Abstract
Arsenic Trioxide (As2O3) is one of the most effective agents in the treatment of acute promyelocytic leukemia (APL), but has no significant efficacy in other forms of AML. The mechanisms of relative resistance of non-APL cells are not well understood, but emerging evidence suggests that activation of negative feedback regulatory loops and pathways contributes to such resistance. We provide evidence that a signaling cascade involving the kinase RSK1 is engaged in a negative feedback manner during arsenic-treatment of cells and exhibits regulatory effects on growth and survival of AML cells in response to treatment with As2O3. Our data demonstrate that pharmacological inhibition or molecular disruption of expression of RSK1 enhances As2O3-dependent apoptosis and/or growth inhibition of AML cells. Importantly, combination of a pharmacological inhibitor of RSK and As2O3 results in enhanced suppression of primary AML leukemic progenitors. Altogether, our findings suggest an important regulatory role for RSK1 in the generation of the effects of As2O3 in AML cells. They also raise the potential of RSK1 targeting in combination with As2O3 as a novel approach to promote antileukemic responses.
Keywords: :
Introduction
Arsenic trioxide (As2O3) has major clinical activity in the treatment of refractory acute promyelocytic leukemia (APL), at generally well tolerated doses.Citation1-Citation4 Its introduction in the treatment of this form of acute myeloid leukemia (AML) has had a dramatic impact in the management and outcome of this disease,Citation1-Citation4 but has not been extended to the treatment of other hematological malignancies. There have been trials to test its efficacy in other non-APL subtype refractory or relapsed AML cases, but in these cases arsenic trioxide had no significant clinical activity.Citation5 Such lack of responses in non-APL cases may well be a reflection of the relative resistance and the requirement for high concentrations for arsenic-induced apoptosis.Citation6 Other recent evidence has suggested that beyond apoptosis, induction of autophagy may be a major mechanism for the generation of the antileukemic properties of arsenic trioxide,Citation7-Citation9 underscoring the complexity and diversity of mechanisms that may account for leukemic cell resistance to its effects.
The MEK-ERK pathway is constitutively activated in the majority of primary AML cases and has been the focus of clinical-translational interest for the treatment of AML.Citation10 MEK is downstream of the RAS/RAF pathway,Citation10 which is activated in AML by RAS mutations, as well as mutations or overexpression of upstream receptor tyrosine kinases such as FLT3.Citation11,Citation12 The downstream effector of MEK, ERK1/2, is active in the majority of patients with AML.Citation13 An important group of ERK substrates includes the RSK (90 kDa ribosomal S6 kinase) family of kinases (RSK 1–4), whose activities regulate cellular effectors that promote cell growth and survival.Citation14
In previous work we had shown that the p38 MAPK pathway and various upstream and downstream effectors of this pathway act as negative feedback regulators for As2O3-induced antileukemic responses,Citation15-Citation19 while others have shown that targeting MEK/ERK pathways promotes the pro-apoptotic effects of arsenic trioxide in multiple myeloma cells.Citation20 RSK1 is a kinase activated downstream of the MEK/ERK and the PI3′K-PDK1 pathways and mediates important downstream signals.Citation21
In the present study we examined the effects of As2O3-treatment of AML cells on the activation of RSK1 and the generation of arsenic-dependent antileukemic responses. Our data demonstrate that during treatment of different AML cell lines with arsenic trioxide there is phosphorylation/activation of RSK1. Combinations of an RSK1 inhibitor with arsenic trioxide were found to result in more potent suppression of leukemic progenitor colony formation than each agent along, suggesting that RSK1 is activated in a negative feedback regulatory manner to counteract arsenic-dependent antileukemic responses. Similarly, increased arsenic-dependent antileukemic effects in vitro were seen in cells in which RSK1 was knocked down. Altogether, our findings identify RSK1 as a potentially important target to enhance the antileukemic properties of arsenic trioxide.
Results
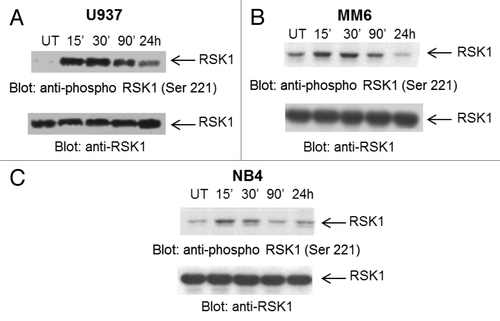
In initial studies we examined the effects of As2O3 treatment on the phosphorylation status of RSK1 in different acute myeloid leukemia cell lines. When the U937 (), MM6 () or NB4 () AML cell lines were treated with As2O3, we found that there was time-dependent induction of phosphorylation of RSK1 (). Such phosphorylation was rapid, occurring within 15 min of treatment of cells. Maximum phosphorylation was detected at approximately 15 to 30 min and the signal subsequently declined, although it was still detectable at 90 min (). The finding that As2O3 induces phosphorylation of RSK1 in various AML cell lines raised the possibility that this kinase may be activated during arsenic-treatment of cells in a negative-feedback regulatory manner, similarly to what has been previously shown for other arsenic-induced negative feedback pathways.Citation15-Citation19,Citation22-Citation24
Figure 1. As2O3 induces phosphorylation of RSK1. U937 (A), MM6 (B) and NB4 (C) cells were treated with As2O3 for the indicated times. (Upper panels) Total cell lysates were resolved by SDS-PAGE and immunoblotted with an antibody against the phosphorylated form of RSK1 on Ser 221. (Lower panels) Equal amounts of cell lysates from the same experiments shown in the upper panels were analyzed separately by SDS-PAGE and immunoblotted with an anti-RSK1 antibody, as indicated.
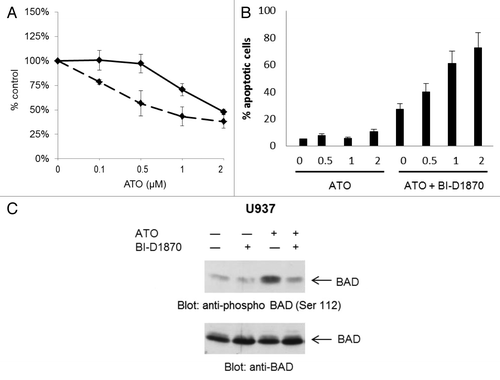
To examine the role of RSK1 in the generation of As2O3-induced functional responses, experiments were performed in which U937 leukemia cells were treated in the presence or absence of As2O3 and/or the RSK inhibitor BI-D1870 and cell viability was determined by MTT assays. The combination of As2O3 with BI-D1870 significantly inhibited cell proliferation/viability when compared with either As2O3 or BI-D1870 alone (). Similarly, when induction of apoptosis was assessed by propidium iodide/annexin V staining, we found that the combination As2O3 with BI-D1870 significantly increased the percentage of apoptotic cells as compared with treatment with either As2O3 or BI-D1870 alone (). The pro-apoptotic member of the Bcl-2 family,Citation25 BAD, is a known substrate of RSK1 and undergoes phosphorylation at Ser112, resulting in an anti-apoptotic signal.Citation26 Treatment of U937 cells with arsenic trioxide induced phosphorylation of BAD at Ser112. Such phosphorylation was inhibited by co-treatment of the cells with BI-D1870, strongly suggesting that such phosphorylation is RSK1-dependent () and indicating a mechanism for the enhancement of apoptosis by the arsenic/BI-D1870 combination.
Figure 2. Inhibition of RSK enhances As2O3-mediatedsuppression of growth and apoptosis. (A) U937 cells were treated with the indicated doses of As2O3 in the presence (dashed line) or absence (continuous line) of the RSK inhibitor BI-D1870 (1 µM) for four days. Cell viability was determined by MTT assays. Data are expressed as percent of untreated controls for the different conditions. The means ± SE of the values from 3 experiments are shown. (B) U937 cells were treated with the indicated concentrations of As2O3 in the absence or presence of BI-D1870 (2µM). Apoptosis was determined by flow cytometry studies for propidium iodide/annexin V staining. The means ± SE of the values from 3 experiments are shown. (C) U937 cells were cultured in the presence or absence of As2O3 for 60 min, in the presence or absence of BI-D1870 (2 µM). Total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-phospho-Ser 112-BAD antibody. The same blot was then stripped and re-probed with an anti-BAD antibody.
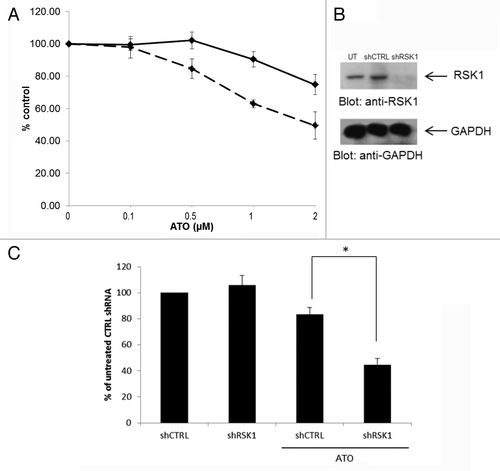
We next assessed the effects of combinations of As2O3 with BI-D1870 on primitive leukemic precursors in clonogenic assays in methylcellulose. BI-D1870 significantly enhanced the suppressive effects of As2O3 on U937-derived primitive leukemic precursor (CFU-L) colony formation (). Moreover, consistent with these findings, shRNA knockdown of RSK1 in U937 cells () resulted in more potent inhibitory effects on U937-derived leukemic CFU-L progenitors ().
Figure 3. Pharmacological or molecular targeting of RSK1 enhances As2O3 -dependent suppression of AML leukemic precursors. (A) U937 cells were cultured with the indicated concentrations of As2O3, in the presence (dashed line) or absence (continuous line) of BI-D1870 (1 µM) and leukemic progenitor colony formation was assessed in clonogenic assays in methylcellulose. Data are expressed as % control colony formation for each condition (untreated or BI-D1870 treated). The means ± SE of the values from 3 experiments are shown. (B) Knockdown of RSK1 in U937 cells using RSK1 specific shRNA. (C) U937 cells transfected with RSK1 shRNA or control shRNA were cultured in the presence or absence of As2O3 0.5 µM and leukemic progenitor colony formation was assessed in clonogenic assays in methylcellulose. The means ± SE of the values from 3 experiments are shown. (* p < 0.05, using a paired two-tailed t-test).
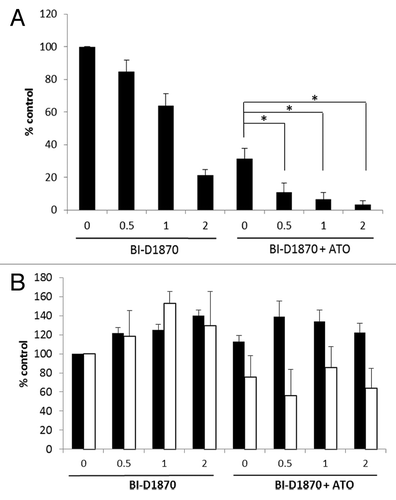
In subsequent studies we evaluated the effects of pharmacological inhibition of RSK1 on primary leukemic progenitors from AML patients. Peripheral blood mononuclear cells from six patients with AML were isolated, and the effects of the RSK inhibitor BI-D1870 on primitive leukemic precursors (CFU-L) were determined. As shown in , BI-D1870 exhibited potent suppressive effects on primary leukemia progenitors and such effects were enhanced by the co-treatment with As2O3 (). Importantly, no significant suppressive effects in response to BI-D1870 used alone or in combination with As2O3 (0.5 μM) treatment were seen on normal CD34+ derived myeloid progenitors (CFU-GM) (). Similarly, BI-D1870 at concen-trations of 0.5 to 2.0 μM did not suppress normal erythroid BFU-E colony formation, although there was some inhibition seen by As2O3 (0.5 μM) alone or in combination of BI-D1870 (). Thus, combined targeting of RSK1 with arsenic trioxide treatment results in enhanced in vitro responses against primitive leukemic progenitors, but not normal myeloid hematopoietic progenitors, suggesting an approach to enhance the antileukemic effects of arsenic by combinations with RSK1 inhibitors.
Figure 4. Generation of antileukemic responses by pharmacological inhibition of RSK. (A) Peripheral blood mononuclear cells from patients with AML were plated in a methylcellulose culture assay system with the indicated concentrations of BI-D1870 and in the presence or absence of As2O3 (0.5 µM), as indicated. Data are expressed as percent control of leukemic CFU-blast (CFU-L) colony formation for control untreated cells. Means ± SE of the values from six experiments using different patient samples are shown. (* p < 0.05, using a paired two-tailed t-test). (B) Normal human, CD34+ hematopoietic progenitor cells were plated in a methylcellulose culture assay system with the indicated concentrations of BI-D1870 and in the presence or absence of As2O3 (0.5µM), as indicated. Data are expressed as percent control of normal myeloid (CFU-GM) (shaded bars) or erythroid (BFU-E) (open bars) hematopoietic progenitor colony formation for control untreated cells. Means ± SE of the values from 3 experiments are shown.
Discussion
There has been extensive prior evidence that activation of the MEK/ERK pathway in AML cells is important for the transmission of proliferative signals that promote leukemogenesis and that targeting MEK/ERK pathways and effectors may provide a unique approach for the treatment of leukemias.Citation27-Citation31 A particular component of this pathway, the RSK family of proteins, has been recently the focus of attention, as there is evidence that this group of MEK/ERK effectors play key regulatory roles in malignant cell proliferation and survival.Citation14,Citation32 Beyond the 4 RSK isoforms (RSK 1–4), this family includes two other kinase members, MSK1 and MSK2, whose structures are distinct, but structurally related to the RSK isoforms.Citation32
There is a substantial amount of emerging evidence that the functions of members of the RSK family of kinases are important for control of cell proliferation, cell cycle progression, transcriptional regulation, protein synthesis and cell survival.Citation32 At the same time there is an accumulating list of proteins that appear to function as substrates for these kinases and ultimate mediators of biological responses.Citation32 There is also emerging evidence for important roles for members of the RSK family in tumorigenesis and leukemogenesis.Citation32 Notably, a recent study demonstrated that RSK2 is essential for FLT3-ITD leukemic transformation, although it was found to be dispensable for BCR-ABL leukemogenesis.Citation33 Various downstream regulatory events may account for the transforming/pro-neoplastic activities of members of the RSK family, including regulatory effects on p27kip1;Citation32,Citation34 phosphorylation and subsequent degradation of Mad1, a suppressor of Myc activity;Citation34,Citation35 and regulatory effects on the mTOR pathway via phosphorylation of Tsc-2Citation36 or Raptor in the mTORC1 complex.Citation37 Notably, mTOR complexes and downstream mediators are often dysregulated in hematological malignancies and, therefore, have become important targets for drug development.Citation38-Citation40 The combination of RSK inhibition with the specific inhibition of the mTOR axis warrants further investigation.
In the present study we provide evidence that RSK1 is activated by As2O3 during treatment of AML cells in a negative feedback regulatory manner. Our data also demonstrate that pharmacological inhibition of RSK results in enhanced As2O3-dependent suppression of proliferation and increased apoptosis of leukemia cell lines. Furthermore, molecular silencing of RSK1 was found to result in significant suppressive effects on U937-derived primitive leukemic progenitors. Pharmacological inhibition of RSK activity also enhanced the suppressive effects of As2O3 on primary leukemic (CFU-L) progenitors from AML patients, underscoring the relevance of this family of kinases in leukemogenesis. Such enhanced suppression was not seen on normal human hematopoietic precursors, suggesting specificity toward leukemia cells and further raising the possibility for clinical-translational approaches involving RSK1 targeting to promote arsenic-dependent antileukemic effects.
It is of particular interest that among other negative feedback induced by arsenic is another member of the broad RSK family, Msk1.Citation18 Also, we have previously reported that the mTOR pathway is another arsenic-inducible negative-feedback pathway,Citation22,Citation23 while others have shown in other systems that mTOR activation is also regulated by RSK kinases under certain circumstances.Citation36,Citation37 This suggests some redundancy in the patterns of induction of negative feedback regulatory signals and raises the possibility that simultaneous targeting of more than one negative feedback loops will be likely necessary to fully overcome leukemic cell resistance to As2O3 in vitro and in vivo. Nevertheless, this remains to be directly established in future studies. Independently of whether this hypothesis proves to be correct, our findings clearly establish that the downstream substrate of the MEK-ERK pathway, RSK1, is a contributor to the negative regulation of As2O3-responses in leukemia cells and should be considered a potential therapeutic target to enhance the antileukemic action of As2O3 in AML and possibly other leukemias.
Materials and Methods
Cells and reagents
The U937, MM6 and NB4 human cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. Arsenic trioxide (As2O3) was purchased from Sigma. Antibodies against RSK1 and BAD and the phosphorylated forms of RSK1 (Ser-221) and BAD (Ser112) were obtained from Cell Signaling Technology, Inc. Antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from EMD Millipore. The RSK inhibitor, BI-D1870 was obtained from Symansis. The MEK1/2 inhibitor U0126 and the PI3K inhibitor LY294002 were purchased from Calbiochem/EMD Millipore. Lentiviral human RSK1 and non-targeted control shRNAs were purchased from Santa Cruz Biotechnology, Inc.
Cell lysis and immunoblotting
Cells were treated with the indicated doses of As2O3 for the indicated times and subsequently lysed in the phosphorylation lysis buffer as previously described.Citation22-Citation24 In the experiments in which pharmacological inhibitors were used, the cells were pre-treated for 60 min with the inhibitors at the indicated final concentrations of inhibitors and subsequently treated for the indicated times with As2O3, in the continuous presence of the inhibitors, prior to cell lysis in phosphorylation lysis buffer. Immunoblotting using an enhanced chemiluminescence (ECL) method was done as previously described.Citation41-Citation45
Cell proliferation/viability assays
Cells were treated with the indicated doses of As2O3, in the presence or absence of BI-D1870 (0.5–4 μM), for seven days. Cell proliferation/viability assays using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide (MTT) method were performed as in our previous studies.Citation46
Evaluation of apoptosis
Cells were exposed to the indicated doses of As2O3 for the indicated time periods. Flow cytometric assays to evaluate apoptosis by propidium iodide/annexin V staining were done as previously described.Citation47
Hematopoietic cell progenitor assays
Peripheral blood from patients with AML was collected after obtaining consent approved by the Institutional Review Board of Northwestern University. The effects of arsenic trioxide on the growth of leukemic progenitors (CFU-L) were assessed by clonogenic assays in methylcellulose, as in previous studies.Citation46-Citation48
Acknowledgments
This work was supported by National Institutes of Health grants CA121192, CA155566 and CA77816 and by a grant from the Department of Veterans Affairs (to L.C.P.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, et al. Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood 2010; 116:3751 - 7; http://dx.doi.org/10.1182/blood-2010-02-269621; PMID: 20705755
- Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol 2001; 19:3852 - 60; PMID: 11559723
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89:3354 - 60; PMID: 9129042
- Niu C, Yan H, Yu T, Sun HP, Liu JX, Li XS, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 1999; 94:3315 - 24; PMID: 10552940
- Parmar S, Rundhaugen LM, Boehlke L, Riley M, Nabhan C, Raji A, et al. Phase II trial of arsenic trioxide in relapsed and refractory acute myeloid leukemia, secondary leukemia and/or newly diagnosed patients at least 65 years old. Leuk Res 2004; 28:909 - 19; http://dx.doi.org/10.1016/j.leukres.2003.12.012; PMID: 15234567
- Platanias LC. Biological responses to arsenic compounds. J Biol Chem 2009; 284:18583 - 7; http://dx.doi.org/10.1074/jbc.R900003200; PMID: 19363033
- Goussetis DJ, Altman JK, Glaser H, McNeer JL, Tallman MS, Platanias LC. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J Biol Chem 2010; 285:29989 - 97; http://dx.doi.org/10.1074/jbc.M109.090530; PMID: 20656687
- Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324 - 31; http://dx.doi.org/10.1182/blood-2010-01-261040; PMID: 20574048
- Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. [Epub ahead of print] Blood 2012; 120:3555 - 62; http://dx.doi.org/10.1182/blood-2012-01-402578; PMID: 22898604
- Milella M, Precupanu CM, Gregorj C, Ricciardi MR, Petrucci MT, Kornblau SM, et al. Beyond single pathway inhibition: MEK inhibitors as a platform for the development of pharmacological combinations with synergistic anti-leukemic effects. Curr Pharm Des 2005; 11:2779 - 95; http://dx.doi.org/10.2174/1381612054546842; PMID: 16101455
- Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 1999; 93:3074 - 80; PMID: 10216104
- Neubauer A, Dodge RK, George SL, Davey FR, Silver RT, Schiffer CA, et al. Prognostic importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemia. Blood 1994; 83:1603 - 11; PMID: 8123851
- Lunghi P, Tabilio A, Dall’Aglio PP, Ridolo E, Carlo-Stella C, Pelicci PG, et al. Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia 2003; 17:1783 - 93; http://dx.doi.org/10.1038/sj.leu.2403032; PMID: 12970778
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 2008; 9:747 - 58; http://dx.doi.org/10.1038/nrm2509; PMID: 18813292
- Verma A, Mohindru M, Deb DK, Sassano A, Kambhampati S, Ravandi F, et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. J Biol Chem 2002; 277:44988 - 95; http://dx.doi.org/10.1074/jbc.M207176200; PMID: 12239215
- Giafis N, Katsoulidis E, Sassano A, Tallman MS, Higgins LS, Nebreda AR, et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res 2006; 66:6763 - 71; http://dx.doi.org/10.1158/0008-5472.CAN-05-3699; PMID: 16818652
- Dolniak B, Katsoulidis E, Carayol N, Altman JK, Redig AJ, Tallman MS, et al. Regulation of arsenic trioxide-induced cellular responses by Mnk1 and Mnk2. J Biol Chem 2008; 283:12034 - 42; http://dx.doi.org/10.1074/jbc.M708816200; PMID: 18299328
- Kannan-Thulasiraman P, Katsoulidis E, Tallman MS, Arthur JS, Platanias LC. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J Biol Chem 2006; 281:22446 - 52; http://dx.doi.org/10.1074/jbc.M603111200; PMID: 16762916
- McNeer JL, Goussetis DJ, Sassano A, Dolniak B, Kroczynska B, Glaser H, et al. Arsenic trioxide-dependent activation of thousand-and-one amino acid kinase 2 and transforming growth factor-beta-activated kinase 1. Mol Pharmacol 2010; 77:828 - 35; http://dx.doi.org/10.1124/mol.109.061507; PMID: 20159944
- Lunghi P, Giuliani N, Mazzera L, Lombardi G, Ricca M, Corradi A, et al. Targeting MEK/MAPK signal transduction module potentiates ATO-induced apoptosis in multiple myeloma cells through multiple signaling pathways. Blood 2008; 112:2450 - 62; http://dx.doi.org/10.1182/blood-2007-10-114348; PMID: 18583568
- Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci 2008; 13:4258 - 75; http://dx.doi.org/10.2741/3003; PMID: 18508509
- Altman JK, Yoon P, Katsoulidis E, Kroczynska B, Sassano A, Redig AJ, et al. Regulatory effects of mammalian target of rapamycin-mediated signals in the generation of arsenic trioxide responses. J Biol Chem 2008; 283:1992 - 2001; http://dx.doi.org/10.1074/jbc.M705227200; PMID: 18048359
- Yoon P, Giafis N, Smith J, Mears H, Katsoulidis E, Sassano A, et al. Activation of mammalian target of rapamycin and the p70 S6 kinase by arsenic trioxide in BCR-ABL-expressing cells. Mol Cancer Ther 2006; 5:2815 - 23; http://dx.doi.org/10.1158/1535-7163.MCT-06-0263; PMID: 17121928
- Platanias LC. Biological responses to arsenic compounds. J Biol Chem 2009; 284:18583 - 7; http://dx.doi.org/10.1074/jbc.R900003200; PMID: 19363033
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell 2010; 37:299 - 310; http://dx.doi.org/10.1016/j.molcel.2010.01.025; PMID: 20159550
- Yang X, Liu L, Sternberg D, Tang L, Galinsky I, DeAngelo D, et al. The FLT3 internal tandem duplication mutation prevents apoptosis in interleukin-3-deprived BaF3 cells due to protein kinase A and ribosomal S6 kinase 1-mediated BAD phosphorylation at serine 112. Cancer Res 2005; 65:7338 - 47; http://dx.doi.org/10.1158/0008-5472.CAN-04-2263; PMID: 16103085
- McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Bäsecke J, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008; 22:708 - 22; http://dx.doi.org/10.1038/leu.2008.27; PMID: 18337766
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773:1263 - 84; http://dx.doi.org/10.1016/j.bbamcr.2006.10.001; PMID: 17126425
- Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008; 22:686 - 707; http://dx.doi.org/10.1038/leu.2008.26; PMID: 18337767
- Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Bäsecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011; 25:1080 - 94; http://dx.doi.org/10.1038/leu.2011.66; PMID: 21494257
- Chung E, Kondo M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol Res 2011; 49:248 - 68; http://dx.doi.org/10.1007/s12026-010-8187-5; PMID: 21170740
- Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J 2012; 441:553 - 69; http://dx.doi.org/10.1042/BJ20110289; PMID: 22187936
- Elf S, Blevins D, Jin L, Chung TW, Williams IR, Lee BH, et al. p90RSK2 is essential for FLT3-ITD- but dispensable for BCR-ABL-induced myeloid leukemia. Blood 2011; 117:6885 - 94; http://dx.doi.org/10.1182/blood-2010-10-315721; PMID: 21527514
- Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2003; 278:49254 - 60; http://dx.doi.org/10.1074/jbc.M306614200; PMID: 14504289
- Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A 2008; 105:6584 - 9; http://dx.doi.org/10.1073/pnas.0802785105; PMID: 18451027
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 2004; 101:13489 - 94; http://dx.doi.org/10.1073/pnas.0405659101; PMID: 15342917
- Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol 2008; 18:1269 - 77; http://dx.doi.org/10.1016/j.cub.2008.07.078; PMID: 18722121
- Hagner PR, Schneider A, Gartenhaus RB. Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood 2010; 115:2127 - 35; http://dx.doi.org/10.1182/blood-2009-09-220020; PMID: 20075156
- Redig AJ, Vakana E, Platanias LC. Regulation of mammalian target of rapamycin and mitogen activated protein kinase pathways by BCR-ABL. Leuk Lymphoma 2011; 52:Suppl 1 45 - 53; http://dx.doi.org/10.3109/10428194.2010.546919; PMID: 21299459
- Beauchamp EM, Platanias LC. The evolution of the TOR pathway and its role in cancer. [Epub ahead of print] Oncogene 2012; http://dx.doi.org/10.1038/onc.2012.567; PMID: 23246968
- Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3′-kinase. J Biol Chem 1995; 270:15938 - 41; http://dx.doi.org/10.1074/jbc.270.27.15938; PMID: 7608146
- Verma A, Deb DK, Sassano A, Uddin S, Varga J, Wickrema A, et al. Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-beta on normal hematopoiesis. J Biol Chem 2002; 277:7726 - 35; http://dx.doi.org/10.1074/jbc.M106640200; PMID: 11773065
- Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, et al. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem 2001; 276:28570 - 7; http://dx.doi.org/10.1074/jbc.M011685200; PMID: 11353767
- Sassano A, Katsoulidis E, Antico G, Altman JK, Redig AJ, Minucci S, et al. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res 2007; 67:4524 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-06-3686; PMID: 17483369
- Sassano A, Lo Iacono M, Antico G, Jordan A, Uddin S, Calogero RA, et al. Regulation of leukemic cell differentiation and retinoid-induced gene expression by statins. Mol Cancer Ther 2009; 8:615 - 25; http://dx.doi.org/10.1158/1535-7163.MCT-08-1196; PMID: 19240159
- Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, et al. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res 2011; 17:4378 - 88; http://dx.doi.org/10.1158/1078-0432.CCR-10-2285; PMID: 21415215
- Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A 2010; 107:12469 - 74; http://dx.doi.org/10.1073/pnas.1005114107; PMID: 20616057
- Vakana E, Altman JK, Glaser H, Donato NJ, Platanias LC. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood 2011; 118:6399 - 402; http://dx.doi.org/10.1182/blood-2011-01-332783; PMID: 22021366