Abstract
Blockade of the renin angiotensin system (RAS) can inhibit tumor growth and this may be mediated via undefined immunomodulatory actions. This study investigated the effects of RAS blockade on liver macrophages (Kupffer cells; KCs) in an orthotopic murine model of colorectal cancer (CRC) liver metastases. Here we showed that pharmacological targeting of the RAS [ANG II (31.25 µg/kg/h i.p.), ANG-(1–7) (24 µg/kg/h i.p.) or the ACE inhibitor; captopril (750 mg/kg/d i.p.)] altered endogenous KC numbers in the tumor-bearing liver throughout metastatic growth. Captopril, and to a lesser extent ANG-(1–7), increased KC numbers in the liver but not tumor. KCs were found to express the key RAS components: ACE and AT1R. Treatment with captopril and ANG II increased the number of AT1R-expressing KCs, although total KC numbers were not affected by ANG II. Captopril (0.1 µM) also increased macrophage invasion in vitro. Additionally, captopril was administered with KC depletion before tumor induction (day 0) or at established metastatic growth (day 18) using gadolinium chloride (GdCl3; 20 mg/kg). Livers were collected at day 21 and quantitative stereology used as a measure of tumor burden. Captopril reduced growth of CRC liver metastases. However, when captopril was combined with early KC depletion (day 0) tumor growth was significantly increased compared with captopril alone. In contrast, late KC depletion (day 18) failed to influence the anti-tumor effects of captopril. The result of these studies suggests that manipulation of the RAS can alter KC numbers and may subsequently influence progression of CRC liver metastases.
Introduction
Colorectal cancer (CRC) liver metastasis is a major health concern worldwide and current treatment modalities for patients remain limited. There is evidence that the local renin angiotensin system (RAS) may provide a novel target to improve patient outcomes. The RAS is deregulated in several cancers and can influence several aspects of tumor growth.Citation1-Citation5 Enzymatic action by the angiotensin converting enzyme (ACE) yields the principal effector peptide of the RAS, Angiotensin II (ANG II). ANG II mediates proliferative, angiogenic, and inflammatory effects through the angiotensin type I receptor (AT1R). The alternative peptide, the ANG-(1–7), is believed to negatively modulate tumor growth.Citation6-Citation8 We previously showed that ACE inhibition (captopril) could significantly slow tumor growth in an orthotopic syngeneic mouse model of CRC liver metastases.Citation9,Citation10
The recent identification of RAS expression in hepatic macrophages (Kupffer cells; KCs) suggests they may be a novel mediator of RAS-induced effects in the liver.Citation11 KCs account for approximately 15% of the total liver cell population and can be identified by its expression of F4/80 (macrophage marker).Citation12 Together with other non-parenchymal cell populations, which include sinusoidal endothelial cells and stellate cells, they play a crucial role in the pathogenesis of liver disease. The role of KCs in the progression of CRC liver metastases is multipotent; direct and indirect activation of KCs results in the production of growth factors and chemotactic cytokines capable of mediating cytotoxic, anti-tumor effects, but also capable of facilitating pro-tumor effects.Citation13-Citation15
The AT1R is highly expressed in tumor-associated macrophages and mice mutant for the AT1R have fewer infiltrating macrophages following tumor induction.Citation16 Furthermore, activated peritoneal macrophages from ACE-knockout mice were deficient in the production of effector factors, such as tumor necrosis factor-α, interleukin-12p40, and CD86, suggesting a significant role for ACE in myelopoiesis.Citation17
Here we investigated the role of the RAS in regulating KC numbers during the development of CRC liver metastases. We also examined the contribution of KCs to the anti-tumor effects of ACE inhibition in a mouse model of CRC liver metastases.
Results
Captopril and ANG-(1–7) increased KC numbers in the liver but not in the tumor
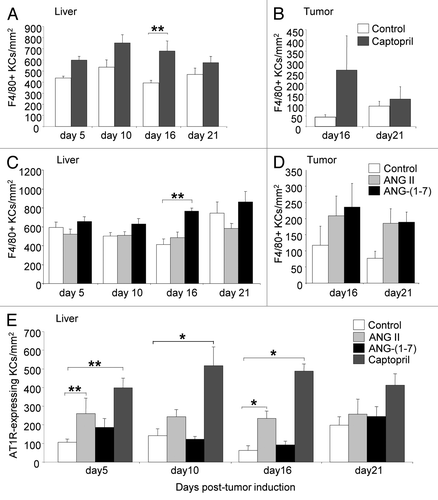
Mice were treated with ANG II, ANG-(1–7), or captopril (ACE inhibitor) from the time of tumor induction (day 0) and culled at day 5, 10, 16, or 21. Control animals received saline. There was a trend for captopril to increase F4/80+ KC numbers in the liver at all the time points examined (day 5, 10 and 16). However, it was only statistically significant at day 16 (exponential growth phase) compared with control (P = 0.019; ). Similar to captopril, ANG-(1–7) infusion increased F4/80+ KC numbers in the liver at day 16 (P = 0.032; ). In contrast, ANG II infusion did not significantly alter total F4/80+ KC numbers in the liver compared with controls at any of the examined time points (). In the tumor tissues an accumulation of KCs was observed especially at the tumor/host interface and near vascular lakes. F4/80+ KC numbers within tumors did not significantly alter following captopril, ANG-(1–7), or ANG II treatment at day 16 or 21 compared with controls ().
Figure 1. The RAS can regulate KC numbers in CRC liver metastases. Animals were treated with ANG II infusion (31.25 µg/kh/h), ANG-(1-7) infusion (24 µg/kh/h), or captopril (750 mg/kg/d). The solubilizing agents (saline) provided a control. Treatments continued from the time of tumor induction to tissue collection at day 5, 10, 16, or 21. Immunohistochemical staining with the F4/80 macrophage marker was used to identify KCs in the (A and C) liver and (B and D) tumor following RAS treatment. (E) Double immunostaining using the F4/80 and AT1R marker identified AT1R-expressing KCs. F4/80 and AT1R-expressing F4/80 cells were quantified as the number of cells per mm2. Results are expressed as mean values ± SEM. N = 6 animals for each group. *P ≤ 0.005, **P < 0.05
KCs express AT1R and ACE
AT1R-expressing KCs were identified using double immunohistochemical staining in both the liver parenchyma and in tumors (black arrow; ). However, not all KCs displayed AT1R expression (pink arrow; ). AT1R-expressing non-F4/80 cells were also observed (gray arrow; ). Most of these cells (possibly tumor cells or cancer-associated fibroblasts) localized to the tumor/host interface and were found to be intermingling with KCs (). Due to the extremely high number and clustered nature of these AT1R-expressing F4/80+ cells, cell counts using our described method could not be performed in the tumor.
Figure 2. KCs express the AT1R and ACE in CRC liver metastases. Double immunostaining using the F4/80 and AT1R marker identified AT1R-expressing KCs in both the liver and tumors (black arrow) (A and B). AT1R-expressing non-F4/80 cells were also observed (gray arrow) (A). Double immunostaining also identified ACE-expressing KCs in both the liver and in tumors (green arrow) (C and D). However, not all KCs displayed AT1R or ACE expression (pink arrow) (A and C). ACE expression was also localized on vascular endothelial cells (blue arrow) (C).
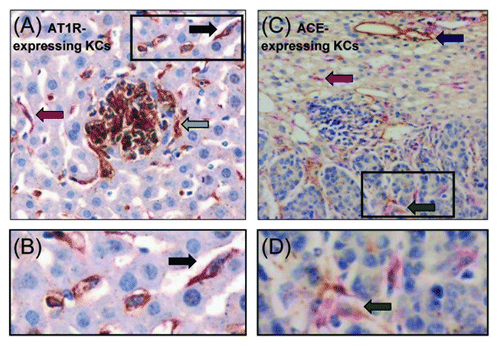
ACE-expressing KCs were also identified in both the liver and in tumors (green arrow; ). Counts of ACE-expressing KCs were not performed due to difficulties in clearly identifying all double-stained cells. ACE expression was also localized to endothelial cells in the liver (blue arrow; ). Very little ACE staining was observed in tumors ().
Captopril and ANG II increased liver AT1R-expressing KC numbers
Captopril increased the number of liver AT1R-positive KCs at days 5, 10 and 16 compared with controls (P = 0.013 d 5; P = 0.003 d 10; P < 0.001 d 16; Figure. 1E). Similarly, AT1R-expressing KC numbers were increased by ANG II infusion in the liver at days 5 (P = 0.016; ) and 16 (P = 0.005; ). ANG-(1–7) however, did not alter AT1R-expressing KC numbers in the liver at any of the time points examined. By day 21 the numbers of AT1R-positive KCs in all 3 treatment groups were at control levels ().
Captopril increased macrophage invasion in vitro
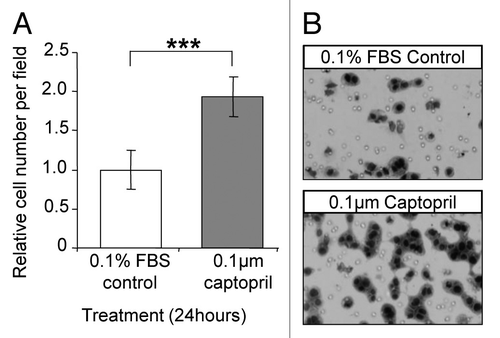
The stimulatory effect of captopril on macrophage invasion was confirmed in vitro using the Boyden Chamber assay. Mouse P388D1 macrophages showed a trend toward increased invasion across fibronectin following captopril treatment at a concentration of 0.1 µM compared with 0.1% FBS/RPMI controls (P = 0.103; ).
Figure 3. Captopril increased macrophage invasion in vitro. (A) P388D1 macrophage cells were allowed to invade through fibronectin for 24 hours under the influence of 0.1 µM captopril or 0.1% FBS/RPMI control (Boyden Chamber assay). (B) Representative images of migrated P388D1 cells in the lower chamber of the membrane. The average number of cells in 25‒35 representative images per membrane was used to quantify the extent of invasion. Results are expressed as mean values ± SEM. N = 6. ***P = 0.10.
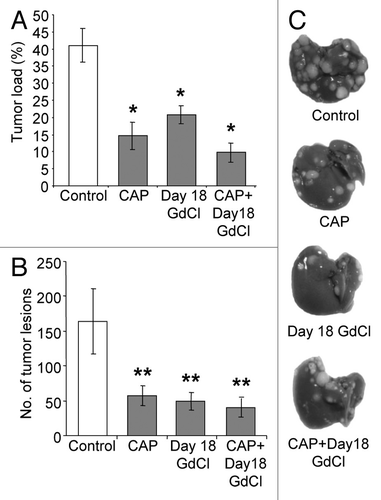
Early KC depletion reduced the anti-tumor effects of Captopril
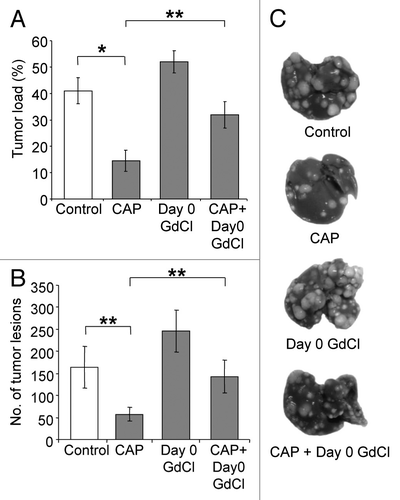
Next we evaluated the contribution of KCs to the anti-tumor effects of ACE inhibition. Captopril was administered with or without KC depletion using gadolinium chloride (GdCl3). GdCl3 was administered before tumor induction (day 0) or at day 18 (tumor plateau phase). Captopril significantly decreased both liver tumor burden (P < 0.001; ) and the number of tumor nodules (P = 0.018; ) compared with the untreated control group. The level of tumor inhibition achieved by captopril treatment in the current experiment is comparable to our previously published studies.Citation9,Citation10 In this study early KC depletion (day 0) increased tumor burden and the number of tumor nodules compared with untreated controls, however, this was not statistically significant (). The combined treatment of captopril and early KC depletion reduced the captopril effect by significantly increasing both tumor burden (P = 0.002; ) and tumor nodules (P = 0.013; ) compared with captopril alone.
Figure 4. Early KC depletion reduced the anti-tumor effects of ACE inhibition by captopril. Animals were administered gadolinium chloride (GdCl) before tumor induction, with or without captopril (CAP). Solubilizing agent (saline) provided a control. Livers were collected at day 21. Quantitative stereological methods were used to determine (A) the percentage of tumor volume over total tumor-bearing liver volume and (B) the number of tumor nodules per liver. Results suggest captopril can reduce the stimulatory effect of early KC depletion but not completely reverse it. (C) Representative macroscopic images of treated tumor-induced livers.Results are expressed as mean values ± SEM. N = 6 animals for each group, *P ≤ 0.001, **P < 0.01.
Late KC depletion did not alter the anti-tumor effects of captopril
KC depletion in established CRC liver metastases (day 18) decreased tumor burden (P < 0.001; ), suggesting that KCs have a pro-tumor function at this stage. This anti-tumor effect was of the same magnitude as seen when mice were treated the captopril alone. A decrease in tumor load was observed when late KC depletion was combined with captopril treatment compared with untreated controls (P < 0.001 tumor burden; P = 0.007 tumor nodules; ). Combined treatment slightly decreased the tumor burden compared with single treatments, however this was not statistically significant ().
Figure 5. Late KC depletion did not alter the anti-tumor effects of ACE inhibition by captopril.Animals were administered gadolinium (GdCl) at day 18, with or without captopril (CAP). Solubilizing agent (saline) provided a control. Livers were collected at day 21. Quantitative stereological methods were used to determine (A) the percentage of tumor volume over total tumor-bearing liver volume and (B) the number of tumor nodules per liver. (C) Representative macroscopic images of treated tumor-induced livers. Results are expressed as mean values ± SEM. N = 6 animals for each group, *P ≤ 0.001 and **P < 0.05 when compared to controls.
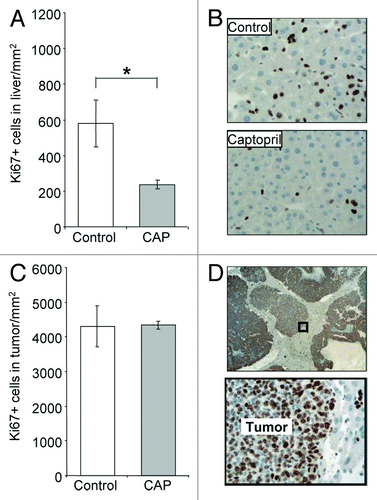
Captopril decreased proliferating cells in the liver but not in the tumor
Captopril significantly decreased proliferating cells (Ki67+ cells) in the liver compared with untreated controls (P < 0.050; ). Histological examination suggested that a majority of these proliferating cells were hepatocytes (). In contrast, the number of proliferating cells in the tumor following captopril treatment remained at control levels (). Similarly, no change was observed in the percentage of tumor necrosis (data not shown).
Figure 6. Captopril decreased proliferating cells in the liver but not in the tumor.Captopril (CAP) was administered to animals daily from the time of tumor induction to tissue collection at day 21. Solubilizing agent (saline) provided a control. Proliferating cells were identified using the Ki67 marker, and quantified as the number of positively stained cells per mm2 in the (A) liver and (C) tumor. (B and D) Representative images of Ki67+ staining. Proliferating cells were stained brown; a majority appeared to be hepatocytes in the liver and cancer cells in the tumor. Results are expressed as mean values ± SEM. N = 6 animals for each group, *P < 0.05.
Discussion
The RAS can affect the growth and progression of several tumor types,Citation7,Citation18-Citation21 including CRC liver metastases.Citation10 Although the RAS has well documented roles in angiogenesis and cell proliferation, it also contributes to the regulation of macrophage function and migration.Citation17,Citation22-Citation24 This work aimed to determine if modification of the liver macrophage population (KCs) in response to alterations of the RAS could contribute to the effects of RAS blockade on the growth of CRC liver metastases.
ACE inhibition with captopril has previously been shown to decrease tumor burden in CRC liver metastases,Citation10 and this effect was confirmed here. We also observed a sustained increase in KC numbers in the liver following captopril treatment; from cancer cell seeding to the exponential tumor growth phase. However, this increase was no longer evident by the tumor plateau phase (day 21). In vitro, we also showed a stimulatory effect of captopril on the invasive capacity of mouse macrophages. It is possible that the effect of captopril may be mediated by an increase in anti-tumor KC activity during early metastatic growth. To date, a number of studies have shown that ACE inhibitors reduce monocyte/macrophage infiltration; however, this is predominately in experimental models of atherosclerosis and hypertension where disease progression is particularly exacerbated by pro-inflammatory factors.Citation25-Citation28 To our knowledge, this is the first study to show the promotion of macrophage invasion in a tumor setting by captopril. Changes in disease model can result in opposing effects of the RAS on monocyte/macrophage infiltration,Citation29,Citation30 and illustrates the complex nature of RAS-derived immunomodulatory effects.
Similar to captopril, ANG-(1–7) infusion also increased macrophage numbers in the liver. ACE inhibition can increase local and plasma levels of ANG-(1–7).Citation31 Thus, the effects of captopril on KC numbers may result from an underlying increase in ANG-(1–7). Similar to captopril, ANG-(1–7) infusion can inhibit tumor growth and angiogenesis.Citation6-Citation8 Unlike ANG-(1–7) or captopril treatment, ANG II did not alter KC numbers in the liver parenchyma. The reduction in tumor growth by captopril could be a synergistic immunomodulatory effect of decreased local ANG II levels by ACE inhibition accompanied by increased levels of counter-regulatory peptide, ANG-(1–7).
In contrast to the liver parenchyma, pharmacological manipulation of the RAS did not affect KC numbers within the tumor. Given that KCs in the liver parenchyma are a major component of the tumor-bearing liver, their paracrine effects may be exploited by stromal and tumor cells to support growth and invasive properties. Future studies understanding functional differences between liver and tumor macrophages would be important. Additionally, our results illustrate the importance of the microenvironment in determining immune responses. The tumor microenvironments may alter the phenotypes of infiltrating immune cells or increase the recruitment of particular macrophage subsets which are beneficial to tumor progression. Differences in KC numbers between the liver and tumor found in our study indicate the potential to alter either liver or tumor immune response independently. This independence is particularly important when considering the benefits of selectively targeting tumors with anti-cancer therapies.
In accordance with past studies, KCs express the key RAS components ACE and AT1R.Citation11 The number of AT1R-expressing KCs in the liver was increased by captopril and ANG II treatment. ANG II is known to mediate tumor promoting attributes through the AT1R, including proliferative, angiogenic, and inflammatory effects.Citation3,Citation4,Citation10,Citation16,Citation32-Citation34 Therefore, it is unlikely an increase in AT1R-positive KCs contribute to the inhibitory effects of captopril. An increase in AT1R-expressing KCs following ACE inhibition may instead reflect a compensatory mechanism to enable an amplified response to limited ANG II. The RAS can compensate for disturbances to the system in order to maintain balance.Citation35,Citation36
Current research attributes a predominantly anti-tumor phenotype to KCs during the early stages of tumor establishment.Citation37-Citation39 Our study in agreement with these reports indicates early KC depletion to have a tumor stimulatory effect. The anti-tumor activity of KCs at this early stage is attributed to their phagocytic properties,Citation40 while captopril exerts its anti-tumor effect through inhibition of angiogenesis, tumor proliferation and reduction in inflammation.Citation10,Citation41-Citation44 Here we showed that when captopril was combined with early KC depletion (day 0) tumor growth was significantly increased compared with captopril treatment. Our results could be explained by these mechanisms; KC depletion results in increased seeding of metastases and although their growth is delayed by captopril treatment, by the endpoint in this study they contribute significantly to the tumor burden compared with captopril treatment alone.
The tumor inhibitory effect of captopril at established metastatic growth does not appear to be associated with KC function as depletion of KCs at this stage failed to impact upon the anti-tumor effects of captopril. Once tumors become established, key growth and angiogenic factors in the tumor microenvironment induce tumor-associated macrophages (TAMs) to assist in tumor growth and metastasis.Citation45-Citation50 Captopril may affect TAMs differently to early anti-tumor macrophages. It is also possible that captopril may only have effects on the early stages of tumor development and these effects are reflected in the delayed progression observed in later stages. A study of tumor growth kinetics may allow the temporal effects of captopril to be better understood.
While tumor cell proliferation at day 21 was unaffected by captopril treatment, hepatocyte proliferation significantly decreased compared with untreated controls. This suggests that the mechanism of tumor growth retardation does not involve an inhibition of cancer cell proliferation. Hepatocyte growth factor (HGF) is a key stimulator of hepatocyte proliferation after liver injuryCitation51 and is produced by cells of mesenchymal origin including KCs.Citation52 Our results suggest that captopril may reduce liver damage by limiting tumor growth and perhaps the subsequent release of HGF by TAMs. Captopril was found to reduce inflammation in liver regenerationCitation53 and it is possible that it has a similar effect in the tumor bearing liver, thus reduced inflammation is reflected in reduced hepatocyte proliferation.
The results presented here demonstrate an immunomodulatory role of the RAS. Blockade of ACE with captopril increased KC infiltration in the tumor-bearing liver in vivo and invasion in vitro as well as appearing to alter the function of early, anti-tumor KCs during tumor progression. Understanding this immunomodulatory potential may allow us to optimize the anti-tumor effects of ACE inhibitors.
Materials and Methods
In vivo model and cell lines
The mouse colorectal cancer (MoCR) cell line used for in vivo experiments was harvested from a dimethylhydrazine-induced colon carcinoma in a CBA mouse at a stage known to metastasize to the liver.Citation54 CRC liver metastases were induced as described previously.Citation10,Citation54 Briefly, a suspension of 2.5 × 104 MoCR cells was injected into the spleen of six- to eight-week-old male CBA mice and, after three minutes, the spleen was removed to confine metastases to the liver. In this model, angiogenesis is established by day 10, with a slow growth phase from days 10–16, a rapid growth phase until day 19, followed by a plateau phase.Citation54 A minimum of six animals per group were used. All experiments were approved by the Austin Health Animal Ethics Committee and conform to the provisions of the Declaration of Helsinki in 1995. Liver samples were collected and fixed in fresh 4% PFA.
RAS treatment of colorectal liver metastases
Treatments included the RAS ligands ANG II (Auspep, 2078; 31.25 µg/kg/h) or ANG-(1–7) (Auspep, 2588; 24 µg/kg/h) infusion (Alzet® osmotic mini pumps 1004). Captopril (ACE inhibitor) was given as daily i.p. injections of 750 mg/kg (Sigma-Aldrich, 21751). Doses were based on previously published studies.Citation10,Citation55-Citation57 The solubilizing agent (saline) provided a control. Treatments continued from the time of tumor induction to tissue collection at day 5, 10, 16, or 21.
ACE inhibition and KC depletion of colorectal liver metastases
Captopril (Sigma-Aldrich, 21751; 750 mg/kg) was administered with or without KC depletion using gadolinium chloride (GdCl3; 20 mg/kg). Captopril or vehicle (saline) control was given as daily i.p. injections. GdCl3 or vehicle (saline) control was administered by two successive doses (6 to 24 h apart) via tail-vein injection before tumor induction (day 0) or at day 18 (tumor plateau phase). GdCl3 at this dose was previously determined to result in significant depletion of KCs.Citation58-Citation61 Captopril treatment continued from the time of tumor induction to tissue collection at day 21.
Tumor burden
Quantitative stereological analysis was performed on whole fixed livers collected at endpoints to determine tumor burden. The fixed livers were transversely sliced into 1.5 mm sections with a multi blade fractionator and an image of liver sections taken using Lumenera Infinity4 digital CCD camera. Tumor area (mm2) and the number of tumor nodules per liver were assessed using Image-Pro plus 6.0. The areas were converted to volume measurements based on the thickness of each tissue slice (1.5 mm) and the proportion of liver sampled. This was subsequently used to calculate the percentage of tumor burden in the liver.
Immunohistochemical staining and analyses
AT1R (rabbit polyclonal against human, Santa Cruz, sc-1173), ACE (rabbit polyclonal against human, Santa Cruz, sc-20791), KC (F4/80; monoclonal rat anti-mouse, ATCC no HB-198, culture supernatant) and proliferation (Ki67; rat monoclonal anti-mouse, Thermoscientific, RM-9106-S1) were assessed in PFA-fixed paraffin embedded tissues. AT1R was used at a concentration of 1 µg/ml, ACE at 1 µg/ml, F4/80 at a 1:20 000 dilution and Ki67 at 1:100 dilution. Non-immunized rabbit IgG (Santa Cruz, sc-2027), at an equivalent concentration to the primary antibody, was used as a negative control. For F4/80, proteinase K (1 mg/ml) antigen retrieval at 37 °C was required. Endogenous peroxidases were blocked with 3% H2O2 and non-specific binding inhibited with 10% normal goat serum (Zymed, 01–6201). Slides were incubated with primary antibodies at 37 °C for 1 h and then 4 °C overnight. Slides were then incubated with the secondary antibody (Dako Envision Plus goat anti-rabbit HRP secondary 4011) and positive staining visualized with diaminobenzidine (DAB). F4/80 required incubation with a linking polyclonal rabbit anti-rat biotinylated antibody (Dako Envision Plus) at 37 °C prior to the secondary antibody. Slides were counterstained with Mayer's hematoxylin.
AT1R- or ACE-expressing macrophages were identified by double staining using the AT1R or ACE with DAB followed by the F4/80 marker coupled with Vulcan Fast Red detection (Applied Medical).
For quantitation of staining, representative images from immunohistochemically stained sections were captured using a digital light microscope (Nikon Coolscope®, Nikon Corporation) at a magnification between 40× and 400× . Images were captured using a pattern which ensured unbiased selection and no overlap. Depending on the animal and its tumor load, 30‒40 total images across 5‒10 tumors were taken for analysis. F4/80, AT1R-expressing F4/80 and Ki67 were assessed as the number of positively stained cells per high power area of tumor or liver (ImagePro-Plus Version 6.0).
Cell migration/invasion assay
Cell migration/invasion was determined using the Boyden Chamber assay described by Ishizu et al.Citation62 with the following modification. Membranes (8-µm pore size, Becton Dickinson) were coated with 3 µg of fibronectin on the lower surfaces and placed into a 24-well plate containing 600 µl/well of serum-free RPMI with 0.1% BSA. Cultured cells (P388D1 murine macrophages; 2.5 × 104/100 µl/chamber) were added to the upper chambers. Cells were allowed to migrate for 24 h under the influence of 0.1 µM captopril diluted in 0.1% FBS/RPMI at 37 °C in a 5% CO2 atmosphere. P388D1 cells treated with 0.1% FBS/RPMI served as negative controls. The remaining non-migrated cells were removed from the upper chamber before the membranes were fixed and stained with Quick Dip (Fronine). A random pattern that covered the entire membrane was used to capture representative digital images at 200× magnifications using the Coolscope (Nikon Coolscope®, Nikon Corporation). The average number of cells in 25‒35 representative images per membrane was used to quantify the extent of invasion.
Statistical analysis
Quantitative data are presented as means ± SEM. Statistical analyses were conducted using SPSS (Statistical Package for the Social Sciences, version 17). Data sets were tested for normality using the Kolmogorov-Smirnov test. Parametric data was analyzed using ANOVA with post-hoc comparison (Tukey method). Non-parametric data was analyzed using the Mann–Whitney U test. Adjusted P ≤ 0.05 was considered statistically significant and 0.05 < P ≤ 0.10 considered a significant trend.
| Abbreviations: | ||
| RAS | = | renin angiotensin system |
| KC | = | Kupffer cell |
| CRC | = | colorectal cancer |
| ANG | = | angiotensin |
| ACE | = | angiotensin converting enzyme |
| GdCl3 | = | gadolinium chloride |
Acknowledgments
This work was supported by Cancer Australia's Priority-driven Collaborative Cancer Research Scheme (co-funded by Cancer Australia and Cure Cancer Australia Foundation) and by the Cancer Council of Victoria. EIA was supported by an NHMRC Post-doctoral Training Award. SWW and JN were supported by an Australian Rotary Health Research Fund PhD Scholarship. TF aided in editing the manuscript. SWW performed experimental and statistical analysis and drafted the manuscript. EIA and SWW contributed equally to experimental design and protocol optimization. JN contributed to the RAS treatment and tissue collection. EIA and CC reviewed the manuscript and aided in the development of the concepts tested.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis 2008; 29:1675 - 84; http://dx.doi.org/10.1093/carcin/bgn171; PMID: 18632755
- De Paepe B, Verstraeten VL, De Potter CR, Vakaet LA, Bullock GR. Growth stimulatory angiotensin II type-1 receptor is upregulated in breast hyperplasia and in situ carcinoma but not in invasive carcinoma. Histochem Cell Biol 2001; 116:247 - 54; PMID: 11685554
- Rhodes DR, Ateeq B, Cao Q, Tomlins SA, Mehra R, Laxman B, et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc Natl Acad Sci U S A 2009; 106:10284 - 9; http://dx.doi.org/10.1073/pnas.0900351106; PMID: 19487683
- Suganuma T, Ino K, Shibata K, Kajiyama H, Nagasaka T, Mizutani S, et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res 2005; 11:2686 - 94; http://dx.doi.org/10.1158/1078-0432.CCR-04-1946; PMID: 15814650
- Uemura H, Hasumi H, Ishiguro H, Teranishi J, Miyoshi Y, Kubota Y. Renin-angiotensin system is an important factor in hormone refractory prostate cancer. Prostate 2006; 66:822 - 30; http://dx.doi.org/10.1002/pros.20407; PMID: 16482568
- Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7). Carcinogenesis 2004; 25:2045 - 52; http://dx.doi.org/10.1093/carcin/bgh236; PMID: 15284177
- Menon J, Soto-Pantoja DR, Callahan MF, Cline JM, Ferrario CM, Tallant EA, et al. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res 2007; 67:2809 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-06-3614; PMID: 17363603
- Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther 2009; 8:1676 - 83; http://dx.doi.org/10.1158/1535-7163.MCT-09-0161; PMID: 19509262
- Neo JH, Ager EI, Angus PW, Zhu J, Herath CB, Christophi C. Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer 2010; 10:134; http://dx.doi.org/10.1186/1471-2407-10-134; PMID: 20380732
- Neo JH, Malcontenti-Wilson C, Muralidharan V, Christophi C. Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model of colorectal cancer liver metastases. J Gastroenterol Hepatol 2007; 22:577 - 84; http://dx.doi.org/10.1111/j.1440-1746.2006.04797.x; PMID: 17376054
- Leung PS, Suen PM, Ip SP, Yip CK, Chen G, Lai PB. Expression and localization of AT1 receptors in hepatic Kupffer cells: its potential role in regulating a fibrogenic response. Regul Pept 2003; 116:61 - 9; http://dx.doi.org/10.1016/S0167-0115(03)00192-7; PMID: 14599716
- Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol 2006; 12:7413 - 20; PMID: 17167827
- van der Bij GJ, Oosterling SJ, Meijer S, Beelen RH, van Egmond M. Therapeutic potential of Kupffer cells in prevention of liver metastases outgrowth. Immunobiology 2005; 210:259 - 65; http://dx.doi.org/10.1016/j.imbio.2005.05.020; PMID: 16164033
- Paschos KA, Majeed AW, Bird NC. Role of Kupffer cells in the outgrowth of colorectal cancer liver metastases. Hepatol Res 2010; 40:83 - 94; http://dx.doi.org/10.1111/j.1872-034X.2009.00578.x; PMID: 19788686
- Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 2006; 26:1175 - 86; http://dx.doi.org/10.1111/j.1478-3231.2006.01342.x; PMID: 17105582
- Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 2003; 112:67 - 75; PMID: 12840060
- Lin C, Datta V, Okwan-Duodu D, Chen X, Fuchs S, Alsabeh R, et al. Angiotensin-converting enzyme is required for normal myelopoiesis. FASEB J 2011; 25:1145 - 55; http://dx.doi.org/10.1096/fj.10-169433; PMID: 21148418
- Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett 2001; 495:197 - 200; http://dx.doi.org/10.1016/S0014-5793(01)02377-8; PMID: 11334891
- Kikkawa F, Mizuno M, Shibata K, Kajiyama H, Morita T, Ino K, et al. Activation of invasiveness of cervical carcinoma cells by angiotensin II. Am J Obstet Gynecol 2004; 190:1258 - 63; http://dx.doi.org/10.1016/j.ajog.2003.12.013; PMID: 15167827
- Stoeltzing O, Liu W, Reinmuth N, Parikh A, Ahmad SA, Jung YD, et al. Angiogenesis and antiangiogenic therapy of colon cancer liver metastasis. Ann Surg Oncol 2003; 10:722 - 33; http://dx.doi.org/10.1245/ASO.2003.07.019; PMID: 12900362
- Uemura H, Ishiguro H, Nakaigawa N, Nagashima Y, Miyoshi Y, Fujinami K, et al. Angiotensin II receptor blocker shows antiproliferative activity in prostate cancer cells: a possibility of tyrosine kinase inhibitor of growth factor. Mol Cancer Ther 2003; 2:1139 - 47; PMID: 14617787
- Danilov SM, Sadovnikova E, Scharenborg N, Balyasnikova IV, Svinareva DA, Semikina EL, et al. Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp Hematol 2003; 31:1301 - 9; http://dx.doi.org/10.1016/j.exphem.2003.08.018; PMID: 14662338
- Geara AS, Azzi J, Jurewicz M, Abdi R. The renin-angiotensin system: an old, newly discovered player in immunoregulation. Transplant Rev (Orlando) 2009; 23:151 - 8; http://dx.doi.org/10.1016/j.trre.2009.04.002; PMID: 19539879
- Ruiz-Ortega M, Bustos C, Hernández-Presa MA, Lorenzo O, Plaza JJ, Egido J. Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-kappa B activation and monocyte chemoattractant protein-1 synthesis. J Immunol 1998; 161:430 - 9; PMID: 9647253
- Abd Alla J, Langer A, Elzahwy SS, Arman-Kalcek G, Streichert T, Quitterer U. Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis. J Biol Chem 2010; 285:23496 - 505; http://dx.doi.org/10.1074/jbc.M110.117481; PMID: 20504763
- Clozel M, Kuhn H, Hefti F, Baumgartner HR. Endothelial dysfunction and subendothelial monocyte macrophages in hypertension. Effect of angiotensin converting enzyme inhibition. Hypertension 1991; 18:132 - 41; http://dx.doi.org/10.1161/01.HYP.18.2.132; PMID: 1653185
- Hernández-Presa M, Bustos C, Ortego M, Tuñon J, Renedo G, Ruiz-Ortega M, et al. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997; 95:1532 - 41; http://dx.doi.org/10.1161/01.CIR.95.6.1532; PMID: 9118522
- Kowala MC, Grove RI, Aberg G. Inhibitors of angiotensin converting enzyme decrease early atherosclerosis in hyperlipidemic hamsters. Fosinopril reduces plasma cholesterol and captopril inhibits macrophage-foam cell accumulation independently of blood pressure and plasma lipids. Atherosclerosis 1994; 108:61 - 72; http://dx.doi.org/10.1016/0021-9150(94)90037-X; PMID: 7980708
- Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 2007; 292:F330 - 9; http://dx.doi.org/10.1152/ajprenal.00059.2006; PMID: 16804106
- Wenzel UO, Thaiss F, Helmchen U, Stahl RA, Wolf G. Angiotensin II infusion ameliorates the early phase of a mesangioproliferative glomerulonephritis. Kidney Int 2002; 61:1020 - 9; http://dx.doi.org/10.1046/j.1523-1755.2002.00192.x; PMID: 11849457
- Santos RA, Ferreira AJ. Angiotensin-(1-7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens 2007; 16:122 - 8; http://dx.doi.org/10.1097/MNH.0b013e328031f362; PMID: 17293687
- Fujita M, Hayashi I, Yamashina S, Itoman M, Majima M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem Biophys Res Commun 2002; 294:441 - 7; http://dx.doi.org/10.1016/S0006-291X(02)00496-5; PMID: 12051731
- Imai N, Hashimoto T, Kihara M, Yoshida S, Kawana I, Yazawa T, et al. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab Invest 2007; 87:189 - 98; http://dx.doi.org/10.1038/labinvest.3700504; PMID: 17318197
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35:881 - 900; http://dx.doi.org/10.1016/S1357-2725(02)00271-6; PMID: 12676174
- Bumpus FM. Angiotensin I and II. Some early observations made at the Cleveland Clinic Foundation and recent discoveries relative to angiotensin II formation in human heart. Hypertension 1991; 18:Suppl III122 - 5; http://dx.doi.org/10.1161/01.HYP.18.5_Suppl.III122; PMID: 1937674
- Lindpaintner K, Jin M, Wilhelm MJ, Suzuki F, Linz W, Schoelkens BA, et al. Intracardiac generation of angiotensin and its physiologic role. Circulation 1988; 77:I18 - 23; PMID: 3286043
- Heuff G, Oldenburg HS, Boutkan H, Visser JJ, Beelen RH, Van Rooijen N, et al. Enhanced tumour growth in the rat liver after selective elimination of Kupffer cells. Cancer Immunol Immunother 1993; 37:125 - 30; http://dx.doi.org/10.1007/BF01517045; PMID: 8319242
- Pearson HJ, Anderson J, Chamberlain J, Bell PR. The effect of Kupffer cell stimulation or depression on the development of liver metastases in the rat. Cancer Immunol Immunother 1986; 23:214 - 6; http://dx.doi.org/10.1007/BF00205652; PMID: 3024833
- Rushfeldt C, Sveinbjørnsson B, Seljelid R, Smedsrød B. Early events of hepatic metastasis formation in mice: role of Kupffer and NK-cells in natural and interferon-gamma-stimulated defense. J Surg Res 1999; 82:209 - 15; http://dx.doi.org/10.1006/jsre.1998.5532; PMID: 10090831
- Timmers M, Vekemans K, Vermijlen D, Asosingh K, Kuppen P, Bouwens L, et al. Interactions between rat colon carcinoma cells and Kupffer cells during the onset of hepatic metastasis. Int J Cancer 2004; 112:793 - 802; http://dx.doi.org/10.1002/ijc.20481; PMID: 15386374
- Amirshahrokhi K, Ghazi-khansari M, Mohammadi-Farani A, Karimian G. Effect of captopril on TNF-α and IL-10 in the livers of bile duct ligated rats. Iran J Immunol 2010; 7:247 - 51; PMID: 21189448
- Attoub S, Gaben AM, Al-Salam S, Al Sultan MA, John A, Nicholls MG, et al. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann N Y Acad Sci 2008; 1138:65 - 72; http://dx.doi.org/10.1196/annals.1414.011; PMID: 18837885
- Miguel-Carrasco JL, Zambrano S, Blanca AJ, Mate A, Vázquez CM. Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. J Inflamm (Lond) 2010; 7:21; http://dx.doi.org/10.1186/1476-9255-7-21; PMID: 20462420
- Volpert OV, Ward WF, Lingen MW, Chesler L, Solt DB, Johnson MD, et al. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest 1996; 98:671 - 9; http://dx.doi.org/10.1172/JCI118838; PMID: 8698858
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006; 124:263 - 6; http://dx.doi.org/10.1016/j.cell.2006.01.007; PMID: 16439202
- Hamada I, Kato M, Yamasaki T, Iwabuchi K, Watanabe T, Yamada T, et al. Clinical effects of tumor-associated macrophages and dendritic cells on renal cell carcinoma. Anticancer Res 2002; 22:6C 4281 - 4; PMID: 12553070
- Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol 2000; 7:263 - 9; http://dx.doi.org/10.1046/j.1442-2042.2000.00190.x; PMID: 10910229
- Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol 1999; 30:48 - 60; http://dx.doi.org/10.1016/S0168-8278(99)80007-5; PMID: 9927150
- Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer 1999; 79:991 - 5; http://dx.doi.org/10.1038/sj.bjc.6690158; PMID: 10070902
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004; 4:71 - 8; http://dx.doi.org/10.1038/nrc1256; PMID: 14708027
- Nakamura T. Structure and function of hepatocyte growth factor. Prog Growth Factor Res 1991; 3:67 - 85; http://dx.doi.org/10.1016/0955-2235(91)90014-U; PMID: 1838014
- Noji S, Tashiro K, Koyama E, Nohno T, Ohyama K, Taniguchi S, et al. Expression of hepatocyte growth factor gene in endothelial and Kupffer cells of damaged rat livers, as revealed by in situ hybridization. Biochem Biophys Res Commun 1990; 173:42 - 7; http://dx.doi.org/10.1016/S0006-291X(05)81018-6; PMID: 2147853
- Koh SL, Ager E, Malcontenti-Wilson C, Muralidharan V, Christophi C. Blockade of the renin-angiotensin system improves the early stages of liver regeneration and liver function. J Surg Res 2013; 179:66 - 71; http://dx.doi.org/10.1016/j.jss.2012.09.007; PMID: 23110972
- Kuruppu D, Christophi C, Bertram JF, O’Brien PE. Characterization of an animal model of hepatic metastasis. J Gastroenterol Hepatol 1996; 11:26 - 32; http://dx.doi.org/10.1111/j.1440-1746.1996.tb00006.x; PMID: 8672738
- Ager EI, Wen SW, Chan J, Chong WW, Neo JH, Christophi C. Altered efficacy of AT1R-targeted treatment after spontaneous cancer cell-AT1R upregulation. BMC Cancer 2011; 11:274; http://dx.doi.org/10.1186/1471-2407-11-274; PMID: 21703011
- Hirano T, Ran J, Adachi M. Opposing actions of angiotensin II type 1 and 2 receptors on plasma cholesterol levels in rats. J Hypertens 2006; 24:103 - 8; http://dx.doi.org/10.1097/01.hjh.0000198030.30095.98; PMID: 16331107
- Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension 2005; 46:732 - 7; http://dx.doi.org/10.1161/01.HYP.0000182660.74266.6d; PMID: 16172434
- Hardonk MJ, Dijkhuis FW, Hulstaert CE, Koudstaal J. Heterogeneity of rat liver and spleen macrophages in gadolinium chloride-induced elimination and repopulation. J Leukoc Biol 1992; 52:296 - 302; PMID: 1522388
- Kono H, Fujii H, Asakawa M, Yamamoto M, Maki A, Matsuda M, et al. Functional heterogeneity of the kupffer cell population is involved in the mechanism of gadolinium chloride in rats administered endotoxin. J Surg Res 2002; 106:179 - 87; http://dx.doi.org/10.1006/jsre.2002.6434; PMID: 12127824
- Maher JM, Aleksunes LM, Dieter MZ, Tanaka Y, Peters JM, Manautou JE, et al. Nrf2- and PPAR alpha-mediated regulation of hepatic Mrp transporters after exposure to perfluorooctanoic acid and perfluorodecanoic acid. Toxicol Sci 2008; 106:319 - 28; http://dx.doi.org/10.1093/toxsci/kfn177; PMID: 18757529
- Mizgerd JP, Molina RM, Stearns RC, Brain JD, Warner AE. Gadolinium induces macrophage apoptosis. J Leukoc Biol 1996; 59:189 - 95; PMID: 8603991
- Ishizu K, Sunose N, Yamazaki K, Tsuruo T, Sadahiro S, Makuuchi H, et al. Development and characterization of a model of liver metastasis using human colon cancer HCT-116 cells. Biol Pharm Bull 2007; 30:1779 - 83; http://dx.doi.org/10.1248/bpb.30.1779; PMID: 17827739