Abstract
Breast tumor kinase (Brk)/protein tyrosine kinase-6 (PTK-6) is a nonreceptor PTK commonly expressed at high levels in breast cancer. Brk interacts closely with members of the human epidermal growth factor receptor (HER) family in breast cancer but the functional role of this interaction remains to be determined. Here, we provide novel mechanistic insights into the role of Brk in regulating cell survival and epithelial-to-mesenchymal transition (EMT) in the context of HER2-positive breast cancer cells. Overexpression of HER2 in MCF7 breast cancer cells (MCF7HER2) led to a higher level of Brk protein and concomitantly reduced Src Y416-phosphorylation, and the cells became mesenchymal in morphology. An in vivo selection of MCF7HER2 cells in nude mice resulted in a subline, termed EMT1, that exhibited not only mesenchymal morphology but also enhanced migration potential. Compared with MCF7HER2 cells, EMT1 cells maintained a similar level of HER2 protein but had much higher level of activated HER2, and the increase in Brk protein and the decrease in Src Y416-phosphorylation were less in EMT1 cells. EMT1 cells exhibited increased sensitivity to both pharmacological inhibition of HER2 and knockdown of Brk than did MCF7HER2 cells. Knockdown of Brk induced apoptosis and partially reversed the EMT phenotype in EMT1 cells. Overexpression of a constitutively active STAT3, a known substrate of Brk, overcame Brk knockdown-induced effects in EMT1 cells. Together, our findings support a new paradigm wherein Brk plays both a complementary and a counterbalancing role in cooperating with HER2 and Src to regulate breast cancer cell survival and EMT.
Introduction
Invasion and metastasis are the primary causes of death from breast cancer and their successful inhibition is therefore expected to significantly improve breast cancer prognosis.Citation1 Previous studies have clearly shown that the human epidermal growth factor receptor-2 (HER2), a ligandless receptor tyrosine kinase overexpressed in approximately 25% of breast cancers, plays an important role in breast cancer invasion and metastasis and that its expression correlates with poor clinical prognosis.Citation2-Citation5 The nonreceptor protein tyrosine kinase Src has been shown to coordinate with HER2 in the development of HER2-mediated malignant phenotypes and resistance to HER2-targeted therapy.Citation6 It remains interesting to identify additional molecular markers that are critical for HER2-mediated invasion and metastasis in breast cancer.
Breast tumor kinase (Brk), also known as protein tyrosine kinase 6 (PTK6), is another nonreceptor protein tyrosine kinase originally cloned from a human metastatic breast tumor and later found to be highly expressed in approximately two thirds of all breast cancers.Citation7,Citation8 Brk shares 46% homology with c-Src and possesses SH3, SH2, and kinase domains in an arrangement similar to those of Src (SH3-SH2-catalytic)Citation9-Citation11; however, Brk lacks the Src-characteristic N-terminal myristoylation consensus sequences for fatty acylation and membrane anchorage of Src family proteins, and its SH2 and SH3 domains are atypical.Citation12 Brk is thus considered to be a member of a distinct nonreceptor tyrosine kinase family known as the Frk family, which includes Frk, Brk, Srm, and Sik and is distantly related to Src family kinases.Citation13
Compared with the HER family members and Src, Brk is much less well-studied for its role and function in breast cancer. Brk is known to have both kinase-dependent and kinase-independent (i.e., molecular scaffolding or adaptor) functions.Citation14,Citation15 Several Brk substrates and interacting proteins have been identified,Citation16-Citation27 including EGFR, which we recently reported.Citation27 In addition to its interaction with EGFR,Citation27,Citation28 Brk functionally interacts with other members of the human EGFR family: Brk enhances EGF-stimulated HER3 phosphorylation by increasing the recruitment of phosphatidylinositol 3-kinase (PI3K) to HER3 and regulates heregulin-induced activation of ERK5 and p38 MAPK in breast cancer cells.Citation8,Citation29 In tissues from patients with HER2-overexpressing invasive ductal breast carcinomas, Brk is often simultaneously overexpressed with HER2.Citation30,Citation31 Brk can enhance HER2-induced activation of Ras/MAPK signaling and cyclin E/cdk2 in HER2-positive breast cancer cells.Citation31 In a syngeneic mouse model, Brk cooperated with HER2 to increase the proliferative potential of HER2-positive tumors in vivo and conferred resistance in HER2-positive breast cancer cells to lapatinib, a HER2/EGFR dual inhibitor.Citation31 These findings suggest a functional biological link between Brk and HER2 in promoting breast cancer cell proliferation; however, the mechanisms of the interaction between Brk and HER2 are largely unknown. In particular, an explicit elucidation of a definitive role of Brk in HER2-regulated breast cancer cell invasion and metastasis and breast cancer cell survival remains elusive.
In the current study, we addressed this question by investigating the role of Brk-HER2-Src interactions in the regulation of breast cancer survival and epithelial-to-mesenchymal transition (EMT) in the context of HER2-positive breast cancer cells. We adopted a unique approach to recapture the HER2-mediated invasiveness and metastatic potential of breast cancer cells using in vivo selection of HER2-transfected breast cancer cells in nude mice. Here, we report our findings from testing of our hypothesis that Brk interacts with HER2 and Src in the regulation of breast cancer cell survival and EMT.
Results
Characterization of MCF7 cells overexpressing HER2 (MCF7HER2) and an invasive subline of MCF7HER2 cells generated from in vivo selection
To recapture malignant phenotypes of HER2-overexpressing breast cancer cells, we started with MCF7 breast cancer cells with experimentally elevated HER2 (MCF7HER2) and selected sublines of MCF7HER2 cells for enhanced invasion and metastasis potentials in vivo (). The subline derived from the tumor with the strongest IVIS positivity in the anterior region of a mouse, termed EMT1, was used in current study. Compared with the control vector-transfected MCF7neo cells, which had a typical epithelial cell phenotype of tightly connected (cuboidal/cobblestone-shaped) cells, MCF7HER2 were more fibroblast-like (elongated spindle-shaped) characterized by a loss of cell adhesion and by dispersion (, left columns in the upper and lower panels). The EMT1 subline showed even more drastic changes in morphological features (, left columns in the upper and lower panels). Immunofluorescent staining showed a marked decrease in E-cadherin expression (, upper panel) and an increase in fibronectin expression (, lower panel) in MCF7HER2 cells, and these changes were more drastic in EMT1 subline cells. In addition, western blotting showed changes in other EMT markers, such as a decrease in β-catenin and an increase in N-cadherin, in MCF7HER2 and EMT1 cells (). Compared with MCF7neo cells, MCF7HER2 and EMT1 cells also showed increased expression of SNAI 1 but not Twist; both are well-known transcription factors that regulate the EMT phenotype.Citation32,Citation33 These observations indicate that MCF7HER2 and EMT1 cells have undergone epithelial-to-mesenchymal transition.
Figure 1. Generation of invasive sublines of MCF7HER2 cells in nude mice. Bioluminescent MCF7HER2 cells were inoculated into 4 nude mice via injection through the tail vein. The mice were monitored periodically for colonization of the bioluminescent cells inside the bodies of the mice. Shown in the figure are photos of the mice taken under a bright light (top) and with Xenogen’s in vivo imaging system (IVIS) after intraperitoneal administration of luciferin, a substrate of luciferase.
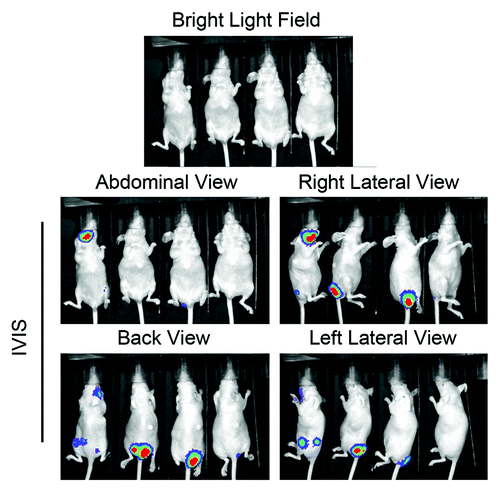
Figure 2. Phenotype changes in MCF7 breast cancer cells following overexpression of HER2 and in vivo selection.(A) Changes in cell morphology and expression levels of E-cadherin and fibronectin. MCF7neo, MCF7HER2, and EMT1 cells were subjected to immunofluorescent staining with antibodies directed against E-cadherin and fibronectin and counterstained with DAPI. Representative areas visualized under phase contrast lens and fluorescent light are shown. (B) Changes in EMT markers. Lysates of MCF7neo, MCF7HER2, and EMT1 cells were prepared and analyzed by western blotting using the indicated antibodies. (C) Changes in the levels of HER2 and HER2 downstream cell signaling substrates. Lysates of MCF7neo, MCF7HER2, and EMT1 cells were prepared and analyzed by western blotting using the indicated antibodies. (D) Differential responses of MCF7neo, MCF7HER2 and EMT1 cells to lapatinib and doxorubicin treatment. The cells were exposed to varying concentrations of lapatinib or doxorubicin for 48 h. MTT assays were performed at end of the treatments. The optical density (OD) values of the treated groups were normalized as a percentage of the OD value of untreated or vehicle treated groups of corresponding cell lines. p < 0.01 between MCF7neo cells and MCF7HER2 or EMT1 cells treated with lapatinib at all doses; p < 0.01 between MCF7neo cells and MCF7HER2 treated with doxorubicin at doses greater than 0.125 nM and between MCF7neo cells and EMT1 cells treated with doxorubicin at doses greater than 1 nM.
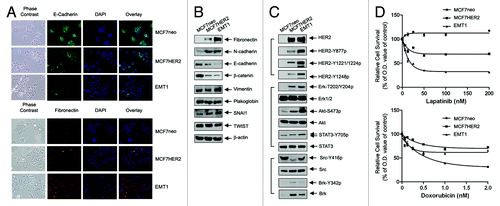
We found that, although the level of HER2 protein is similar in EMT1 and MCF7HER2 cells, the levels of several site-specific tyrosine-phosphorylated HER2 proteins, which are activation-specific, are significantly increased in EMT1 cells (). These increases were accompanied by marked increases in the levels of activation-specific phosphorylation of several well-known HER2 downstream substrates, particularly in the levels of phosphorylated STAT3 (STAT3-Y705) and Akt (Akt-S473). Consistent with the finding, cell growth and survival assays showed that EMT1 cells were more sensitive than MCF7HER2 cells to treatment with lapatinib, a HER2/EGFR small tyrosine kinase inhibitor, whereas MCF7neo cells were insensitive (, upper panel). The increased sensitivity to lapatinib appears to be specific to the changes in HER2-mediated cell signaling, because EMT1 and MCF7HER2 cells exhibited increased resistance to a nonspecific chemotherapeutic agent, doxorubicin, whereas MCF7neo were sensitive to the agent (, lower panel).
Interestingly, in contrast to the increase in HER2 activity, the level of activation-specific Y416-phosphorylated Src was markedly reduced in MCF7HER2 cells and moderately reduced in EMT1 cells (). Nevertheless, the pattern of change in the level of Y416-phosphorylated Src showed an inverse relationship with the change in the levels of total Brk protein and Y342-phosphoryalted Brk among these three isogenic cell lines. These findings suggest that a novel role of Brk in substituting Src to cooperate with HER2 for HER2-mediated functions in MCF7HER2 and EMT1 cells.
Role of Brk in HER2-mediated breast cancer cell survival and migration potential
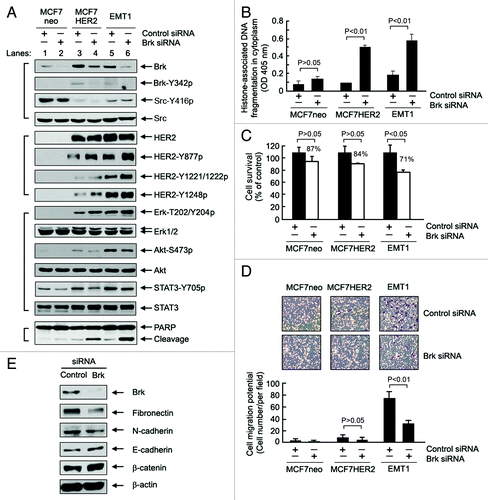
We next examined the role of Brk in HER2-mediated cell signaling and survival and migration potential by knocking down Brk expression with small interfering RNA (siRNA) in MCF7neo, MCF7HER2, and EMT1 cells (). These experiments produced several major findings. First, knockdown of Brk increased the levels of activation-specific HER2 phosphorylation on several tyrosine residues in both MCF7HER2 (lane 3 vs. lane 4) and EMT1 (lane 5 vs. lane 6) cells after 48 h. The basal level of HER2 in MCF7 cells is very low; thus, no noticeable changes were found. Second, the knockdown of Brk also led to a compensatory increase in activation-specific phosphorylation of Erk, but not that of Akt and STAT3, in these cells. Third, very importantly, the knockdown of Brk in MCF7HER2 and EMT1 cells resulted in cell death, as shown by the appearance of cleaved PARP, a marker of apoptosis. Of note, the induction of PARP cleavage was greater in EMT1 cells than in MCF7HER2 cells; in contrast, no obvious PARP cleavage was seen in MCF7neo cells after knockdown of Brk. These findings suggest that Brk is important for cell survival in MCF7HER2 and EMT1 cells, and that because EMT1 cells acquired increased HER2 activity after in vivo selection, Brk is more critical for the survival of EMT1 cells than that of MCF7HER2 cells, despite the increase in Brk level was less in EMT1 cells than in MCF7HER2 cells (lane 5 vs. lane 3). Using an enzyme-linked immunosorbent assay (ELISA) that quantitatively measures the levels of histone-associated DNA fragmentation in the cytoplasm after induction of apoptosis, we further confirmed that the induction of apoptosis by knockdown of Brk was greater in EMT1 cells than in MCF7HER2 cells (). The compensatory increases in the levels of phosphorylated HER2 and phosphorylated Erk after Brk knockdown support the conclusion that Brk is important for maintaining the survival of MCF7HER2 and EMT1 cells.
Figure 3. Role of Brk in cell survival and migration potential of MCF7 breast cancer cells acquired after overexpression of HER2 and in vivo selection. (A) MCF7neo, MCF7HER2, and EMT1 cells were transiently transfected with Brk siRNA or control siRNA for 48 h. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies. (B) The same cell lysates in A were analyzed by an ELISA for quantitative determination of the levels of apoptosis. (C) MCF7neo, MCF7HER2, and EMT1 cells were transiently transfected with Brk-specific siRNA or control siRNA, as described in (A) and then cultured for 3 additional days. An MTT assay was performed to measure the relative number of surviving cells after knockdown of Brk with Brk-specific siRNA. (D) MCF7neo, MCF7HER2, and EMT1 cells were transfected with Brk-specific siRNA or control siRNA for 48 h, as in (A) and then seeded into a Boyden transwell chamber. The average numbers of cells per microscopic field that penetrated through the transwell membrane during a 24 h period were counted and plotted. Representative photomicrograph areas are shown. (E) EMT1 cells were transiently transfected with Brk siRNA or control siRNA for 48 h. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies.
Consistent with the findings from the apoptosis assays after Brk knockdown, a cell growth and survival assay showed that the percentage of surviving cells was significantly lower for EMT1 and MCF7HER2 cells than for MCF7neo cells after transient knockdown of Brk in a 5-d cell culture ().
A notable feature of EMT1 cells is their markedly increased migration potential. shows that MCF7neo cells exhibit a minimal level of cell migration potential, as measured by Boyden’s chamber assay. Overexpression of HER2 in MCF7HER2 cells only marginally increased the migration potential, even though changes in EMT markers in MCF7HER2 cells are indicative of a mesenchymal phenotype. The EMT1 cells, however, exhibited a marked increase in migration potential compared with MCF7neo and MCF7HER2 cells. This difference could be due to the quantitative difference in levels of the EMT markers between EMT1 cells and MCF7HER2 cells (). Similar to the impact of Brk level on the survival of EMT1 cells, knockdown of Brk significantly reduced the migration potential of the cells (), which was accompanied by decrease in the levels of mesenchymal markers (fibronectin and N-cadherin) and increase in the levels of epithelial markers (E-cadherin and β-catenin) ().
Taken together, the data indicate that Brk plays important roles in regulating the survival and migration potential recaptured in EMT1 cells that were derived from MCF7HER2 cells in vivo.
Role of STAT3 in mediating Brk-regulated functions in the context of HER2-overexpressing cells
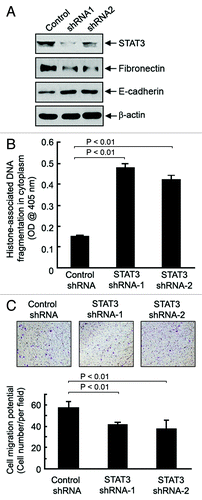
STAT3 is a known substrate of Brk.Citation19 Many STAT3-targeted genes are key components in the regulation of cell survival and migration.Citation34-Citation37 The increase in STAT3 Y705-phosphorylation in EMT1 and MCF7HER2 cells, and the decrease in STAT3 Y705-phosphorylation when Brk was knocked down as shown in , prompted us to hypothesize that STAT3 is a major downstream molecular player that mediates the dependence on Brk for survival and increased migration potential of EMT1 cells. shows that the expression of two distinct STAT3 small hairpin RNA (shRNA) constructs knocked down STAT3 to slightly different levels in EMT1 cells. Knockdown of STAT3 led to a marked increase in the level of apoptosis () and a decrease in the migration potential of EMT1 cells (). The patterns were similar to the effects of Brk knockdown in the same cells ( and ). It is noteworthy that the knockdown of STAT3 was also accompanied by a decreased level of fibronectin and an increased level of E-cadherin (), suggesting that the decrease in the migration potential of EMT1 cells after knockdown of STAT3 is a functional consequence, although the accompanying apoptosis may also contribute to the decreased migration potential of the cells.
Figure 4. Role of STAT3 in cell survival and migration potential of EMT1 cells. (A) EMT1 cells were transiently transfected with one of two STAT3 shRNA constructs or control shRNA construct for 72 h. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies. (B) The same cell lysates in (A) were analyzed using ELISA for quantitative determination of the levels of apoptosis. C, EMT1 cells treated as described in A were seeded into a Boyden transwell chamber. The average numbers of cells per microscopic field that penetrated through the transwell membrane during a 24 h period were counted and plotted. Representative photomicrograph areas are shown.
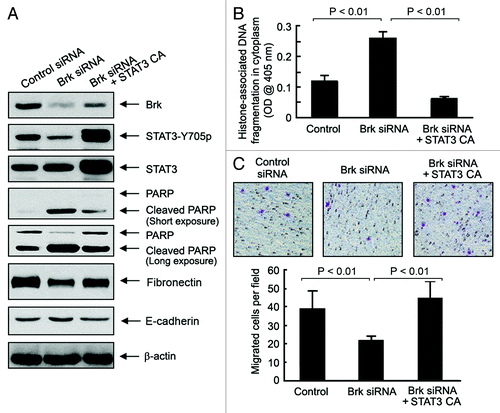
To further confirm the role of STAT3 as an important regulator of cell survival and migration downstream of Brk in EMT1 cells, we examined whether expression of a constitutively active STAT3 can protect EMT1 cells from Brk knockdown-induced apoptosis and inhibition of migration potential (). Overexpression of a constitutively active STAT3, which led to a rise in the level of Y705-phosphorylated STAT3, protected the cells from Brk knockdown-induced apoptosis, as shown by reduced PARP cleavage, compared with the effect of Brk silencing alone. This result was confirmed by an independent quantitative apoptosis ELISA for the level of histone-associated DNA fragmentation in the cytoplasm (). The overexpression of the constitutively active STAT3 also prevented Brk knockdown-induced decrease in fibronectin and increase in E-cadherin () and restored Brk knockdown-induced inhibition of migration potential in EMT1 cells ().
Figure 5. Constitutively active STAT3-mediated protection against Brk knockdown-induced apoptosis and inhibition of cell migration potential. (A) EMT1 cells were transiently transfected with a control siRNA or a Brk-specific siRNA, with and without a constitutively active (CA) STAT3 cDNA construct, for 48 h. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies. (B) The same cell lysates in A were analyzed using ELISA for quantitative determination of the levels of apoptosis. (C) EMT1 cells treated as described in A were seeded into a Boyden transwell chamber. The average numbers of cells per microscopic field that penetrated through the transwell membrane during a 24 h period were counted and plotted. Representative photomicrograph areas are shown.
Knockdown of Brk induces apoptosis in breast cancer cells with naturally high expression of Brk and HER2
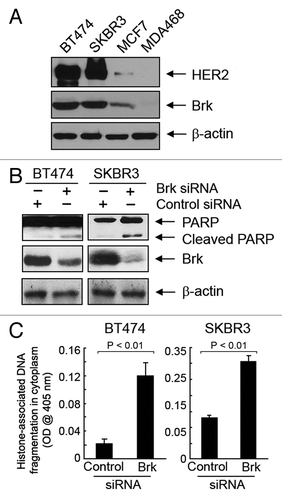
To confirm these novel findings in other breast cancer cells with naturally occurring high levels of HER2 and Brk, we knocked down Brk with siRNA in BT474 and SKBR3 cells, both of which contain high levels of HER2 and Brk compared with low HER2-expressing breast cancer cell lines such as MCF7 and MDA468 cells (). As expected, knockdown of Brk expression in BT474 and SKBR3 cells induced PARP cleavage and increased the level of histone-associated DNA fragmentation ().
Figure 6. Induction of apoptosis by knockdown of Brk in naturally HER2-overexpressing breast cancer cell lines. (A) Cell lysates of indicated breast cancer cell lines were prepared and analyzed by western blotting using the indicated antibodies. (B) BT474 and SKBR3 cells were transiently transfected with Brk siRNA or control siRNA for 48 h. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies. (C) The same cell lysates as in (B) were analyzed using ELISA for quantitative determination of the levels of apoptosis.
Discussion
In this paper, we report three major findings from our studies using an in vivo selected subline (EMT1) of MCF7 breast cancer cells with experimentally elevated high levels of HER2. First, we observed that compared with MCF7 and MCF7HER2 cells, the EMT1 cells are highly mesenchymal and invasive. Second, we found that there exist complicated complementary and counterbalancing relationships between the levels of activation-specific phosphorylation of HER2 and Src and the level of Brk protein for regulating the mesenchymal and invasive phenotype and cell survival in EMT1 cells. Third, we demonstrated that STAT3, which can be phosphorylated and activated by Brk,Citation19 plays a critical role in mediating the acquisition of the EMT1 cell phenotype and in regulating cell survival. These data suggest a novel role of Brk through coordinating with HER2 and Src in regulating breast cancer cell survival and EMT phenotype.
EMT is a reversible cell phenotype regulated by the interplay of extracellular signals, such as growth factors or cytokines, and their downstream effector molecules within the cells. Acquisition of the EMT phenotype in cancer cells can lead to increased motility of the cells, which is associated with invasion and metastasis.Citation38,Citation39 By uncovering the molecular mechanisms that govern EMT in breast cancer, one could presumably prevent metastasis by converting an invasive phenotype into a noninvasive one using target-specific interventions. Thus, inactivating EMT process by targeting relevant signaling pathways and molecular interactions through novel approaches could prevent cancer progression and metastasis, which is paramount for reducing breast cancer mortality.
As a member of EGFR family, HER2 is a classic mitogen for epithelial cells; therefore, it is debatable whether overexpression of HER2 alone can have a direct role in driving the transition of epithelial cells into a mesenchymal phenotype. Our data show that overexpression of HER2 in MCF7 cells induced typical changes in cell morphology and changes in the pattern of markers characteristic of EMT; however, the cell migration potential, which is one of the most important functional changes associated with the acquirement of the EMT phenotype, was not significantly increased in these cells until the cells were selected in vivo. This finding suggests that involvement of additional mechanisms and interaction with components in tumor microenvironment might be needed for cancer cells to acquire the increased migration potential.
Src has been shown to be an important co-target for HER2-targeted therapy for breast cancerCitation6; however, it is noteworthy that, in our cell model, we found the level of activation-specific Src Y416-phosphorylation was reduced whereas the levels of Brk total protein and activation-specific Brk Y342-phosphorylation were upregulated after overexpression of HER2 in MCF7 cells. Although the underlying mechanisms were not explored in the current study, the finding suggests that the three tyrosine kinases (HER2, Src and Brk) have complementary functions. Thus, Brk might be another ideal co-target for HER2-targeted therapy for breast cancer. Indeed, our data showed that EMT1 cells were more sensitive than MCF7HER2 cells to Brk knockdown-induced induction of apoptosis and inhibition of cell migration, suggesting that Brk is critically important for the phenotypes acquired by EMT1 cells.
Additionally, we found that, while HER2-regulated cell signaling can lead to the upregulation of Brk protein, knockdown of Brk can also lead to the upregulation of HER2 activity, as shown by the increased phosphorylation of HER2 at several sites; this increase was more apparent in EMT1 cells than in MCF7HER2 cells, even though the Brk level was less elevated in EMT1 cells than in MCF7HER2 cells. The increase in HER2 phosphorylation is likely a compensatory response of the cells to knockdown of Brk, which is consistent with our conclusion that the functions of Brk and HER2 are complementary. Because the increase in HER2 phosphorylation occurs at multiple sites, the compensatory response may be related to the mechanisms underlying HER2 homo- or heterodimerization with other HER family proteins, such as EGFR or HER3, which are known to lead to HER2 phosphorylation.Citation5
The fact that STAT3 is a known substrate of Brk and the finding that overexpression of constitutively active STAT3 can protect EMT1 cells from Brk knockdown-induced effects indicate that STAT3 is a major molecular player downstream of the interaction between HER2 and Brk. This finding suggests that HER2 may regulate the phosphorylation of STAT3 in part via Brk, because the increase in HER2 activity as a compensatory response to knockdown of Brk in EMT1 cells failed to elevate the level of activation-specific phosphorylation of STAT3 (note: the compensatory increase in HER2 activity also failed to elevate the level of activation-specific phosphorylation of Akt but it did elevate that of Erk). This observation may partially explain the reports in the literature of a lack of a significantly direct correlation between activated STAT3 and HER2 in specimens from breast cancer patients.Citation40
In summary, our findings suggest a new signaling circuit model in breast cancer wherein Brk is upregulated by HER2 and coordinates with HER2 and Src for maintaining the acquired phenotypes. Because silencing of Brk can induce apoptosis and inhibit the migration potential of HER2-overexpressing cells, Brk may participate in or may be required for HER2-induced oncogenic activities. Targeting Brk may therefore improve the response of breast cancer patients to HER2-targeted therapy.
Materials and Methods
Reagents
Antibodies directed against total Akt, serine 473 (S473)-phosphorylated Akt, threonine 202/tyrosine 204 (T202/Y204)-phosphorylated extracellular signal-regulated kinase (Erk), Y1248-phosphorylated HER2, poly(ADP-ribose) polymerase (PARP), Y705-phosphorylated STAT3 (STAT3-Y705), and total STAT3 were obtained from Cell Signaling Technology, Inc. Antibodies against total Erk and Brk were purchased from Santa Cruz Biotechnology, Inc. Antibodies against HER2 were purchased from Calbiochem/EMD Chemicals. All other chemicals were purchased from Sigma-Aldrich Corp. The constitutively active STAT3 plasmid was a kind gift from Dr. Suyun Huang’s laboratory at The University of Texas MD Anderson Cancer Center.
Breast cancer cell lines and culture
The MCF7, MDA468, BT474 and SKBR3 breast cancer cell lines were originally purchased from American Type Culture Collection. MCF7HER2 cells were created by experimentally elevating HER2, as described previously.Citation41 All cell lines were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS), 2 mmol/L glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin and cultured in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Generation of EMT1 subline of MCF7HER2 cells in vivo
MCF7HER2 cells infected with a recombinant firefly luciferase lentivirus were inoculated into immunocompromised nude mice via tail vein injection (2 × 106 cells in a volume of 0.1 ml serum-free medium). Colonization of these bioluminescent MCF7HER2 cells inside the mice was determined using Xenogen’s in vitro imaging system (IVIS). Three of four nude mice inoculated with luciferase-positive MCF7HER2 cells had several IVIS-positive tumors in various parts of their bodies approximately one month after inoculation. These tumors were surgically removed and minced into several small pieces for primary culture in the presence of neomycin (1000 μg/ml), which spares neomycin-resistance HER2-positive MCF7HER2 cells. The resultant pooled cell sublines were found to be 100% HER2-positive, as measured by fluorescence-activated cell sorting analysis using a HER2-specific antibody. Short tandem repeat profiling by PCR confirmed that the genetic background of the EMT1 subline matched that of MCF7 cells. The animal work was approved by institutional animal care and use committee of M D Anderson Cancer Center.
Western blot analysis
Cells were lysed in a lysis buffer containing 50 mmol/L TRIS-HCl, pH 7.4, 150 mmol/L NaCl, 0.5% NP40, 50 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride, 25 μg/mL leupeptin, and 25 μg/mL aprotinin and clarified by centrifugation (14,000 g for 30 min at 4°C). The protein concentration of the cell lysates was determined using the Bradford Coomassie blue method (Pierce Chemical Corp.). Whole-cell lysates were separated by sodium dodecyl sulfate (SDS)-PAGE, transferred onto nitrocellulose by western blotting, and probed with various primary antibodies and horseradish peroxidase–labeled secondary antibodies. The signals were visualized with an enhanced chemiluminescence detection kit (GE Healthcare).
SiRNA/shRNA and transfection
Brk siRNA oligonucleotide duplexes (sense strand: AAGGUGAUUUCUCGAGACAAC dTdT; anti-sense strand: GUUGUCUCGAGAAAUCACCUUdTdT) were purchased from Dharmacon/Thermo Fisher Scientific. Constructs containing STAT3 shRNA (shRNA1 targeting sequence: GCTGACTACACTGGCAGAGAAACTCTTGG; shRNA2 targeting sequence: TGGCTGACTGGAAGAGGCGGCAACAGATT) were purchased from OriGene. Transfection of the siRNA oligonucleotide duplexes and shRNA constructs was performed in a six-well plate (1 × 105 cells/per well) with Lipofectamine 2000 (Invitrogen, Inc.), using the methods recommended by the manufacturer. Using western blotting with specific antibodies, knockdown of Brk with siRNA was examined 48 h after siRNA transfection, and knockdown of STAT3 shRNA constructs was examined 72 h after shRNA transfection.
Immunofluorescent staining of cells
Cells were grown on sterile glass coverslips overnight in a 37°C culture incubator. Prior to immunofluorescent staining, the cells were fixed in pre-chilled -20°C methanol for 5 min and then incubated with 10% normal serum in phosphate-buffered saline (PBS) at 37°C for 30 min to block non-specific binding of IgG. The cells were then incubated with the desired primary antibodies in PBS with 1.5% normal serum at 4°C overnight. After washing the cells twice with PBS, fluorescence-conjugated secondary antibody and 4',6-diamidino-2-phenylindole (DAPI) were added onto the coverslips, and the cells were incubated in the dark at room temperature for 1.5 h. Fluorescently stained cells were examined under a fluorescence microscope.
Transwell chamber assay
Cell migration potential was measured with a Boyden transwell chamber consisting of upper inserts with 8-µm-pore-size filter membranes at the bottom of the inserts and lower wells in 24-well cell culture plates (Corning Life Sciences). Cells (3.5 × 105 cells in 0.2 mL) suspended in serum-free medium with 0.1% bovine serum albumin were seeded into the inserts of the chambers. The inserts were then placed over the wells filled with 0.5 mL 10% FBS culture medium and incubated in a 37°C incubator for 24 h. Cells that had not penetrated the filter membrane in the inserts were wiped off with cotton swabs, and the cells on the underside of the filter membrane were fixed and stained with the HEMA-3 kit (Fisher Diagnostics). Cells in 10 different microscope fields of each filter were counted. Each treatment group was set in triplicate inserts/wells.
Apoptosis assay
After treatment, the cells were measured for apoptosis using an ELISA kit (Roche Diagnostics Corp.) that quantitatively measures cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) and by western blotting with an antibody that recognizes both uncleaved and cleaved PARP, as previously described.Citation42,Citation43
Cell growth and survival assay
Cells were cultured in 24-well plates with 0.5 ml medium per well at 37°C in a CO2 incubator. After transient transfection of the cells with Brk siRNA or control siRNA for 48 h, the cells were incubated for an additional 2 h after the addition of 50 μL/well of 10 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). The cells were then lysed in a lysis buffer (500 μL/well) containing 20% SDS in dimethyl formamide/H2O (1:1, v/v; pH 4.7) at 37°C for at least 6 h. The relative number of surviving cells in each group was determined by measuring the optical density (OD) of the cell lysates at an absorbance wavelength of 570 nm. The OD value in each treatment group was then normalized to that of untreated cells as a percentage of the OD value of the control cells and plotted against the treatments.
| Abbreviations: | ||
| Brk | = | breast tumor kinase |
| EGFR | = | epidermal growth factor receptor |
| EMT | = | epithelial-to-mesenchymal transition |
| HER2 | = | human epidermal growth factor receptor-2 |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| OD | = | optical density |
| PI3K | = | phosphatidylinositol 3-kinase |
| PTK-6 | = | protein tyrosine kinase-6 |
| STAT3 | = | signal transducer and activator of transcription 3 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by US DOD CDMRP Awards W81XWH-06–1-0544 and W81XWH-07–1-0526 (to Z.F.), The Breast Cancer Research Foundation (to Z.F.), NIH grant 5R01CA129036 (to Z.F.), and Cancer Center Support Grant CA016672 from the US National Cancer Institute. We thank Dr. Suyun Huang of the Department of Neurosurgery at The University of Texas MD Anderson Cancer Center for her kind gift of STAT3 constitutively active plasmid.
Related Research Data
References
- Weigelt B, Peterse JL, van ’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5:591 - 602; http://dx.doi.org/10.1038/nrc1670; PMID: 16056258
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235:177 - 82; http://dx.doi.org/10.1126/science.3798106; PMID: 3798106
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244:707 - 12; http://dx.doi.org/10.1126/science.2470152; PMID: 2470152
- Eccles SA. The role of c-erbB-2/HER2/neu in breast cancer progression and metastasis. J Mammary Gland Biol Neoplasia 2001; 6:393 - 406; http://dx.doi.org/10.1023/A:1014730829872; PMID: 12013529
- Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology 2001; 61:Suppl 2 1 - 13; http://dx.doi.org/10.1159/000055396; PMID: 11694782
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 2011; 17:461 - 9; http://dx.doi.org/10.1038/nm.2309; PMID: 21399647
- Barker KT, Jackson LE, Crompton MR. BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene 1997; 15:799 - 805; http://dx.doi.org/10.1038/sj.onc.1201241; PMID: 9266966
- Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res 2007; 67:4199 - 209; http://dx.doi.org/10.1158/0008-5472.CAN-06-3409; PMID: 17483331
- Lee ST, Strunk KM, Spritz RA. A survey of protein tyrosine kinase mRNAs expressed in normal human melanocytes. Oncogene 1993; 8:3403 - 10; PMID: 8247543
- Mitchell PJ, Barker KT, Martindale JE, Kamalati T, Lowe PN, Page MJ, et al. Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 1994; 9:2383 - 90; PMID: 8036022
- Siyanova EY, Serfas MS, Mazo IA, Tyner AL. Tyrosine kinase gene expression in the mouse small intestine. Oncogene 1994; 9:2053 - 7; PMID: 8208550
- Qiu H, Miller WT. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J Biol Chem 2002; 277:34634 - 41; http://dx.doi.org/10.1074/jbc.M203877200; PMID: 12121988
- Serfas MS, Tyner AL. Brk, Srm, Frk, and Src42A form a distinct family of intracellular Src-like tyrosine kinases. Oncol Res 2003; 13:409 - 19; PMID: 12725532
- Harvey AJ, Crompton MR. Use of RNA interference to validate Brk as a novel therapeutic target in breast cancer: Brk promotes breast carcinoma cell proliferation. Oncogene 2003; 22:5006 - 10; http://dx.doi.org/10.1038/sj.onc.1206577; PMID: 12902983
- Harvey AJ, Crompton MR. The Brk protein tyrosine kinase as a therapeutic target in cancer: opportunities and challenges. Anticancer Drugs 2004; 15:107 - 11; http://dx.doi.org/10.1097/00001813-200402000-00002; PMID: 15075665
- Haegebarth A, Heap D, Bie W, Derry JJ, Richard S, Tyner AL. The nuclear tyrosine kinase BRK/Sik phosphorylates and inhibits the RNA-binding activities of the Sam68-like mammalian proteins SLM-1 and SLM-2. J Biol Chem 2004; 279:54398 - 404; http://dx.doi.org/10.1074/jbc.M409579200; PMID: 15471878
- Derry JJ, Richard S, Valderrama Carvajal H, Ye X, Vasioukhin V, Cochrane AW, et al. Sik (BRK) phosphorylates Sam68 in the nucleus and negatively regulates its RNA binding ability. Mol Cell Biol 2000; 20:6114 - 26; http://dx.doi.org/10.1128/MCB.20.16.6114-6126.2000; PMID: 10913193
- Lukong KE, Larocque D, Tyner AL, Richard S. Tyrosine phosphorylation of sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. J Biol Chem 2005; 280:38639 - 47; http://dx.doi.org/10.1074/jbc.M505802200; PMID: 16179349
- Liu L, Gao Y, Qiu H, Miller WT, Poli V, Reich NC. Identification of STAT3 as a specific substrate of breast tumor kinase. Oncogene 2006; 25:4904 - 12; http://dx.doi.org/10.1038/sj.onc.1209501; PMID: 16568091
- Weaver AM, Silva CM. Signal transducer and activator of transcription 5b: a new target of breast tumor kinase/protein tyrosine kinase 6. Breast Cancer Res 2007; 9:R79; http://dx.doi.org/10.1186/bcr1794; PMID: 17997837
- Mitchell PJ, Sara EA, Crompton MR. A novel adaptor-like protein which is a substrate for the non-receptor tyrosine kinase, BRK. Oncogene 2000; 19:4273 - 82; http://dx.doi.org/10.1038/sj.onc.1203775; PMID: 10980601
- Shen CH, Chen HY, Lin MS, Li FY, Chang CC, Kuo ML, et al. Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res 2008; 68:7779 - 87; http://dx.doi.org/10.1158/0008-5472.CAN-08-0997; PMID: 18829532
- Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol Cell Biol 2004; 24:10558 - 72; http://dx.doi.org/10.1128/MCB.24.24.10558-10572.2004; PMID: 15572663
- Vasioukhin V, Tyner AL. A role for the epithelial-cell-specific tyrosine kinase Sik during keratinocyte differentiation. Proc Natl Acad Sci U S A 1997; 94:14477 - 82; http://dx.doi.org/10.1073/pnas.94.26.14477; PMID: 9405638
- Zhang P, Ostrander JH, Faivre EJ, Olsen A, Fitzsimmons D, Lange CA. Regulated association of protein kinase B/Akt with breast tumor kinase. J Biol Chem 2005; 280:1982 - 91; http://dx.doi.org/10.1074/jbc.M412038200; PMID: 15539407
- Qiu H, Zappacosta F, Su W, Annan RS, Miller WT. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene 2005; 24:5656 - 64; http://dx.doi.org/10.1038/sj.onc.1208721; PMID: 15870689
- Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, et al. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene 2012; 31:4372 - 83; http://dx.doi.org/10.1038/onc.2011.608; PMID: 22231447
- Kamalati T, Jolin HE, Mitchell PJ, Barker KT, Jackson LE, Dean CJ, et al. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J Biol Chem 1996; 271:30956 - 63; http://dx.doi.org/10.1074/jbc.271.48.30956; PMID: 8940083
- Kamalati T, Jolin HE, Fry MJ, Crompton MR. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene 2000; 19:5471 - 6; http://dx.doi.org/10.1038/sj.onc.1203931; PMID: 11114724
- Born M, Quintanilla-Fend L, Braselmann H, Reich U, Richter M, Hutzler P, et al. Simultaneous over-expression of the Her2/neu and PTK6 tyrosine kinases in archival invasive ductal breast carcinomas. J Pathol 2005; 205:592 - 6; http://dx.doi.org/10.1002/path.1720; PMID: 15685689
- Xiang B, Chatti K, Qiu H, Lakshmi B, Krasnitz A, Hicks J, et al. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc Natl Acad Sci U S A 2008; 105:12463 - 8; http://dx.doi.org/10.1073/pnas.0805009105; PMID: 18719096
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927 - 39; http://dx.doi.org/10.1016/j.cell.2004.06.006; PMID: 15210113
- Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 2002; 3:155 - 66; http://dx.doi.org/10.1038/nrm757; PMID: 11994736
- Bromberg J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res 2000; 2:86 - 90; http://dx.doi.org/10.1186/bcr38; PMID: 11250696
- Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. Am J Pathol 2004; 165:1449 - 60; http://dx.doi.org/10.1016/S0002-9440(10)63403-7; PMID: 15509516
- Bromberg J. Stat proteins and oncogenesis. J Clin Invest 2002; 109:1139 - 42; PMID: 11994401
- Gao SP, Bromberg JF. Touched and moved by STAT3. Sci STKE 2006; 2006:pe30; http://dx.doi.org/10.1126/stke.3432006pe30; PMID: 16835434
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7:131 - 42; http://dx.doi.org/10.1038/nrm1835; PMID: 16493418
- Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172:973 - 81; http://dx.doi.org/10.1083/jcb.200601018; PMID: 16567498
- Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res 2007; 9:R32; http://dx.doi.org/10.1186/bcr1680; PMID: 17531096
- Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z. Sensitization of breast cancer cells to radiation by trastuzumab. Mol Cancer Ther 2003; 2:1113 - 20; PMID: 14617784
- Liu B, Fang M, Schmidt M, Lu Y, Mendelsohn J, Fan Z. Induction of apoptosis and activation of the caspase cascade by anti-EGF receptor monoclonal antibodies in DiFi human colon cancer cells do not involve the c-jun N-terminal kinase activity. Br J Cancer 2000; 82:1991 - 9; PMID: 10864208
- Li X, Luwor R, Lu Y, Liang K, Fan Z. Enhancement of antitumor activity of the anti-EGF receptor monoclonal antibody cetuximab/C225 by perifosine in PTEN-deficient cancer cells. Oncogene 2006; 25:525 - 35; PMID: 16170346