to alternate between glycolysis and OXPHOS in response to physiological needs. It is becoming increasingly understood that cancer cells can develop resistance to inhibition of one metabolic pathway through upregulation of other alternate metabolic pathways. Combinational approaches that target the increased metabolic flexibility of cancer cells are thus worth exploring. Bezielle (BZL101), a candidate oral drug that is distinguished from other similar agents due to its ability to inhibit both glycolysis and OXPHOS, has shown promising efficacy and excellent safety in the early phase clinical trials for advanced breast cancer.Citation71 Similarly, the combined use of 2-DG and metformin in a recent study led to significant cell death and tumor growth inhibition, associated with decreases in cellular ATP.Citation72 These studies indicate that deprivation of tumor bio-energetics by dual inhibition of energy pathways might be an effective novel therapeutic approach for a board spectrum of human tumors. In conclusion, altered energy metabolism in cancer cells provides a unique opportunity to develop novel and more effective anticancer therapies.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This manuscript is supported by grants from National Natural Sciences Foundation of China (81072146), by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and by US Public Health Service (R01CA135038).
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer 2002; 2:683 - 93; http://dx.doi.org/10.1038/nrc882; PMID: 12209157
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27:441 - 64; http://dx.doi.org/10.1146/annurev-cellbio-092910-154237; PMID: 21985671
- Schornack PA, Gillies RJ. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 2003; 5:135 - 45; PMID: 12659686
- Smallbone K, Gavaghan DJ, Gatenby RA, Maini PK. The role of acidity in solid tumour growth and invasion. J Theor Biol 2005; 235:476 - 84; http://dx.doi.org/10.1016/j.jtbi.2005.02.001; PMID: 15935166
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85 - 95; http://dx.doi.org/10.1038/nrc2981; PMID: 21258394
- Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer 1997; 80:1046 - 51; http://dx.doi.org/10.1002/(SICI)1097-0142(19970915)80:6<1046::AID-CNCR6>3.0.CO;2-7; PMID: 9305704
- Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, et al. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn J Cancer Res 2002; 93:1123 - 8; http://dx.doi.org/10.1111/j.1349-7006.2002.tb01214.x; PMID: 12417042
- Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P, et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol 2007; 59:495 - 505; http://dx.doi.org/10.1007/s00280-006-0291-9; PMID: 16906425
- Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 2011; 208:313 - 26; http://dx.doi.org/10.1084/jem.20101470; PMID: 21242296
- Pedersen PL. Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr 2008; 40:123 - 6; http://dx.doi.org/10.1007/s10863-008-9165-7; PMID: 18780167
- Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett 2001; 173:83 - 91; http://dx.doi.org/10.1016/S0304-3835(01)00667-X; PMID: 11578813
- Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011; 2:393 - 400; PMID: 21789790
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011; 334:1278 - 83; http://dx.doi.org/10.1126/science.1211485; PMID: 22052977
- Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med 2012; 209:217 - 24; http://dx.doi.org/10.1084/jem.20111487; PMID: 22271574
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence and restricted patient survival in human cervical cancers. Cancer Res 2000; 60:916 - 21; PMID: 10706105
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107:2037 - 42; http://dx.doi.org/10.1073/pnas.0914433107; PMID: 20133848
- Yamaguchi R, Janssen E, Perkins G, Ellisman M, Kitada S, Reed JC. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS One 2011; 6:e24102; http://dx.doi.org/10.1371/journal.pone.0024102; PMID: 21949692
- Bhardwaj V, Rizvi N, Lai MB, Lai JC, Bhushan A. Glycolytic enzyme inhibitors affect pancreatic cancer survival by modulating its signaling and energetics. Anticancer Res 2010; 30:743 - 9; PMID: 20392992
- El Sayed SM, El-Magd RM, Shishido Y, Chung SP, Diem TH, Sakai T, et al. 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J Bioenerg Biomembr 2012; 44:61 - 79; http://dx.doi.org/10.1007/s10863-012-9409-4; PMID: 22318356
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99:989 - 94; http://dx.doi.org/10.1038/sj.bjc.6604554; PMID: 18766181
- Kuo W, Lin J, Tang TK. Human glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3 cells and induces tumors in nude mice. Int J Cancer 2000; 85:857 - 64; http://dx.doi.org/10.1002/(SICI)1097-0215(20000315)85:6<857::AID-IJC20>3.0.CO;2-U; PMID: 10709108
- Varshney R, Dwarakanath B, Jain V. Radiosensitization by 6-aminonicotinamide and 2-deoxy-D-glucose in human cancer cells. Int J Radiat Biol 2005; 81:397 - 408; http://dx.doi.org/10.1080/09553000500148590; PMID: 16076755
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 2007; 292:C125 - 36; http://dx.doi.org/10.1152/ajpcell.00247.2006; PMID: 16971499
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev 2007; 26:299 - 310; http://dx.doi.org/10.1007/s10555-007-9064-0; PMID: 17415526
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 2000; 275:21797 - 800; http://dx.doi.org/10.1074/jbc.C000023200; PMID: 10823814
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 2004; 64:3892 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-03-2904; PMID: 15172999
- Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, Eaton JW, et al. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 2006; 25:7225 - 34; http://dx.doi.org/10.1038/sj.onc.1209709; PMID: 16715124
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321:1807 - 12; http://dx.doi.org/10.1126/science.1164382; PMID: 18772396
- Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 2009; 30:7 - 11; http://dx.doi.org/10.1002/humu.20937; PMID: 19117336
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324:261 - 5; http://dx.doi.org/10.1126/science.1170944; PMID: 19359588
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462:739 - 44; http://dx.doi.org/10.1038/nature08617; PMID: 19935646
- Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J 2007; 274:1393 - 418; http://dx.doi.org/10.1111/j.1742-4658.2007.05686.x; PMID: 17302740
- Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol 2006; 175:913 - 23; http://dx.doi.org/10.1083/jcb.200512100; PMID: 17158952
- Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol 2010; 30:1303 - 18; http://dx.doi.org/10.1128/MCB.01101-09; PMID: 20100866
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 2011; 43:1045 - 51; http://dx.doi.org/10.1016/j.biocel.2011.01.023; PMID: 21300172
- Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010; 9:2412 - 22; http://dx.doi.org/10.4161/cc.9.12.11989; PMID: 20562527
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 14; http://dx.doi.org/10.4161/cc.9.17.12731; PMID: 20818174
- Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008; 118:3930 - 42; PMID: 19033663
- Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011; 10:1271 - 86; http://dx.doi.org/10.4161/cc.10.8.15330; PMID: 21512313
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011; 17:1498 - 503; http://dx.doi.org/10.1038/nm.2492; PMID: 22037646
- Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010; 9:4297 - 306; http://dx.doi.org/10.4161/cc.9.21.13817; PMID: 21051947
- Riccio A, Del Prato S, Vigili de Kreutzenberg S, Tiengo A. Glucose and lipid metabolism in non-insulin-dependent diabetes. Effect of metformin. Diabete Metab 1991; 17:180 - 4; PMID: 1936473
- Deng YT, Huang HC, Lin JK. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol Carcinog 2010; 49:141 - 51; PMID: 19777565
- Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Lett 1994; 339:40 - 4; http://dx.doi.org/10.1016/0014-5793(94)80380-3; PMID: 8313978
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008; 27:4324 - 35; http://dx.doi.org/10.1038/onc.2008.69; PMID: 18372923
- Xiao D, Powolny AA, Singh SV. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species-dependent apoptosis in human breast cancer cells. J Biol Chem 2008; 283:30151 - 63; http://dx.doi.org/10.1074/jbc.M802529200; PMID: 18768478
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006; 5:493 - 506; http://dx.doi.org/10.1038/nrd2060; PMID: 16732220
- Eagle H. Nutrition needs of mammalian cells in tissue culture. Science 1955; 122:501 - 14; http://dx.doi.org/10.1126/science.122.3168.501; PMID: 13255879
- Kovacević Z, Morris HP. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res 1972; 32:326 - 33; PMID: 4400467
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012; 15:110 - 21; http://dx.doi.org/10.1016/j.cmet.2011.12.009; PMID: 22225880
- Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35:427 - 33; http://dx.doi.org/10.1016/j.tibs.2010.05.003; PMID: 20570523
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005; 8:311 - 21; http://dx.doi.org/10.1016/j.ccr.2005.09.008; PMID: 16226706
- Carvalho MA, Zecchin KG, Seguin F, Bastos DC, Agostini M, Rangel AL, et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer 2008; 123:2557 - 65; http://dx.doi.org/10.1002/ijc.23835; PMID: 18770866
- Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM, et al. Selective inhibition of fatty acid synthase for lung cancer treatment. Clin Cancer Res 2007; 13:7139 - 45; PMID: 18056164
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104:19345 - 50; http://dx.doi.org/10.1073/pnas.0709747104; PMID: 18032601
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007; 178:93 - 105; http://dx.doi.org/10.1083/jcb.200703099; PMID: 17606868
- Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle 2010; 9:3921 - 32; http://dx.doi.org/10.4161/cc.9.19.13139; PMID: 20935507
- Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One 2009; 4:e4715; http://dx.doi.org/10.1371/journal.pone.0004715; PMID: 19262748
- Lessner HE, Valenstein S, Kaplan R, DeSimone P, Yunis A. Phase II study of L-asparaginase in the treatment of pancreatic carcinoma. Cancer Treat Rep 1980; 64:1359 - 61; PMID: 7471124
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67:6745 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-06-4447; PMID: 17638885
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521 - 34; http://dx.doi.org/10.1016/j.cell.2008.11.044; PMID: 19203585
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762 - 5; http://dx.doi.org/10.1038/nature07823; PMID: 19219026
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18:207 - 19; http://dx.doi.org/10.1016/j.ccr.2010.08.009; PMID: 20832749
- Moreadith RW, Lehninger AL. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J Biol Chem 1984; 259:6215 - 21; PMID: 6144677
- Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res 2008; 10:R84; http://dx.doi.org/10.1186/bcr2154; PMID: 18922152
- Lieberman BP, Ploessl K, Wang L, Qu W, Zha Z, Wise DR, et al. PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4-fluoroglutamine. J Nucl Med 2011; 52:1947 - 55; http://dx.doi.org/10.2967/jnumed.111.093815; PMID: 22095958
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009; 325:1555 - 9; http://dx.doi.org/10.1126/science.1174229; PMID: 19661383
- Chen V, Staub RE, Fong S, Tagliaferri M, Cohen I, Shtivelman E. Bezielle selectively targets mitochondria of cancer cells to inhibit glycolysis and OXPHOS. PLoS One 2012; 7:e30300; http://dx.doi.org/10.1371/journal.pone.0030300; PMID: 22319564
- Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther 2011; 10:2350 - 62; http://dx.doi.org/10.1158/1535-7163.MCT-11-0497; PMID: 21992792