Abstract
The epidermal growth factor receptor (EGFR) is a central regulator of tumor progression in human cancers. Cetuximab is an anti-EGFR monoclonal antibody that has been approved for use in oncology. Despite clinical success the majority of patients do not respond to cetuximab and those who initially respond frequently acquire resistance. To understand how tumor cells acquire resistance to cetuximab we developed a model of resistance using the non-small cell lung cancer line NCI-H226. We found that cetuximab-resistant (CtxR) clones manifested strong activation of EGFR, PI3K/AKT and MAPK. To investigate the role of AKT signaling in cetuximab resistance we analyzed the activation of the AKT pathway effector molecules using a human AKT phospho-antibody array. Strong activation was observed in CtxR clones for several key AKT substrates including c-jun, GSK3β, eIF4E, rpS6, IKKα, IRS-1 and Raf1. Inhibition of AKT signaling by siAKT1/2 or by the allosteric AKT inhibitor MK-2206 resulted in robust inhibition of cell proliferation in all CtxR clones. Moreover, the combinational treatment of cetuximab and MK-2206 resulted in further decreases in proliferation than either drug alone. This combinatorial treatment resulted in decreased activity of both AKT and MAPK thus highlighting the importance of simultaneous pathway inhibition to maximally affect the growth of CtxR cells. Collectively, our findings demonstrate that AKT activation is an important pathway in acquired resistance to cetuximab and suggests that combinatorial therapy directed at both the AKT and EGFR/MAPK pathways may be beneficial in this setting.
Introduction
The epidermal growth factor receptor (EGFR) is a member of the HER family of receptor tyrosine kinases (RTKs), which consists of the EGFR (ErbB1/HER1), HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4). All family members contain an extracellular ligand-binding domain (domains I, II, III and IV), a single membrane-spanning region, a juxtamembrane nuclear localization signal (NLS) and a cytoplasmic tyrosine kinase domain (TKD). EGFR activation stimulates many complex intracellular signaling pathways that are tightly regulated by the presence and identity of ligand, the heterodimer composition and the availability of phosphotyrosine-binding proteins. The two primary signaling pathways activated by EGFR include the RAS/RAF/MEK/ERK and the PI3K/AKT axis; however, SRC tyrosine kinases, PLCγ, PKC and STAT activation and downstream signaling have also been well documented.Citation1 Tumor cell proliferation, survival, invasion and angiogenesis can ultimately be promoted through activation of these pathways. Aberrant expression or activity of the EGFR has been identified as an important biological factor in many human epithelial cancers including head and neck squamous cell carcinoma (HNSCC), non-small cell lung cancer (NSCLC), colorectal cancer (CRC), breast cancer, pancreatic cancer and brain cancer.
Cetuximab (ICM-225, Erbitux™) is a human/murine chimeric monoclonal antibody that works by binding to extracellular domain III of EGFR. This interaction partially blocks the ligand-binding domain and sterically hinders the correct extended conformation of the dimerization arm on domain II.Citation2 Thus, cetuximab inhibits both ligand binding and the proper positioning of the EGFR dimerization domain, preventing dimerization with other HER family members. Cetuximab has exhibited promising antitumor activity in clinical trials as a monotherapy or use in combination with chemotherapy and/or radiation, particularly in the settings of metastatic CRC (mCRC)Citation3-Citation8 and HNSCC.Citation9-Citation13 However, EGFR inhibition by either monoclonal antibodies or small molecule tyrosine kinase inhibitors only demonstrate anti-tumor activity in ~10−20% of cancer patients as reported in several pivotal clinical studies involving different solid tumor types.Citation14 Over the past several years researchers have observed high levels of intrinsic and acquired resistance to EGFR monoclonal antibody therapy, stimulating a new field of EGFR research.Citation15
The serine-threonine kinase AKT was initially identified as the proto-oncogene of the v-AKT oncogenic murine thymoma virus.Citation16 AKT has three isoforms: AKT1, AKT2 and AKT3. AKT1 and AKT2 are expressed in most tissue types while AKT3 expression is generally restricted to neuronal tissue and the testes.Citation17 The three isoforms share over 80% homology and are characterized by three conserved functional domains: an N-terminal pleckstrin homology (PH) domain that regulates intracellular trafficking of the protein, a central catalytic domain and a C-terminal regulatory domain. Activation of all three AKT isoforms is dependent on the activity of phosphatidylinositol 3-kinase (PI3K).Citation18 PI3K is stimulated by a variety of signals, including growth factor and G protein–coupled receptors localized on the cell surface. Activation of PI3K results in the generation of 3′- phosphorylated phosphatidylinositols in the cell membrane, which recruit AKT and other PH domain–containing proteins to the cell membrane. Localization of AKT on the inner leaflet of the cell membrane brings it into close proximity to the serine-threonine kinase phosphoinositide-dependent kinase-1 (PDK1), which phosphorylates AKT at the Thr308 residue of its catalytic domain. The activated conformation of AKT is further stabilized by phosphorylation at the Ser473 residue, either by the mammalian target of rapamycin complex 2 (mTORC2) in response to growth factor stimulation or by DNA-dependent protein kinase (DNA-PK) after DNA damage.Citation19,Citation20 Additionally, various PI3K independent activators of AKT have also been discovered.Citation21 In turn, AKT phosphorylates several cellular proteins, including glycogen synthase kinase 3α (GSK3α), GSK3β, forkhead box O transcription factors (FoxO), MDM2, BCL-2-interacting mediator of cell death (BIM) and BCL-2 associated agonist of cell death (BAD) to facilitate cell survival and cell cycle entry (For a review see ref. Citation22).Citation22 AKT activity is negatively regulated primarily by phosphatases that dephosphorylate phosphatidylinositols at the cell membrane (phosphatase and tensin homolog [PTEN], SHP2).
More than 50 substrates of AKT have been identified.Citation18,Citation23-Citation25 Through these and other effectors, AKT regulates a variety of cellular processes, including proliferation, survival, motility, angiogenesis, differentiation and metabolism/glucose homeostasis. Thus, inhibition of AKT activity is an attractive target for cancer therapies. Currently, several AKT inhibitors are in clinical development for treating cancers. MK-2206 is an orally active allosteric AKT inhibitor that is under development for the treatment of solid tumors. While MK-2206 is equally potent toward purified recombinant human AKT1 (IC50, 5 nmol/L) and AKT2 proteins (IC50, 12 nmol/L), it is approximately 5-fold less potent against human `AKT3 (IC50, 65 nmol/L).Citation26
Previously we established cetuximab resistant (CtxR) clones from the NSCLC cell line NCI-H226 by exposing these cells to increasing concentrations of cetuximab over a 6-mo time course.Citation27 Total protein levels and activation of EGFR in CtxR clones were upregulated, as well as the phosphorylation of MAPK and AKT compared with cetuximab-sensitive (CtxS) parental control cells.Citation27 In this report we investigated if CtxR clones acquired a dependency on AKT signaling and whether they would be sensitive to the AKT inhibitor MK-2206 alone or in combination with cetuximab. Individual clones with acquired resistance to cetuximab were treated with MK-2206 resulting in decreased activation of AKT, along with a decrease in the activation of the downstream signaling molecules in the AKT pathway. This led to decreased proliferation and increased apoptosis in all CtxR clones tested. Moreover, statistically significant decreases in proliferation were noted in combined treatment with cetuximab and MK-2206. The combination of Cetuximab and MK-2206 lead to the growth inhibition of CtxR clones due to reduced signaling by both the MAPK and AKT signaling pathways, suggesting a role for both of these kinases in cetuximab resistance. Taken together, these results suggest that the activation of EGFR and downstream MAPK signaling as well as AKT play a role in cetuximab resistance and that dual targeting of the EGFR and AKT with cetuximab and MK-2206 may provide a strategy to overcome acquired resistance.
Results
NSCLC cell lines with acquired resistance to cetuximab have increased activity of MAPK, AKT and downstream AKT signaling pathways
We previously reported that CtxR clones (HC1, HC4 and HC8) exhibited increased activity of the EGFR, MAPK and AKT relative to the CtxS parental control (HP).Citation27 To determine if CtxR clones exhibited a dependency on AKT signaling we performed proliferation assays using two different non-overlapping small interfering RNAs (siRNA) targeting AKT1/2 (). All three cetuximab-resistant lines displayed growth inhibitory effects at 50 nM with both siAKT1/2(a) and siAKT1/2(b). These data suggest that cells with acquired resistance to cetuximab depend on AKT signaling. Since both siAKT1/2 worked equally well, we chose to focus on siAKT1/2(a) from Cell Signaling for remaining studies.
Figure 1. CtxR clones have increased AKT signaling pathway. (A) CtxR clones are dependent on AKT. CtxR clones were plated and treated with 50 nM of AKT1/2(a) siRNA, 50 nM of AKT1/2(b) siRNA or 50 nM non-targeting siRNA. Cell proliferation was measured at 96 h after treatment using the proliferation assay described in materials and methods. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05. Protein was collected at 96 h after treatment and fractioned by SDS-PAGE and immunoblotted for AKT and phopho-rpS6. α-Tubulin was used as a loading control. (B) Fold increase in expression of phosphorylated proteins in CtxR HC4 cells compared with parental control HP cells by phosphoprotein arrays. CtxS parental cells (HP) and CtxR clones (HC4) were harvested and lysed with the extraction buffer provided as described according to manufacturer’s instructions for phosphoprotein arrays. (C) CtxR overexpress EGFR and have increased AKT signaling pathway. CtxS parental cells (HP) and CtxR clones (HC1, HC4, HC8) were harvested and protein lysates were fractionated on SDS-PAGE followed by immunoblotting for the indicated proteins. α-Tubulin was used as a loading control.
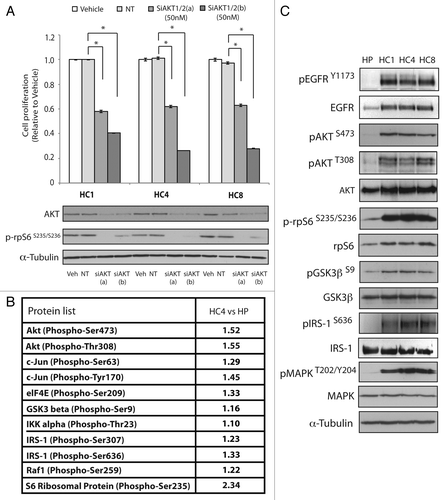
To investigate global activation of AKT signaling pathways in CtxR clones we utilized an AKT specific phosphoprotein antibody array to identify phosphorylated proteins that were upregulated in the CtxR clone HC4 as compared with parental control HP cells. This antibody array includes 137 well-characterized phospho-specific antibodies for proteins in the AKT pathway, each with six replicates. The paired antibodies for the same (but un-phosphorylated) target sites are also included in the array to allow determination of the relative levels of phosphorylation for each AKT substrate. Results from this array platform indicated several AKT substrates including c-Jun, eIF4E, GSK3β, IKKα, IRS-1, Raf-1 and S6 ribosomal protein (rpS6) were upregulated in the HC4 CtxR clone ().
To confirm the AKT specific phosphoprotein array results we analyzed the activity of various AKT effector molecules via western blot analysis in the three CtxR clones HC1, HC4 and HC8 (). We confirmed that the AKT pathway effector molecules rpS6 (serine 235/236), GSK3β (σερινε 9) and IRS-1 (serine 636) were indeed highly active in all three CtxR clones. In addition to activation of MAPK, these results suggest that CtxR clones have enhanced activation of AKT signaling pathways and further, they exhibit dependence on these pathways for enhanced growth potential. Phosphorylation levels of AKT substrate proteins in HC4 cells compared with HP cells are summarized in Table S1.
CtxR cells have increased sensitivity to the allosteric AKT inhibitor MK-2206
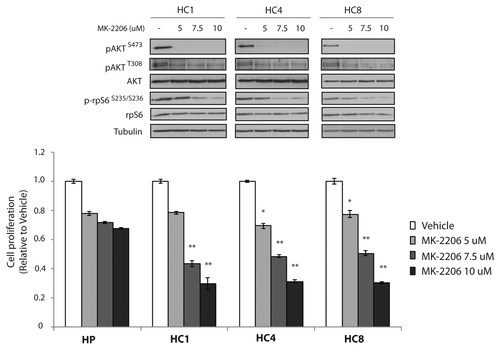
We hypothesized that CtxR clones may be susceptible to AKT inhibitory therapies since these cells remained dependent on the AKT signaling pathway for sustained growth and survival. To test this hypothesis we challenged CtxR clones with the AKT inhibitor MK-2206 (5uM, 7.5uM and 10uM) for 24 h (). MK-2206 is a highly selective, potent non-ATP competitive allosteric AKT inhibitor and is currently undergoing clinical investigation for use in several types of solid tumors. We demonstrate that MK-2206 inhibits the activity of AKT by decreasing the phosphorylation of serine 473 (S473) and threonine 308 (T308) as well as phospho-rpS6 (serine 235/serine 236) (). While phospho-AKT S473 is inhibited with 5uM of MK-2206 there is a dose dependent decrease in phosphorylation of AKT T308 and rpS6. Additionally, MK-2206 treatment demonstrated growth inhibitory effects of all CtxR clones with robust, dose dependent responses. This may be due to the enhanced inhibitory effects of AKT T308 and downstream targets at higher concentrations. Treatment with 7.5uM MK-2206 reduced CtxR cell proliferation rates to approximately 50% compared with vehicle control treatment. MK-2206 treatment had minimal effect on the CtxS parental cells that have very low levels of activation of AKT (). Taken together these results suggest that CtxR cells are dependent on AKT activity for proliferation and MK-2206 is an effective treatment for cells with acquired resistance to cetuximab.
Figure 2. AKT inhibitors, MK-2206 decrease cell proliferation of CtxR clones. MK-2206 significantly inhibits the proliferation of CtxR clones. CtxR clones (HC1, HC4, HC8) and CtxS parental control (HP) were plated and allowed to adhere for 24 h prior to vehicle (DMSO) or MK-2206 treatment: 5 uM, 7.5 uM or 10 uM. Cell growth was measured at 72 h after drug treatment using the growth proliferation assay described in experimental methods and plotted as a percentage of growth relative to vehicle control cells. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05, **p ≤ 0.001. Whole cell protein lysates were collected after 24 h treatment and fractionated on SDS-PAGE followed by immunoblotting for the indicated proteins. α-Tubulin was used as a loading control.
MK-2206 blocks AKT downstream signaling pathway in CtxR cells
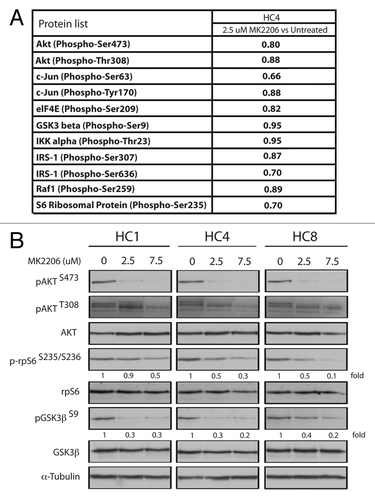
We further explored the mechanisms of cell growth inhibition in CtxR clones by MK-2206. To determine if MK-2206 effects the phosphorylation of other AKT targets in CtxR cells, we probed the same AKT specific phosphoprotein array with protein lysate harvested from the CtxR clone HC4 treated with 2.5 uM MK-2206 for 24 h. Results from this antibody array showed that 2.5 uM of MK-2206 treatment could mildly inhibit multiple downstream AKT targets including c-jun, eIF4E, GSK3β, IKKα, IRS-1, Raf1 and rpS6 (). Since the multiplex array platform the fold changes detected on the array may actually be smaller than the true value, we next validated in all three CtxR clones that the activation of AKT, rpS6 and GSK3β are indeed decreased upon treatment with 2.5 and 7.5 uM of MK-2206 for 24 h (). Treatment with 7.5 uM MK-2206 showed significant decreases on the levels of phosphorylated AKT, rpS6 (50–90%) and GSK3β (60–80%). Total levels of AKT, rpS6 and GSK3β were not affected by MK-2206 treatment (). These results indicate MK-2206 is able to abrogate the activation of AKT as well as its downstream signaling effector molecules, which may suggest why MK-2206 can be effective treatment in cetuximab resistant cell lines. Phosphorylation levels of AKT substrate proteins in HC4 cells with 2.5 uM MK-2206 treatment compared with vehicle control are summarized in Table S1.
Figure 3. AKT downstream signaling molecules are effectively inhibited with MK-2206 treatment in CtxR clones. (A) Fold decrease in expression of phosphorylated proteins treated by MK-2206 in cetuximab-resistant HC4 CtxR cells compared with vehicle control HC4 cells by phosphoprotein arrays. CtxR clones (HC4) were harvested after treatment with 2.5 uM of MK-2206 and lysed with the extraction buffer provided as described according to manufacturer’s instructions for phosphoprotein arrays. (B) CtxR clones (HC1, HC4, HC8) were treated with vehicle (DMSO) or MK-2206 (2.5 or 7.5 uM) for 24 h. Whole cell protein lysates were fractionated on SDS-PAGE followed by immunoblotting for the indicated proteins. α-Tubulin was used as a loading control.
MK-2206 plus cetuximab has greater therapeutic effect than either agent alone
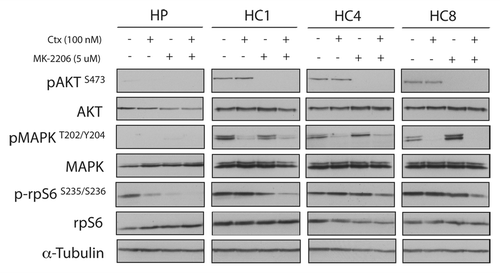
We showed that CtxR cells are dependent on AKT activity for proliferation and MK-2206 is an effective treatment for cells with acquired resistance to cetuximab ( and ). To determine if loss of AKT is important in acquired resistant to cetuximab, we treated CtxR clones with siAKT1/2(a) and cetuximab for 72 h. The combination treatments resulted in significant cell growth inhibition in CtxR clones (). Next, we examined if MK-2206 could have therapeutic benefit in CtxR cells with cetuximab treatment. We performed cell proliferation analysis using vehicle, cetuximab (0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM), MK-2206 (0.1uM, 1 uM, 5 uM and 10 uM) or 100 nM cetuximab plus MK-2206 (0.1 uM, 1 uM, 5 uM and 10 uM) in combination on HP, HC1, HC4 and HC8. Despite complete resistance up to 1000 nM of cetuximab, addition of 100 nM of cetuximab led to a marked statistically significant increase of MK-2206 inhibitory potency over a wide range of MK-2206 doses (). To see if the augmentation of growth inhibition of MK-2206 with cetuximab correlated with increased apoptosis, we performed Annexin-V analysis after treatment with vehicle, 100 nM cetuximab, 5 uM MK-2206 or the combination for 24 h (). MK-2206 treatments resulted in a statistically significant increase in apoptosis of CtxR clones compared with vehicle control. Furthermore, combinatorial treatment (MK-2206 plus cetuximab) in two out of three cell lines induced a mild enhancement of apoptosis as compared with MK-2206 alone. We further investigated which pathways were inhibited by MK-2206 and cetuximab combination treatment. CtxR clones were treated with vehicle, 100 nM cetuximab, 5 uM MK-2206 or combinatorial treatment for 24 h. MAPK phosphorylation level was decreased by cetuximab treatment, while phosphorylation of AKT was inhibited by MK-2206 treatment (). Combination of cetuximab with MK-2206 resulted in an inhibition of both phospho-MAPK and phospho-AKT as well as the downstream effector molecule phospho-rpS6 in all CtxR clones (). Interestingly, treatment with cetuximab or MK-2206 led to modest increases in steady-state expression of phospho-AKT or phospho-MAPK, respectively, in CtxR clones. Overall, these data suggest that MK-2206 and cetuximab combinatorial treatment impact proliferation by the dual targeting of AKT and MAPK, resulting in the downregulation of two prominent signaling pathways.
Figure 4. Combination treatment of siAKT and cetuximab Inhibit cell proliferation in CtxR clones. CtxR clones (HC1, HC4, HC8) and parental controls (HP) were plated and treated with 50 nM of AKT1/2 siRNA(a) or 50 nM non-targeting siRNA. The next day, cells were treated with DMSO or 100 nM cetuximab for 72 h. Growth was measured at 72 h after drug treatment using the proliferation assay as described in the experimental procedures and plotted as a percentage of growth relative to the untreated control cells. Data points are represented as mean ± SEM (n = 4). **p ≤ 0.001. Protein was collected at 72 h after treatment and fractioned by SDS-PAGE and immunoblotted for AKT. α-Tubulin was used as a loading control.
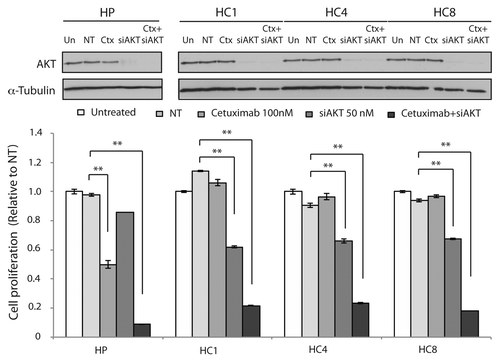
Figure 5. MK-2206 treatment enhanced the susceptibility of CtxR cells to cetuximab by inducing apoptosis. (A) MK-2206 treatment enhanced the susceptibility of CtxR cells to cetuximab. CtxR cells (HC1, HC4, HC8) and parental controls (HP) were treated with DMSO, Ctx (0.1–1000 nM), MK-2206 (0.1–10 uM) or the combination of Ctx 100nM+ MK-2206 (0.1–10 uM), for 72 h. Growth was measured at 72 h after drug treatment using the proliferation assay as described in the experimental procedures and plotted as a percentage of growth relative to the untreated control cells. Data points are represented as mean ± SEM (n = 4). *p ≤ 0.05. (B) MK-2206 plus Cetuximab induced modest apoptosis in CtxR clones. CtxS parental cell line (HP) or CtxR cell lines (HC1, HC4, HC8) were plated and allowed to adhere for 24 h prior to treatment with vehicle (DMSO), cetuximab (100 nM), MK-2206 (5 uM) or the combination (cetuximab+MK-2206) for 24 h prior to Annexin-V analysis via flow cytometry. Annexin-V analysis was described in the materials and methods. Data points are represented as mean ± SEM (n = 3). *p ≤ 0.05. Flow cytometry profile represents Annexin-V-FITC staining in x axis and PI in y axis. The number represents the percentage of cells in each condition.
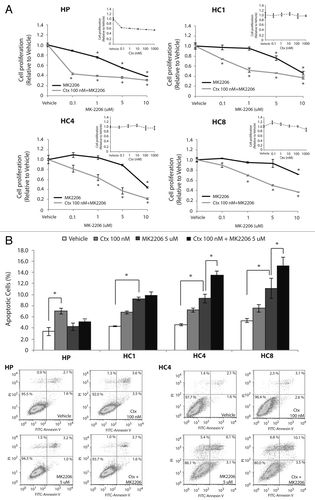
Figure 6. Dual blockade of AKT and EGFR have beneficial effects on AKT and MAPK activity. CtxR cells (HC1, HC4, HC8) were plated and treated with the DMSO (vehicle), 100 nM cetuximab, 5 uM MK-2206 or the combination (cetuximab+MK-2206) for 24 h. Cells were harvested and protein was collected, fractionated by SDS-PAGE and immunoblotted for the indicated proteins. α-Tubulin was used as a loading control.
Discussion
Cetuximab has exhibited promising antitumor activity in clinical trials particularly in the settings of mCRC and HNSCC either as monotherapy or in combination with chemotherapy and/or radiation.Citation15 However, acquired resistance to cetuximab remains a major obstacle for the successful use of this promising molecular targeting agent.Citation28,Citation29 Previously, we developed a model of acquired resistance to cetuximab using the NSCLC NCI-H226.Citation27 Results from these studies indicated that CtxR cell lines had increased expression and activation of the EGFR, MAPK and AKT.Citation27
In this study we investigated (1) if CtxR clones exhibited a dependency on AKT signaling pathways and (2) whether the allosteric AKT inhibitor MK-2206 could be advantageous in the setting of acquired resistance to cetuximab. Gene-silencing studies using siAKT indicated that all CtxR NCI-H226 clones remained addicted to AKT signaling pathways (). Various other researchers have noted the upregulation of AKT signaling pathways in defined subsets of human NSCLC, supporting our current study, which demonstrates the overexpression of AKT signaling pathways in numerous CtxR clones.Citation30-Citation32 We also found that CtxR cells exhibit increased steady-state activity of the EGFR (). Studies by Kim et al. further support our findings by reporting that CtxR HCC827 clones also had increased AKT activation and marked decreased protein levels of PTEN.Citation33 Further, Chen et al. established a pair of cell lines from Huh7 hepatocellular carcinoma cells that are resistant to several tyrosine kinases and Raf kinase inhibitor sorafenib.Citation34 They found that sorafenib resistant cells exhibited upregulation of AKT signaling compared with the sorafenib sensitive parental Huh7 cells. These results indicate that acquired resistance to molecular targeting agents such as cetuximab and sorafenib may share a common mechanism of resistance through the activation of AKT and its downstream signaling pathways. Overall, the data presented in the current study demonstrate that the activation of AKT plays a role in cetuximab resistance and provides a rational strategy through which cetuximab-based treatment may be improved.
To evaluate this concept, we treated our CtxR clones with MK-2206, an AKT-specific inhibitor and assayed growth inhibition. MK-2206 treatment yielded statistically significant cell growth inhibition of all CtxR clones (). AKT antibody array and immunoblot analyses revealed AKT substrates such as phospho-rpS6 and phospho-GSK3β were blocked by MK-2206 treatment (). We demonstrate that higher concentrations of MK-2206 may be necessary to completely inhibit the activation of AKT (T308) and its downstream targets, which may explain why CtxR cells are more sensitive to MK-2206 at higher doses. Hirai et al. determined similar findings in non–small-cell lung cancer cells (NSCLC), with an IC50 ranging between 3.4 and 28.6 uM, while AKT inhibition was detected at much lower concentrations.Citation35 These results suggested that AKT signaling pathways are essential for cell survival in CtxR clones and MK-2206 may be a valuable drug for inhibiting this pathway in a variety of cancers. Sangai et al. also revealed that MK-2206 had a dose-dependent effect on cell signaling and tumor growth. Although AKT phosphorylation was inhibited with clinically relevant doses, dose escalation had a greater effect on downstream effectors.Citation36 Our data indicated that increased amounts of MK-2206 lead to more potent decreases in AKT T308 and rpS6 as well as GSK3β activation ().
Treatment of CtxR clones with AKT siRNA alone and addition to cetuximab also significantly inhibited cell proliferation (). Further, we analyzed if combinatorial MK-2206 and cetuximab therapy would result in greater anti-proliferative activity than either agent alone (). Addition of 100nM of cetuximab led to a marked increase of MK-2206 inhibitory potency over a wide range of MK-2206 doses. Various mechanisms have been proposed for the anti-proliferative effects observed with MK-2206 treatment, including the induction of apoptosis, autophagy and promotion of cell cycle arrest.Citation35,Citation37-Citation40 Cheng et al. reported that the EGFR inhibitor gefitinib could induce approximately 10–17% of glioblastoma cells to undergo apoptosis and MK-2206 treatment enhanced these levels by approximately 10%.Citation40 In the current study, we observed that MK-2206 treatment alone could induce mild levels of apoptosis (approximately 10% in two cetuximab resistant cell lines), while the addition of cetuximab could enhance levels by approximately 5% (). Thus, the anti-proliferative effects observed with MK-2206 treatment alone and in combination with cetuximab in the current study may be due to alternative mechanisms other than apoptosis.
In the current study, MK-2206 and cetuximab treatment demonstrated greater growth inhibitory effects than MK-2206 alone in all CtxR clones. The treatment of CtxR clones with cetuximab had no effect on proliferation but did, however, inhibit the activation of MAPK (). This finding demonstrates that CtxR cells have developed dependency on other growth promoting pathways. When cells were challenged with MK-2206 we observed a growth inhibitory effect that was greatly enhanced with the addition of cetuximab (). We speculate that MK-2206 can augment cetuximab response because this combination inhibits both AKT and MAPK activation () and thereby downregulates two critical pathways of cellular proliferation. This finding highlights the importance of simultaneously inhibiting both AKT and MAPK activation to achieve maximal growth inhibitory potential and suggests that either pathway can compensate for the loss of the other to maintain growth-promoting signals. In the current model of cetuximab resistance, CtxR cells have become dependent on AKT activity to maintain their growth potential, which is effectively targeted with MK-2206, and enhanced through the inhibition of MAPK with cetuximab. This point is further supported by the modest compensatory increased activation of either AKT by cetuximab or MAPK by MK-2206 (). Previous studies have also described similar compensatory activation of AKT upon inhibition of either the MAPK or mTOR1 pathways.Citation41-Citation46 Overall, the concurrent blockade of AKT and MAPK seems to be crucial for the maximal growth inhibition of CtxR clones, a strategy that may be a useful in overcoming cetuximab resistance.
Currently, MK-2206 is undergoing clinical trials in the numerous tumor settings. Yap et al. reported that 33 patients with advanced solid tumors such as colon/rectum, breast, pancreatic and lung received MK-2206 on alternate days. The MK-2206 was well tolerated at biologically active doses that inhibit AKT signaling in this phase I clinical trial.Citation26 Pal et al. also summarized several ongoing phase I studies with advanced solid tumors using MK-2206 or combinations of both cytotoxic agents and targeted therapies with MK-2206 (for a review see ref. Citation45).Citation47 In the current study we demonstrate that AKT and EGFR, through MAPK, cooperate in acquired resistance to cetuximab, suggesting that combinatorial treatment with both cetuximab and MK-2206 or potentially MAPK inhibitors may be an effective strategy for future translational research in the setting of acquired resistance.
Materials and Methods
Cell lines
The human NSCLC line NCI-H226 was purchased from ATCC. The cells were maintained in 10% fetal bovine serum in RPMI-1640 (Mediatech Inc.) with 1% penicillin and streptomycin. The development of cells with acquired resistance to cetuximab has been previously described.Citation27
siRNA and transfection
For siRNAs, CtxR cells (HC1, HC4 and HC8) were transiently transfected with siRNAs siAKT1/2(a) (#6211S, Cell Signaling Technology) or siAKT1/2(b) (sc-43609, Santa Cruz biotechnology, Inc.) using Lipofectamine RNAiMAX according to the manufacturer’s instructions (Invitrogen). The non-targeting siRNA (ON-TARGETplus Non-targeting Pool, #D-001810–10) was obtained from Dharmacon as a control. Cells were then lysed for analysis of protein knockdown by immunoblotting after siRNA transfection.
Compounds
Cetuximab (C225, ErbituxTM) was generously provided by ImClone Systems Inc. MK-2206 was generously provided by Merck. Antibodies
All antibodies were purchased from commercial sources as indicated below: EGFR, pEGFR (Y1173), AKT and HRP-conjugated goat-anti-rabbit IgG and goat-anti-mouse IgG were obtained from Santa Cruz Biotechnology Inc.. pAKT (S473), pAKT (T308), prpS6 (S235/S236), rpS6, pGSK3β (S9), GSK3β, IRS-1, p-MAPK (T202/Y204) and MAPK were obtained from Cell Signaling Technology. pIRS-1 (S636) was purchased by Thermo Scientific. α-tubulin was purchased from Calbiochem.
Cell proliferation assay
Cells were seeded at 2,000 cells per well in 100 ul of media on a 96 well plate, grown for 24 h and then treated with drug for 72 h prior to analysis using the Cell Counting Kit 8 (Dojindo Molecular Technologies). Ten microliters of CCK-8 solution was added to each well and incubated for one hour prior to absorbance analysis (A450 nm with plate reader). The percentage of cell growth was calculated by comparison of the A450 reading from treated vs vehicle control wells. All treatments were performed in quadruplicate.
Immunoblotting analysis
Whole cell protein lysate was obtained by tween-20 lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 0.1% Tween-20, 10% glycerol, 2.5 mM EGTA, 1 mM EDTA, 1 mM DTT, 1mM Na3VO4, 1 mM PMSF, 1 mM BGP and 10 μg/ml of leupeptin and aprotinin). Samples were sonicated and then centrifuged at 15,000 g for 10 min at 4°C. Protein concentrations were determined by Bradford assay (Bio-Rad Laboratories). Equal amounts of protein were fractionated by SDS-PAGE, transferred to a PVDF membrane (Millipore) and analyzed by incubation with the appropriate primary antibody. Proteins were detected via incubation with HRP-conjugated secondary antibodies and ECL Western Blotting Substrate (Promega Cooperation), SuperSignal* West Dura Extended Duration Chemiluminescent Substrate or SuperSignal* West Femto Maximum Sensitivity Chemiluminescent Substrate (Thermo Fisher Scientific).
Annexin-V apoptosis assay
An amount of 800,000 cells were plated in 100 mm plates and after 24 h of incubation treated with either vehicle, 100 nM cetuximab, 5 uM MK-2206 and the combination for 24 h and harvested after trypsinization. Next, cells were washed with PBS, resuspended in binding buffer (BD Biosciences) and stained with FITC Annexin-V (FITC Annexin-V apoptosis detection kit, BD Biosciences). The cells were analyzed by flow cytometry (BD FACScan). FlowJo Software (Tree Star, Inc.) was used to analyze the data. All experimental arms were done in triplicate and displayed as averages with standard error bars.
Phosphoprotein antibody array
Phosphoprotein arrays were obtained from FullMoon Biosystems, Inc.. Cells were seeded in three 15 cm culture dishes and treated 24 h: (1) HP with vehicle, (2) HC4 with vehicle and (3) HC4 with 2.5 uM of MK-2206. Cells were lysed with the extraction buffer provided as described according to manufacturer’s instructions. Antibody array analysis was performed by FullMoon Biosystems.
| Abbreviations: | ||
| BAD | = | BCL-2 associated agonist of cell death |
| BIM | = | BCL-2-interacting mediator of cell death |
| CtxR | = | cetuximab-resistant |
| CtxS | = | cetuximab-sensitive |
| DNA-PK | = | DNA-dependent protein kinase |
| EGFR | = | epidermal growth factor receptor |
| eIF4E | = | eukaryotic translation initiation factor 4E |
| FoxO | = | forkhead box O transcription factors |
| GSK3 | = | glycogen synthase kinase 3 |
| HNSCC | = | head and neck squamous cell carcinoma |
| MAPK | = | mitogen-activated protein kinase |
| mCRC | = | metastatic colorectal carcinoma |
| mTORC2 | = | mammalian target of rapamycin complex 2 |
| NLS | = | nuclear localization signal |
| NSCLC | = | non-small cell lung cancer |
| PDK1 | = | phosphoinositide-dependent kinase-1 |
| PH | = | pleckstrin homology |
| PI3K | = | phosphatidylinositol 3-kinase |
| PTEN | = | phosphatase and tensin homolog |
| SH2 | = | Src Homology 2 |
| rpS6 | = | ribosomal protein S6 |
| TKD | = | tyrosine kinase domain |
Additional material
Download Zip (285 KB)Acknowledgments
This project was supported, in part, by grant P30CA014520 from the National Cancer Institute, by grant RSG-10–193–01-TBG from the American Cancer Society (D.L.W.) and by NIH grant T32 GM08.1061–01A2 from Graduate Training in Cellular and Molecular Pathogenesis of Human Diseases (TMB) and the Clinical and Translational Science Award (CTSA) program, previously through the National Center for Research Resources (NCRR) grant 1UL1RR025011 and now by the National Center for Advancing Translational Sciences (NCATS), grant 9U54TR000021 (D.L.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Submitted
07/16/2012
Revised
02/21/2013
Accepted
03/18/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/nucleus/article/24342.
Related Research Data
References
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 2006; 7:505 - 16; http://dx.doi.org/10.1038/nrm1962; PMID: 16829981
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301 - 11; http://dx.doi.org/10.1016/j.ccr.2005.03.003; PMID: 15837620
- Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040 - 8; http://dx.doi.org/10.1056/NEJMoa071834; PMID: 18003960
- Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005; 23:1803 - 10; http://dx.doi.org/10.1200/JCO.2005.08.037; PMID: 15677699
- Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:2311 - 9; http://dx.doi.org/10.1200/JCO.2007.13.1193; PMID: 18390971
- Borner M, Koeberle D, Von Moos R, Saletti P, Rauch D, Hess V, et al, Swiss Group for Clinical Cancer Research (SAKK), Bern, Switzerland. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first-line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Ann Oncol 2008; 19:1288 - 92; http://dx.doi.org/10.1093/annonc/mdn058; PMID: 18349029
- Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 2009; 27:663 - 71; http://dx.doi.org/10.1200/JCO.2008.20.8397; PMID: 19114683
- Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360:1408 - 17; http://dx.doi.org/10.1056/NEJMoa0805019; PMID: 19339720
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 27; http://dx.doi.org/10.1056/NEJMoa0802656; PMID: 18784101
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006; 354:567 - 78; http://dx.doi.org/10.1056/NEJMoa053422; PMID: 16467544
- Herbst RS, Arquette M, Shin DM, Dicke K, Vokes EE, Azarnia N, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol 2005; 23:5578 - 87; http://dx.doi.org/10.1200/JCO.2005.07.120; PMID: 16009949
- Baselga J, Trigo JM, Bourhis J, Tortochaux J, Cortés-Funes H, Hitt R, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol 2005; 23:5568 - 77; http://dx.doi.org/10.1200/JCO.2005.07.119; PMID: 16009950
- Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA, Eastern Cooperative Oncology Group. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 2005; 23:8646 - 54; http://dx.doi.org/10.1200/JCO.2005.02.4646; PMID: 16314626
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:e73; http://dx.doi.org/10.1371/journal.pmed.0020073; PMID: 15737014
- Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther 2011; 11:777 - 92; http://dx.doi.org/10.4161/cbt.11.9.15050; PMID: 21293176
- Staal SP, Hartley JW, Rowe WP. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci U S A 1977; 74:3065 - 7; http://dx.doi.org/10.1073/pnas.74.7.3065; PMID: 197531
- Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 2004; 3:268 - 75; http://dx.doi.org/10.4161/cbt.3.3.703; PMID: 15034304
- Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296:1655 - 7; http://dx.doi.org/10.1126/science.296.5573.1655; PMID: 12040186
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098 - 101; http://dx.doi.org/10.1126/science.1106148; PMID: 15718470
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30:203 - 13; http://dx.doi.org/10.1016/j.molcel.2008.02.024; PMID: 18439899
- Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol 2012; 227:3178 - 84; http://dx.doi.org/10.1002/jcp.24065; PMID: 22307544
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550 - 62; http://dx.doi.org/10.1038/nrc2664; PMID: 19629070
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010; 28:1075 - 83; http://dx.doi.org/10.1200/JCO.2009.25.3641; PMID: 20085938
- Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol 2010; 346:31 - 56; http://dx.doi.org/10.1007/82_2010_58; PMID: 20517722
- Vasudevan KM, Garraway LA. AKT signaling in physiology and disease. Curr Top Microbiol Immunol 2010; 347:105 - 33; http://dx.doi.org/10.1007/82_2010_66; PMID: 20549472
- Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 2011; 29:4688 - 95; http://dx.doi.org/10.1200/JCO.2011.35.5263; PMID: 22025163
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008; 27:3944 - 56; http://dx.doi.org/10.1038/onc.2008.19; PMID: 18297114
- Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol 2010; 7:493 - 507; http://dx.doi.org/10.1038/nrclinonc.2010.97; PMID: 20551942
- Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets 2010; 10:80 - 95; http://dx.doi.org/10.2174/156800910790980241; PMID: 20088790
- Dobashi Y, Suzuki S, Kimura M, Matsubara H, Tsubochi H, Imoto I, et al. Paradigm of kinase-driven pathway downstream of epidermal growth factor receptor/Akt in human lung carcinomas. Hum Pathol 2011; 42:214 - 26; http://dx.doi.org/10.1016/j.humpath.2010.05.025; PMID: 21040950
- David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, et al. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res 2004; 10:6865 - 71; http://dx.doi.org/10.1158/1078-0432.CCR-04-0174; PMID: 15501963
- Suzuki S, Igarashi S, Hanawa M, Matsubara H, Ooi A, Dobashi Y. Diversity of epidermal growth factor receptor-mediated activation of downstream molecules in human lung carcinomas. Mod Pathol 2006; 19:986 - 98; http://dx.doi.org/10.1038/modpathol.3800619; PMID: 16648865
- Kim SM, Kim JS, Kim JH, Yun CO, Kim EM, Kim HK, et al. Acquired resistance to cetuximab is mediated by increased PTEN instability and leads cross-resistance to gefitinib in HCC827 NSCLC cells. Cancer Lett 2010; 296:150 - 9; http://dx.doi.org/10.1016/j.canlet.2010.04.006; PMID: 20444542
- Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH, Chen PJ, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther 2011; 337:155 - 61; http://dx.doi.org/10.1124/jpet.110.175786; PMID: 21205925
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956 - 67; http://dx.doi.org/10.1158/1535-7163.MCT-09-1012; PMID: 20571069
- Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res 2012; 18:5816 - 28; http://dx.doi.org/10.1158/1078-0432.CCR-12-1141; PMID: 22932669
- Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD, et al. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin Cancer Res 2011; 17:2852 - 62; http://dx.doi.org/10.1158/1078-0432.CCR-10-2544; PMID: 21536547
- Li Z, Yan S, Attayan N, Ramalingam S, Thiele CJ. Combination of an allosteric Akt Inhibitor MK-2206 with etoposide or rapamycin enhances the antitumor growth effect in neuroblastoma. Clin Cancer Res 2012; 18:3603 - 15; http://dx.doi.org/10.1158/1078-0432.CCR-11-3321; PMID: 22550167
- Pant A, Lee II, Lu Z, Rueda BR, Schink J, Kim JJ. Inhibition of AKT with the orally active allosteric AKT inhibitor, MK-2206, sensitizes endometrial cancer cells to progestin. PLoS One 2012; 7:e41593; http://dx.doi.org/10.1371/journal.pone.0041593; PMID: 22911820
- Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 2012; 11:154 - 64; http://dx.doi.org/10.1158/1535-7163.MCT-11-0606; PMID: 22057914
- Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res 2009; 69:565 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-08-3389; PMID: 19147570
- Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res 2010; 70:8736 - 47; http://dx.doi.org/10.1158/0008-5472.CAN-10-0902; PMID: 20959481
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18:683 - 95; http://dx.doi.org/10.1016/j.ccr.2010.11.023; PMID: 21156289
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925; PMID: 16452206
- Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008; 26:1603 - 10; http://dx.doi.org/10.1200/JCO.2007.14.5482; PMID: 18332469
- Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 2011; 30:4567 - 77; http://dx.doi.org/10.1038/onc.2011.164; PMID: 21666717
- Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 2010; 19:1355 - 66; http://dx.doi.org/10.1517/13543784.2010.520701; PMID: 20846000