Abstract
Breast cancer can be classified into different molecular subtypes with varying clinical and pathological characteristics. The basal-like breast cancer subtype represents one of the most aggressive and lethal types of breast cancer, and due to poor mechanistic understanding, it lacks targeted therapy. Many basal-like breast cancer patient samples display alterations of established drivers of cancer development, including elevated expression of EGFR, p53 inactivating mutations and loss of expression of the tumor suppressor PTEN; however, their contribution to human basal-like breast cancer pathogenesis remains ill-defined. Using non-transformed human mammary epithelial cells, we set out to determine whether altering EGFR, p53 and PTEN in different combinations could contribute to basal-like breast cancer progression through transformation of cells. Altering PTEN in combination with either p53 or EGFR in contrast to any of the single alterations caused increased growth of transformed colonies in soft agar. Concomitantly modifying all three genes led to the highest rate of cellular proliferation and the greatest degree of anchorage-independent colony formation. Results from our effort to engineer a model of BBC expressing alterations of EGFR, p53 and PTEN suggest that these changes are cooperative and likely play a causal role in basal-like breast cancer pathogenesis. Consideration should be given to targeting EGFR and restoring p53 and PTEN signaling simultaneously as a strategy for treatment of this subtype of breast cancer.
Introduction
Breast cancer (BC) is a heterogeneous disease characterized by at least five molecular subtypes: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, normal-like and basal-like.Citation1 Basal-like breast cancer (BBC), one of the most aggressive and lethal subtypes, comprises 15–20% of all BC cases.Citation1,Citation2 BBC itself is very heterogeneous, and according to the multi-platform analysis performed by the Cancer Genome Atlas Network, BBC expresses the highest frequency of mutations along with HER2-positive BC.Citation3,Citation4 Because BBC generally lacks expression of estrogen receptor (ER), progesterone receptor (PR) and HER2, no targeted therapy is available for effective treatment.Citation1,Citation5 Hence, human mammary cell model systems that faithfully represent the alterations that are prevalent in BBC would be beneficial to delineate important molecular mechanisms that drive this subtype of BC.
This study focuses on alterations in three onco-proteins that occur at high frequency in BBC, namely epidermal growth factor receptor (EGFR), p53 and the phosphatase and tensin homolog deleted on chromosome 10 (PTEN).Citation1,Citation4,Citation6,Citation7 EGFR is a tyrosine kinase trans-membrane receptor that can activate both the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. EGFR can be altered in multiple cancers and overexpressed in up to 70% of BBC cases.Citation8,Citation9 PTEN, a negative regulator of the PI3K pathway, affects proliferation, apoptosis, migration, genomic instability and metabolism.Citation10-Citation15 Decreased PTEN expression, through loss of heterozygosity, chromosomal rearrangement and/or epigenetic silencing, is observed in over 50% of all BBC cases and in 80% of familial BBCs.Citation7,Citation16-Citation21 A recent paper suggests that PI3K/AKT pathway alteration, which can be controlled by either EGFR and/or PTEN, is the highest in BBC among all BC subtypes.Citation4 Additionally, the tumor suppressor p53, which is a regulator of apoptosis, DNA damage response, cell cycle and metabolism, has inactivating mutations in over 80% of BBCs.Citation1,Citation4,Citation21-Citation23 Furthermore, recent data now suggests that the TP53 pathway activity is altered within most, if not all, BBCs.Citation4 Taken together, EGFR, p53 and PTEN are altered at high frequency in a large subset of BBC cases, indicating the potential for these genes to cooperate in BBC initiation and/or progression in those tumors.
The MCF10A cell line is a powerful human mammary cell culture model system for studying the genetic insults that can lead to breast cancer. MCF10A cells are human, spontaneously immortalized, untransformed, non-tumorigenic, mostly diploid and lack CDKN2A.Citation17,Citation24-Citation26 These cells depend on growth factors and hormones for growth and survival, form polarized acini-like spheroids in a suspension of extracellular matrix (matrigel), grow in monolayers and do not grow in suspension in soft agar unless they are transformed by expression of one or more onco-proteins.Citation27,Citation28 For example, the disruption of proper acini formation in MCF10A cells in matrigel has been shown to occur following knock-down of BRCA1 or after the expression of mutant alleles of PIK3CA, HER2, PCDH8, only serine/threonine kinase AKT/PKB, overexpression of wild type human growth hormone (hGH), or co-expression of EGFR with c-SRC.Citation29-Citation35 MCF10A cells have been shown to grow in an anchorage-independent manner in soft agar without extracellular matrix following overexpression of mutant H-RAS, HER2/NeuT (rat c-neu with single activating point mutation) or FGFR1.Citation36-Citation38 Furthermore, MCF10A cells cluster closely with the BBC subtype in gene expression profiling studies and express markers commonly associated with a basal epithelial phenotype such as high-molecular weight cytokeratins and p63.Citation39-Citation41 They lack estrogen and progesterone expression and overexpress the MYC oncogene, which is characteristically upregulated in BBC, making these cells potentially suitable for modeling BBC tumor progression.Citation42-Citation44
Studies examining the effects of EGFR, p53 and PTEN mutations in combination have not been performed in mammary epithelial cells. However, studies examining each alteration alone have been investigated. MCF10A cells expressing a knock-in mutant EGFR (delE746-A750) displayed increased levels of total and phosphorylated EGFR, but had no advantage in growth or in their ability to form colonies in soft agar.Citation45 MCF10A cells knocked out for p53 by Weiss et al. were able to grow in the absence of EGF.Citation46 Overexpression of a truncated dominant-negative p53 (p53DD) did not affect proliferation rate in MCF10A cells in media supplemented with growth factors.Citation47 Also, either knock down of p53 or the expression of dominant negative mutant p53 (G245S) in MCF10A cells led to near-normal acini structures with incompletely cleared lumen due to decreased apoptosis.Citation48 MCF10A cells lacking PTEN exhibited increased signaling, enhanced growth on plastic, resistance to anoikis, but no increase in growth in soft agar.Citation49 Therefore, in the above studies in MCF10A cells, single manipulations of EGFR, p53 or PTEN could affect different cellular properties important for tumorigenesis but none led to transformation of mammary cells as measured by the ability to grow in an anchorage independent manner. Here, we show that EGFR overexpression together with inactivation of p53 and PTEN, alterations that are frequently observed in BBC, cooperate in cellular transformation.
Results
Single modifications of PTEN, EGFR and p53 in MCF10A affect signaling
We used MCF10A cells to construct a model system to reflect changes seen in BBCs. To do so, we stably expressed EGFR, p53 and PTEN alterations in these cells to study their combinatorial effects on transformation (). To develop a triple modified model of BBC, we first compared the effects of altering EGFR, p53 or PTEN alone. To examine the effects of EGFR, we stably overexpressed wildtype EGFR via retroviral transduction in MCF10A cells and we showed that total EGFR protein level was significantly increased (). To model the effect of p53 mutation, we stably overexpressed dominant negative p53 (p53DD) in MCF10A cells via retroviral transduction. The p53DD is a C-terminal fragment of p53 that forms functionally impaired multimers with the wildtype endogenous p53 to abrogate sequence-specific DNA binding.Citation50 Multiple groups have shown the effectiveness of p53DD in cancer models.Citation54,Citation55 The ectopic expression of p53DD was verified using an antibody to the C-terminal region in the truncated mutant (). Next, to model PTEN inactivation, we utilized MCF10A cells that harbored a targeted deletion of exon 2 of PTEN (MCF10A-PTEN−/−).Citation49 We validated that MCF10A-PTEN−/− cells express no PTEN protein as compared with the parental line (). Thus, single alterations to EGFR, TP53 or PTEN genes engineered in MCF10A cells can model changes that are found in a large proportion of BBC tumors, allowing us to analyze for their phenotypic effects in mammary cells.
Figure 1. Modeling basal-like breast cancer through altering EGFR, p53 and PTEN in MCF10A cells. (A) Studies analyzing loss of PTEN, overexpression of EGFR or loss of p53 function have not shown an oncogenic effect individually in mammary epithelial cells. Given the high incidence of these alterations in basal-like breast cancer (BBC), we hypothesize that loss of PTEN, overexpression of EGFR, and loss of p53 function together can cooperate to transform MCF10A cells for a model of BBC. (B) EGFR stable overexpression alone and an empty control cassette were expressed in MCF10A cells. Total and phospho-tyrosine EGFR antibodies were probed to show EGFR was overexpressed and active as a kinase. (C) Expression of an N-terminal deletion mutant p53DD to abrogate p53 function or empty control cassette in MCF10A cells. Lysates were probed with an antibody targeting the remaining C-terminal domain of p53. Functional validation of this p53DD was shown by lower p21 level in the p53DD sample. (D) To study the effect of loss of PTEN, we utilized MCF10A-PTEN−/− cells deleted for exon 2 and we confirmed that PTEN protein was not present compared with the parental cells. For (C-D), whole protein lysates were harvested after overnight starvation with basal media only. Lysates were probed for vinculin or actin as loading controls.
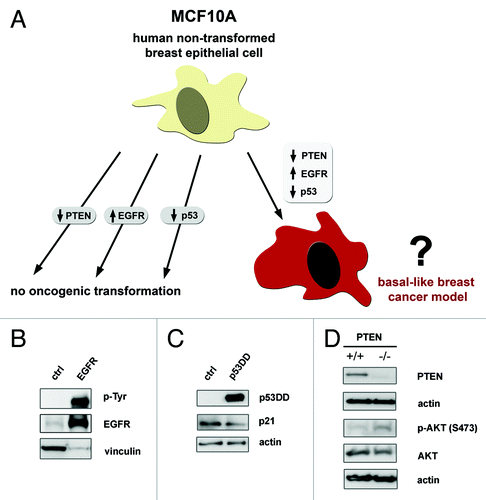
To verify that the changes to EGFR, p53 or PTEN had the predicted functional effects, we investigated signaling in our models. Cells expressing EGFR, p53DD or PTEN loss were starved overnight and total protein lysates were analyzed by western blot. We found that the transduced EGF receptor was functionally active by the increased total tyrosine phosphorylation on EGFR in the cells overexpressing EGFR as compared with control cells (). Consistent with an effective dominant-negative disruption of p53 function, the p53DD infected cells displayed a decreased level of p21, a critical cell cycle downstream effector of p53 activation (). AKT activation at serine-473 was increased by PTEN mutation (). Therefore, we observed signaling changes that were expected as the result of overexpressing EGFR, p53DD or losing PTEN in our system.
EGF-independent growth is feasible only in PTEN-null cells and not in EGFR- or p53DD-expressing MCF10As
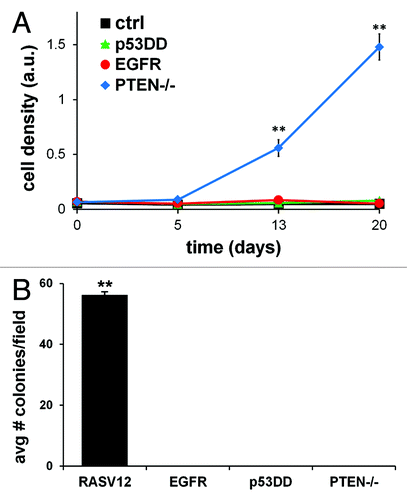
We characterized the growth properties of MCF10A cells harboring overexpression of EGFR, p53DD or loss of PTEN to compare each alteration’s effect on proliferation under starved conditions. Non-transformed cells are dependent on growth factor signaling for their ability to proliferate; thus, in the absence of the appropriate mitogenic signals, they do not grow.Citation56 Proliferation was determined by assessing the relative cell accumulation at different time points, as previously described.Citation57 Cells were maintained in media devoid of any growth supplement for a period of 20 d. When cells expressing different single alterations were compared for their ability to grow, we observed distinct differences in their proliferation capacity. Consistent with prior observations,Citation49 loss of PTEN allowed cells to proliferate without growth factor stimulation (). MCF10A cells expressing EGFR or p53DD did not proliferate under identical conditions. Therefore, whereas losing PTEN expression alone was an instigator for cell growth in the absence of growth factors, exogenous expression of either EGFR or p53DD was not.
Figure 2. In vitro tumorigenic properties of single-modified MCF10A cells. (A) MCF10A cells expressing EGFR, p53DD, PTEN loss or empty control were plated in 48 well plates in quadruplicate and grown without growth factors for a period of 20 d. Cells were stained with crystal violet dye at days 0, 5, 13 and 20 and analyzed for growth rate differences. The means ± SD for the four experiments are shown and statistical analysis was calculated by chi-square test. (B) Soft agar assay of MCF10A cells tested for anchorage-independent growth of each single alteration in MCF10A cells. MCF10A cells overexpressing constitutively active mutant HRAS (RASV12) was utilized as an internal positive control for this assay. The means ± SD for the three experiments are shown and statistical analysis was calculated by chi-square test.
Growth in soft agar is not permissible by altering EGFR, p53 or PTEN individually
Anchorage-independent growth in soft agar is a property of transformed cells that best correlates with tumorigenesis.Citation58,Citation59 Therefore, we sought to determine the anchorage-independent capacity of any single alteration in MCF10A BBC model cells to grow in soft agar. MCF10A cells are non-transformed cells and by themselves do not to form colonies in soft agar, as previously established.Citation60 MCF10A cells that overexpressed oncogenic H-RasV12, as our positive control cells, induced colonies in soft agar (), as previously reported.Citation36 However, none of the singly-modified MCF10A cells grew as colonies in soft agar. This is in agreement with previous studies reporting that neither an EGFR activating mutant nor a PTEN-null mutant can induce colony formation when expressed in MCF10A cells.Citation33,Citation49 It has also been shown that p53DD expression in hTERT-immortalized human mammary epithelial cells do not grow in soft agar.Citation55 Individually, none of these three alterations in MCF10A cells confer anchorage-independent growth property in soft agar.
Loss of PTEN affects proliferation regardless of presence of EGFR or p53DD
After analyzing the signaling and growth phenotype of cells expressing only one alteration at a time, we engineered cells to express two alterations to study their combinatorial effect for BBC modeling. We stably overexpressed wildtype EGFR and/or p53DD in MCF10A or MCF10A-PTEN−/− cells to generate the three new experimental cell lines, herein named EGFR-p53DD, p53DD-PTEN−/− and EGFR-PTEN−/−. Additionally, we stably expressed the control vectors as our control line, herein named ctrl. PTEN−/− cells expressing EGFR or p53DD grew better than EGFR-p53DD or ctrl cells, neither of which grew under starvation conditions (). Interestingly, neither EGFR-PTEN−/− nor p53DD-PTEN−/− cells grew significantly better than PTEN−/− cells alone, suggesting that exogenous expression of EGFR or p53DD did not confer a growth advantage under the same conditions. Overall, the increased proliferation due to PTEN loss was not affected by co-expression of either EGFR or p53DD.
Figure 3. Double- and triple- modified cells grow in absence of growth factors and in soft agar. (A) Proliferation of cells was assessed for the double- and triple-modified cells. Cells were plated in quadruplicate and grown in 48-well plates in media lacking growth factors for a period of 20 d before staining cells with crystal violet dye for analysis. As a double-modified control, empty control vectors for both pBABE-puro and pCMV-hyg (herein named ctrl) were expressed in MCF10A cells. The means ± SD for the four experiments are shown and P values were calculated by chi-square test. (B–C) Soft agar colony assay for the double- and triple-modified cells show their ability to form colonies in an anchorage-independent fashion. Each cell line was plated in soft agar in triplicate. Representative photographs of colonies were taken at 4× magnification. The means ± SD for the three experiments for each line are shown and P values were calculated by chi-square test. (D) Signaling was measured for the double- and triple-modified lines. Cell lysates were harvested after overnight starvation and analyzed for EGFR, AKT and ERK signaling. Western blot exposures for each antibody were taken from the same blot and irrelevant intervening lanes removed.
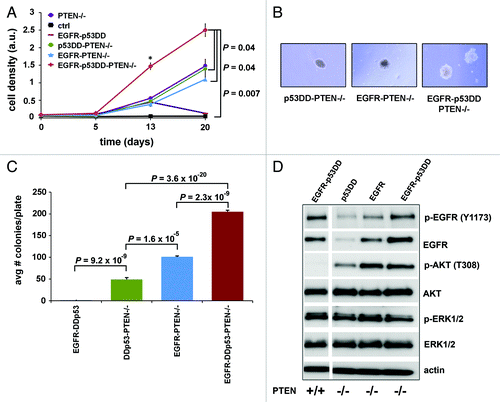
Either EGFR or p53DD cooperates with PTEN mutation for anchorage-independent colony growth in soft agar
Given that MCF10A cells with alterations in EGFR, p53 or PTEN were not able to confer anchorage-independent growth individually, we sought to find out if double combinations of those altered genes would allow for soft agar colony formation. We observed that the double-modified EGFR-PTEN−/− and p53DD-PTEN−/− cells were capable of growing colonies in suspension in soft agar (). On the other hand, EGFR-p53DD did not cooperate to transform cells as measured by growth in soft agar. We saw that the double-modified cells expressing PTEN−/− as one of their alterations grew significantly more colonies when compared with EGFR-p53DD (p = 9.2x10−9 when comparing EGFR-p53DD to p53DD-PTEN−/−). The lack of soft agar growth by EGFR-p53DD cells is consistent with their significantly lower AKT activation at threonine-308 residue compared with EGFR-PTEN−/− and p53DD-PTEN−/− cells that formed colonies (, lane 1 compared with lanes 2 and 3). These experiments demonstrate that although EGFR-p53DD did not cooperate to grow in suspension in soft agar, PTEN loss with either EGFR or p53DD did significantly synergize to grow colonies in an anchorage-independent fashion. Interestingly, EGFR-PTEN−/− cells grew on average twice as many colonies per plate as compared with p53DD-PTEN−/− cells (p = 1.6x10−5; ), consistent with the known effects of EGFR signaling on additional tumor-promoting signaling pathways. This result is consistent with higher phosphorylation and activation of AKT at threonine-308 in EGFR-PTEN−/− cells as compared with p53DD-PTEN−/− cells (, compare lanes 2 and 3). Therefore, the ability of either p53DD or EGFR to potentiate soft agar growth in the context of PTEN mutation suggests that either altered p53 or EGFR have the potential to cooperate with PTEN loss to stimulate tumor progression.
Increased growth by co-expression of PTEN, EGFR and p53 in cells
Next, we investigated whether all three alterations together could show cooperation beyond what was observed with the double changes. Cells stably expressing all three changes, herein called EGFR-p53DD-PTEN−/−, were compared with the other modified cells for their ability to grow in media devoid of all growth factors. Strikingly, we observed enhancement in the rate of growth of EGFR-p53DD-PTEN−/− cells as compared with the various double-modified cell lines. Our triple-modified cells proliferated significantly more than any of the PTEN-null double-modified cells (p = 0.04) or the ctrl cell line (p = 0.007) (). Therefore, we observed the greatest cooperation in proliferative ability when all three alterations were concomitantly present.
We also compared the ability of the triple-modified cells to grow colonies in soft agar relative to the double-modified lines (). Our results with soft agar show that when all three alterations were expressed together, the transformation phenotype became more pronounced (). There was a statistically significant increase in the average number of colonies in EGFR-p53DD-PTEN−/− cells as compared with the double-modified cells capable of growing in soft agar, EGFR-PTEN−/− and p53DD-PTEN−/− cells (p = 2.3 × 10−9 and p = 3.6 × 10−20, respectively; ). Therefore, we showed that concomitant alteration of EGFR, p53 and PTEN maximized the number of colonies in soft agar, a characteristic of transformed cells. The triple modified cells were unable to form tumors in SCID mice (n = 10) after at least 3 mo.
Triple-modified cells had a notable difference in signaling as compared with the double-modified lines. Whereas AKT activation in EGFR-PTEN−/− cells showed a similar degree as the EGFR-p53DD-PTEN−/− cells, EGFR activation was highest in EGFR-p53DD and the triple-modified cells (). Therefore high activation of both the AKT and EGFR signaling was seen only in the triple modified cells. We observed no ERK activation difference between the double- and the triple-modified cells. We conclude that the triple modified line was unique in its activation of both AKT and EGFR signaling as compared with any of the double-modified lines.
Discussion
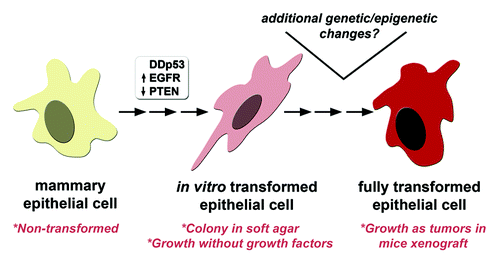
The ultimate goal of human cancer models is to manipulate genes that are altered in human tumors to demonstrate whether their expression can lead to tumorigenesis of normal cells.Citation16,Citation61 Previous human mammary epithelial cancer cell models have provided important insights into critical pathways involved in cancer formation.Citation30,Citation46,Citation49,Citation53,Citation62-Citation64 EGFR, p53 and PTEN alterations frequently occur in a relatively large proportion of the basal-like subtype of breast cancer, but their combinatorial expression has not been examined for cancer initiation in mammary epithelial cells. Therefore, we sought to determine if these three onco-proteins are able to faithfully model initiation of BBC when expressed in MCF10A cells, a well-established model for studying the effects of genes in mammary tumorigenesis.Citation28,Citation30-Citation32,Citation38 Here we showed that EGFR, p53 and PTEN alterations cooperate in MCF10A cells to drive transformation as measured by the ability of cells to grow in an anchorage-independent fashion or on plastic in the absence of growth factors (). Enhanced growth was associated with activation of biochemical signaling on multiple pathways. Our cell line models with mutant PTEN and overexpression of EGFR and/or p53DD that transform MCF10A cells can serve as a foundation for identifying additional driver insults (epigenetic and/or genetic) capable of making the cells more transformed and tumorigenic in mice.
Figure 4. A MCF10A model for BBC transformation. We demonstrate that introducing three highly prevalent somatic alterations (EGFR, p53, PTEN) in BBC biopsies was capable of transforming MCF10A cells as shown by growth in the absence of growth factors and anchorage-independent colony formation in soft agar. Given the high frequency of other somatic genetic and epigenetic changes found in BBC, additional genetic and/or epigenetic changes likely contribute to further transform the EGFR-p53DD-PTEN−/− cell line model in vitro and in vivo.
We demonstrate that cooperation between alteration of EGFR, p53 and PTEN caused transformation of MCF10A cells as measured by causing both the ability to grow in the absence of growth factors and in suspension in soft agar. As cells become transformed, they lose their dependence on growth factors and are able to grow devoid of them.Citation65 As a single event, losing PTEN was the most important alteration in mammary epithelial cells as it was the only alteration to cause them to grow in the absence of mitogenic stimuli. When all three alterations were expressed, our results indicated an additive effect as cells grew the fastest in this triple combination in the absence of growth factors. However, no single alteration was able to induce growth in soft agar. The ability of cells to grow in suspension was only permitted when PTEN inactivation cooperated with either loss of p53 function through p53DD expression or EGFR overexpression. Thus loss of PTEN synergized with p53 or EGFR alteration in this setting. Further synergy in soft agar was observed when all three alterations were expressed together to form 2- to 4-fold more colonies as compared with the double-modified cells. The additive effect observed when EGFR, p53 and PTEN are all altered in the anchorage-independent assay is in accordance with the model for cancer progression where a series of stepwise alterations have to accrue for cells to become transformed and display the different hallmarks of cancer.Citation66,Citation67
The substantial difference in phenotype between EGFR-PTEN−/− and triple-modified cells would indicate an important selective pressure to inactivate p53, which is highly supported by data showing that over 80% of BBCs have mutations in p53 or in a component of its pathway.Citation4 Further work is needed to identify the important target proteins coordinating the advantage gained by the triple-modified cells. We know that altering EGFR, p53 and PTEN pathways can regulate a myriad of crucial functions such as proliferation, apoptosis, protein translation, metabolism, DNA damage response and cell cycle control, which is likely why the triple-modified cells grow better than the double-modified cells.
While no studies have investigated the combined effect of altering PTEN, EGFR and p53 together, prior studies have been performed in which two of these genes were altered in combination. PTEN mutation was shown to cooperate with EGFR activation in human glioblastoma.Citation68 Inactivation of both PTEN and p53 was sufficient to induce invasive prostate cancer in a mouse model but inactivation of just one alteration was not sufficient for tumorigenesis.Citation69 The cooperation between EGFR and p53 was shown to expand a subpopulation of esophageal cells to go through epithelial-to-mesenchymal transition, a phenotype of transformed cells.Citation70 Similar to the above studies in other systems, we have also found evidence that PTEN deletion cooperates with p53 and/or EGFR alteration in mammary epithelial cells.
Our results demonstrate that beyond their individual contributions, the combined alterations of EGFR, p53 and PTEN together cooperate to bestow features of cellular transformation on MCF10A cells. Therefore, our findings suggest that coincident molecular alterations of these three genes, which can be found in human breast tumor biopsies, contribute to BBC pathogenesis. Finally, our triple modified MCF10A cell line system for BBC may provide a useful platform for pharmacological studies to explore the potential therapeutic benefit of simultaneous targeting of these three genes in BBC.
Methods
Cell culture
MCF10A-PTEN−/− (clones 1 and 3) and their parental line were a kind gift from Kurtis Bachman (Glaxo Smith Kline) and Ben Ho Park (Johns Hopkins University).Citation49 Cells were cultured at 37°C and 5% CO2 in DMEM/F12 50:50 (Fisher Scientific) media supplemented with 5% horse serum, 20 ng/ml of EGF, 10 µg/ml insulin, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin and 1% penicillin/streptomycin solution. For signaling, cells were starved in DMEM/F12 media without growth factors overnight and washed twice with cold phosphate buffer saline (PBS) before lysis, as described below. All experiments were performed with two different MCF10A-PTEN−/− clones, which showed similar results.
Retroviral production
Stable cell lines were generated by serial viral infection of MCF10A or MCF10A-PTEN−/− cells with retroviral supernatants expressing pCMV-p53DD-hygromycin or pBABE-EGFR-puromycin. The p53DD vector was a generously kind gift from Moshe Oren (Weizmann Institute of Science).Citation50 The EGFR vector was obtained from Addgene (plasmid 11011).Citation51 Empty vectors for pCMV and pBABE were used as negative controls. Retroviral production in phoenix cells was performed as previously described.Citation52 Cultured viral supernatants were collected after 48 h. Cells were infected with filtered viral supernatants in the presence of 12 µg/ml polybrene (Sigma). Post viral infection, successfully transduced polyclonal population of cells was obtained when selected with the appropriate drugs (hygromycin at 100 µg/ml for p53DD and puromycin at 1 µg/ml for EGFR).
Immunoblotting
Cells were lysed in 1.25 M TRIS-HCl, pH 6.8, 10% β-mercaptoethanol, 10% sodium dodecyl sulfate (SDS), 20% glycerol, 0.5% bromophenol blue solution and 8 M urea. Protein lysates were boiled for five min and 25 µg of total protein was separated on 10% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes. Membranes were blocked and incubated with primary antibodies followed by secondary antibody incubation. The primary antibodies were PTEN (6H2.1, Cascade); EGFR (1005, Santa Cruz); phospho-tyrosine (4G10, Fisher); p53 (Ab-1, Calbiochem); phospho-EGFR Y-1173 (Cell Signaling), AKT, phospho-AKT-S473, phospho-AKT-T308, ERK-p44/42, phospho-ERK-p44/42 (all Cell Signaling) and actin (AC-15, Sigma). Membranes were developed with an enhanced chemiluminescence detection kit (SuperSignal West Pico, Fisher) according to manufacturer’s instructions.
Proliferation assay
MCF10A cells were plated in 48 well plates at a density of 1 × 104 cells per well. At the indicated time points, cells were washed with PBS and fixed and stained with 0.05% crystal violet (JT Baker) in formalin for 20 min. Cells were rinsed three times with PBS to remove excess stain and allowed to dry overnight. Stained cells were analyzed by extracting the cell-associated dye with 1 ml 10% acetic acid per well and shaking plates for 3 h at room temperature. Optical density was measured at 565 nm. Samples were performed in quadruplicates.
Anchorage-independent soft agar growth and transplant assays
For soft agar colony formation assays, 2.5 × 104 MCF10A cells were seeded per 60 mm plate with a bottom layer of 0.6% Bacto agar (BD Biosciences) and a top layer of 0.3% Bacto agar, both in complete media. Colonies were photographed after three weeks of incubation and analyzed. The total number of colonies per cell line was counted per plate. Colonies were counted in at least four different views to calculate the average value. Soft agar was performed in triplicate for each sample. For evaluation of cell growth in mice, 1 × 106 cells were injected into the mammary fat pad of female SCID mice (Harlan Laboratories) at 1:1 ratio with matrigel and monitored by palpation for a minimum of three months. Mice were housed in the mouse facility of the Irving Cancer Research Center and were treated in accordance with Columbia University Institutional Animal Care and Use Committee (IACUC).
Statistical Analysis
Proliferation rate and soft agar assays were analyzed using the chi-square test, with a p-value ≤ 0.05 defined as statistically significant.
| Abbreviations: | ||
| BC | = | breast cancer |
| BBC | = | basal-like breast cancer |
| PTEN | = | phosphatase and tensin homolog deleted on chromosome 10 |
| EGFR | = | epidermal growth factor receptor |
| HER2 | = | human epidermal growth factor receptor 2 |
| p53DD | = | dominant negative p53 |
| PI3K | = | phosphatidylinositol 3-kinase |
Acknowledgments
M.M.P. was awarded and supported by the Department of Defense Predoctoral Breast Cancer Traineeship Award (W81XWH-09–1-0045). This work was also supported by the following grants to R.E.P.: National Cancer Institute (P01CA97403), the Susan G. Komen for the Cure Foundation and The Manhasset Women’s Coalition Against Breast Cancer. We thank the members of the Parsons laboratory for helpful discussions and feedback, particularly Dr Megan Keniry. We thank Dr Marcos M. Pires (Lehigh University) for helpful discussions and help with the manuscript. We thank Dr Matthew Maurer and Dr Ying-ka Ingar Lau (Columbia University) for helpful discussions.
Disclosure of Potential Conflicts of Interest
The authors have no conflict of interest or financial interest to disclose.
Related Research Data
References
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001; 98:10869 - 74; http://dx.doi.org/10.1073/pnas.191367098; PMID: 11553815
- Da Silva L, Clarke C, Lakhani SR. Demystifying basal-like breast carcinomas. J Clin Pathol 2007; 60:1328 - 32; http://dx.doi.org/10.1136/jcp.2006.041731; PMID: 17496191
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012; 486:395 - 9; PMID: 22495314
- Cancer Genome Atlas N, Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61 - 70; http://dx.doi.org/10.1038/nature11412; PMID: 23000897
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13:4429 - 34; http://dx.doi.org/10.1158/1078-0432.CCR-06-3045; PMID: 17671126
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 2003; 100:8418 - 23; http://dx.doi.org/10.1073/pnas.0932692100; PMID: 12829800
- Saal LH, Gruvberger-Saal SK, Persson C, Lövgren K, Jumppanen M, Staaf J, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet 2008; 40:102 - 7; http://dx.doi.org/10.1038/ng.2007.39; PMID: 18066063
- Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol 2006; 19:264 - 71; http://dx.doi.org/10.1038/modpathol.3800528; PMID: 16341146
- Siziopikou KP, Cobleigh M. The basal subtype of breast carcinomas may represent the group of breast tumors that could benefit from EGFR-targeted therapies. Breast 2007; 16:104 - 7; http://dx.doi.org/10.1016/j.breast.2006.09.003; PMID: 17097880
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95:29 - 39; http://dx.doi.org/10.1016/S0092-8674(00)81780-8; PMID: 9778245
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A 1999; 96:6199 - 204; http://dx.doi.org/10.1073/pnas.96.11.6199; PMID: 10339565
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998; 273:13375 - 8; http://dx.doi.org/10.1074/jbc.273.22.13375; PMID: 9593664
- Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005; 7:193 - 204; http://dx.doi.org/10.1016/j.ccr.2005.01.009; PMID: 15710331
- Puc J, Parsons R. PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle 2005; 4:927 - 9; http://dx.doi.org/10.4161/cc.4.7.1795; PMID: 15970699
- Smith U. PTEN--linking metabolism, cell growth, and cancer. N Engl J Med 2012; 367:1061 - 3; http://dx.doi.org/10.1056/NEJMe1208934; PMID: 22970949
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang XM, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 2005; 65:2554 - 9; http://dx.doi.org/10.1158/0008-5472-CAN-04-3913; PMID: 15805248
- Higgins MJ, Beaver JA, Wong HY, Gustin JP, Lauring JD, Garay JP, et al. PIK3CA mutations and EGFR overexpression predict for lithium sensitivity in human breast epithelial cells. Cancer Biol Ther 2011; 11:358 - 67; http://dx.doi.org/10.4161/cbt.11.3.14227; PMID: 21124076
- Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PLM, et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999; 155:1253 - 60; http://dx.doi.org/10.1016/S0002-9440(10)65227-3; PMID: 10514407
- Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol 2001; 14:672 - 6; http://dx.doi.org/10.1038/modpathol.3880371; PMID: 11454999
- Hu XL, Stern HM, Ge L, O’Brien C, Haydu L, Honchell CD, et al. Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol Cancer Res 2009; 7:511 - 22; http://dx.doi.org/10.1158/1541-7786.MCR-08-0107; PMID: 19372580
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006; 295:2492 - 502; http://dx.doi.org/10.1001/jama.295.21.2492; PMID: 16757721
- Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006; 25:5846 - 53; http://dx.doi.org/10.1038/sj.onc.1209876; PMID: 16998499
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009; 9:691 - 700; http://dx.doi.org/10.1038/nrc2715; PMID: 19759539
- Cowell JK, LaDuca J, Rossi MR, Burkhardt T, Nowak NJ, Matsui S. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet Cytogenet 2005; 163:23 - 9; http://dx.doi.org/10.1016/j.cancergencyto.2005.04.019; PMID: 16271952
- Yoon DS, Wersto RP, Zhou WB, Chrest FJ, Garrett ES, Kwon TK, et al. Variable levels of chromosomal instability and mitotic spindle checkpoint defects in breast cancer. Am J Pathol 2002; 161:391 - 7; http://dx.doi.org/10.1016/S0002-9440(10)64194-6; PMID: 12163363
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr., Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 1990; 50:6075 - 86; PMID: 1975513
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr., Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 1990; 50:6075 - 86; PMID: 1975513
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30:256 - 68; http://dx.doi.org/10.1016/S1046-2023(03)00032-X; PMID: 12798140
- Yu JS, Koujak S, Nagase S, Li CM, Su T, Wang X, et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene 2008; 27:4657 - 65; http://dx.doi.org/10.1038/onc.2008.101; PMID: 18408767
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 2005; 65:10992 - 1000; http://dx.doi.org/10.1158/0008-5472.CAN-05-2612; PMID: 16322248
- Muthuswamy SK, Li DM, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol 2001; 3:785 - 92; http://dx.doi.org/10.1038/ncb0901-785; PMID: 11533657
- Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol 2003; 163:315 - 26; http://dx.doi.org/10.1083/jcb.200304159; PMID: 14568991
- Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen GS, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res 2007; 67:4164 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-06-2580; PMID: 17483327
- Zhu T, Starling-Emerald B, Zhang X, Lee KO, Gluckman PD, Mertani HC, et al. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res 2005; 65:317 - 24; PMID: 15665309
- Furuta S, Jiang XZ, Gu BN, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A 2005; 102:9176 - 81; http://dx.doi.org/10.1073/pnas.0503793102; PMID: 15967981
- Ciardiello F, McGeady ML, Kim N, Basolo F, Hynes N, Langton BC, et al. Transforming growth factor-alpha expression is enhanced in human mammary epithelial cells transformed by an activated c-Ha-ras protooncogene but not by the c-neu protooncogene, and overexpression of the transforming growth factor-alpha complementary DNA leads to transformation. Cell Growth Differ 1990; 1:407 - 20; PMID: 1981145
- Martínez-Lacaci I, Kannan S, De Santis M, Bianco C, Kim N, Wallace-Jones B, et al. RAS transformation causes sustained activation of epidermal growth factor receptor and elevation of mitogen-activated protein kinase in human mammary epithelial cells. Int J Cancer 2000; 88:44 - 52; http://dx.doi.org/10.1002/1097-0215(20001001)88:1<44::AID-IJC7>3.0.CO;2-8; PMID: 10962438
- Xian W, Pappas L, Pandya D, Selfors LM, Derksen PW, de Bruin M, et al. Fibroblast growth factor receptor 1-transformed mammary epithelial cells are dependent on RSK activity for growth and survival. Cancer Res 2009; 69:2244 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-08-3398; PMID: 19258500
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515 - 27; http://dx.doi.org/10.1016/j.ccr.2006.10.008; PMID: 17157791
- Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A 1999; 96:9212 - 7; http://dx.doi.org/10.1073/pnas.96.16.9212; PMID: 10430922
- DiRenzo J, Signoretti S, Nakamura N, Rivera-Gonzalez R, Sellers W, Loda M, et al. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer Res 2002; 62:89 - 98; PMID: 11782364
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515 - 27; http://dx.doi.org/10.1016/j.ccr.2006.10.008; PMID: 17157791
- Worsham MJ, Pals G, Schouten JP, Miller F, Tiwari N, van Spaendonk R, et al. High-resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res Treat 2006; 96:177 - 86; http://dx.doi.org/10.1007/s10549-005-9077-8; PMID: 16319984
- Shekhar PVM, Chen ML, Werdell J, Heppner GH, Miller FR, Christman JK. Transcriptional activation of functional endogenous estrogen receptor gene expression in MCF10AT cells: a model for early breast cancer. Int J Oncol 1998; 13:907 - 15; PMID: 9772278
- Di Nicolantonio F, Arena S, Gallicchio M, Zecchin D, Martini M, Flonta SE, et al. Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. Proc Natl Acad Sci U S A 2008; 105:20864 - 9; http://dx.doi.org/10.1073/pnas.0808757105; PMID: 19106301
- Weiss MB, Vitolo MI, Mohseni M, Rosen DM, Denmeade SR, Park BH, et al. Deletion of p53 in human mammary epithelial cells causes chromosomal instability and altered therapeutic response. Oncogene 2010; 29:4715 - 24; http://dx.doi.org/10.1038/onc.2010.220; PMID: 20562907
- Sheen JH, Dickson RB. Overexpression of c-Myc alters G(1)/S arrest following ionizing radiation. Mol Cell Biol 2002; 22:1819 - 33; http://dx.doi.org/10.1128/MCB.22.6.1819-1833.2002; PMID: 11865060
- Zhang YH, Yan WS, Chen XB. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem 2011; 286:16218 - 28; http://dx.doi.org/10.1074/jbc.M110.214585; PMID: 21454711
- Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, et al. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res 2009; 69:8275 - 83; http://dx.doi.org/10.1158/0008-5472.CAN-09-1067; PMID: 19843859
- Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol 1992; 12:5581 - 92; PMID: 1448088
- Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2005; 2:e313; http://dx.doi.org/10.1371/journal.pmed.0020313; PMID: 16187797
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 2003; 3:483 - 95; http://dx.doi.org/10.1016/S1535-6108(03)00088-6; PMID: 12781366
- Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res 2009; 69:6299 - 306; http://dx.doi.org/10.1158/0008-5472.CAN-09-0820; PMID: 19602588
- Zhao JJ, Liu ZN, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 2005; 102:18443 - 8; http://dx.doi.org/10.1073/pnas.0508988102; PMID: 16339315
- Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol 2007; 81:7069 - 76; http://dx.doi.org/10.1128/JVI.00115-07; PMID: 17442716
- McCormick F. Signalling networks that cause cancer. Trends Cell Biol 1999; 9:M53 - 6; http://dx.doi.org/10.1016/S0962-8924(99)01668-2; PMID: 10611683
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593 - 602; http://dx.doi.org/10.1016/S0092-8674(00)81902-9; PMID: 9054499
- Shin SI, Freedman VH, Risser R, Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A 1975; 72:4435 - 9; http://dx.doi.org/10.1073/pnas.72.11.4435; PMID: 172908
- Cifone MA, Fidler IJ. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci U S A 1980; 77:1039 - 43; http://dx.doi.org/10.1073/pnas.77.2.1039; PMID: 6928659
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet 1999; 21:111 - 4; http://dx.doi.org/10.1038/5056; PMID: 9916802
- Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res 2005; 7:171 - 9; http://dx.doi.org/10.1186/bcr1275; PMID: 15987472
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001; 15:50 - 65; http://dx.doi.org/10.1101/gad.828901; PMID: 11156605
- Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A 2009; 106:2835 - 40; http://dx.doi.org/10.1073/pnas.0813351106; PMID: 19196980
- Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One 2010; 5:e9201; http://dx.doi.org/10.1371/journal.pone.0009201; PMID: 20169162
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature 1999; 400:464 - 8; http://dx.doi.org/10.1038/22780; PMID: 10440377
- Farber E. The multistep nature of cancer development. Cancer Res 1984; 44:4217 - 23; PMID: 6467183
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- Pore N, Liu S, Haas-Kogan DA, O’Rourke DM, Maity A. PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Cancer Res 2003; 63:236 - 41; PMID: 12517803
- Chen ZB, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725 - 30; http://dx.doi.org/10.1038/nature03918; PMID: 16079851
- Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 2010; 70:4174 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-09-4614; PMID: 20424117